


Abstract
The heterogeneous collection of nucleosome remodelling and deacetylation (NuRD) complexes can be grouped into the MBD2- or MBD3-containing complexes MBD2–NuRD and MBD3–NuRD. MBD2 is known to bind to methylated CpG sequences in vitro in contrast to MBD3. Although functional differences have been described, a direct comparison of MBD2 and MBD3 in respect to genome-wide binding and function has been lacking. Here, we show that MBD2–NuRD, in contrast to MBD3–NuRD, converts open chromatin with euchromatic histone modifications into tightly compacted chromatin with repressive histone marks. Genome-wide, a strong enrichment for MBD2 at methylated CpG sequences is found, whereas CpGs bound by MBD3 are devoid of methylation. MBD2-bound genes are generally lower expressed as compared with MBD3-bound genes. When depleting cells for MBD2, the MBD2-bound genes increase their activity, whereas MBD2 plus MBD3-bound genes reduce their activity. Most strikingly, MBD3 is enriched at active promoters, whereas MBD2 is bound at methylated promoters and enriched at exon sequences of active genes.
INTRODUCTION
The nucleosome remodelling and deacetylation (NuRD) complex harbours a multi-functional and highly conserved combination of chromatin-modifying activities. Through the MBD2 and MBD3 proteins, with their methyl-CpG-binding domains (MBD), the NuRD complex combines reading of DNA methylation marks with modifying histones [for recent reviews see (1,2)]. Through the combinatorial assembly of similar, paralogous versions of histone deacetylases, nucleosome-remodelling ATPases, metastasis-associated (MTA) factors and others, biological specificity of the NuRD complex is achieved during development, oncogenesis and cancer progression.
Specificity mediated by or associated with MBD2 and MBD3 has been documented on several levels. MBD2-knockout mice are viable and fertile with subtle defects only, whereas MBD3-knockout mice are embryonic lethal (3). On the molecular level, both proteins differ in respect to binding to methylated DNA. MBD2 is able to bind DNA with a 5-methylcytosine (5 mC) modification (4,5), in contrast to MBD3, which binds to 5-hydroxymethylcytosine (5 hmC), but not to 5 mC (4–7). Purification and analysis of NuRD complexes revealed that MBD2 and MBD3 are components of mutually exclusive NuRD complexes, MBD2–NuRD and MBD3–NuRD (8). Despite these and other differences, both factors have been shown to interact with GATAD2A/p66α and GATAD2B/p66β within the NuRD complex (9–11), as well as with CSB (Cockayne Syndrome Protein B) (12), HIC1 (Hypermethylated in Cancer 1) (13) and DOC-1 (Deleted in Oral Cancer 1) (14).
Testing of Hela cell promoter regions revealed preferential MBD2 binding near transcriptional start sites (TSS) (15). Genome-wide binding analysis of NuRD complexes in ES cells, which are primarily MBD3–NuRD complexes, revealed a broad binding near promoters, but with a gap at the TSS (16). In contrast, ChIPseq analysis of MBD3 in ES cells revealed a sharp occupancy at the TSS (7). In addition to these published differences for MBD3–NuRD binding distribution, a direct comparison between MBD2 and MBD3 on the whole genome level is missing.
Because of the increasing information for both functional differences as well as similarities, we wanted to know whether MBD2 and MBD3 differ in respect to chromatin modification and in genome-wide binding. Functional tests revealed a dramatic difference in that MBD2–NuRD, but not MBD3–NuRD, transformed euchromatin into repressed chromatin. Analysis of the genome-wide binding pattern of MBD2 and MBD3 within the same cell type showed a preference for MBD2 to be bound at methylated CpG islands and inactive promoters, whereas MBD3 was found at unmethylated CpG islands and active promoters. Most strikingly, exon sequences of active genes were enriched for MBD2 binding.
MATERIALS AND METHODS
Antibodies
Immunostaining was done using commercial antibodies recognizing Mi2 (Santa Cruz, sc-11378), RbAp46 (Santa Cruz, sc-8272), HDAC1 (Santa Cruz, sc-9397), MBD2 (Santa Cruz, sc-9397) and MBD3 (Santa Cruz, sc-9402). For ChIP H3K9ac (Abcam, 4441), H3K9me3 (Abcam, 8898), GFP (polyclonal rabbit antiserum was raised against full length GFP), MBD2a/b (Sigma Aldrich, M-7318), MBD3 (Bethyl Laboratories A302-528A), normal rabbit control IgG (Abcam, ab46540) antibodies and in addition V5-agarose (Sigma Aldrich, A7345) was used. GFP, MBD2a/b and MBD3 antibodies were used for Western Blot as well.
Luciferase assay
A10 cells were co-transfected with 1 µg of either GFP-LacI, GFP-LacI-VP16, GFP-LacI-MBD2a, GFP-LacI-MBD2b or GFP-LacI-MBD3 and βGAL-vector using TurboFect (Fermentas) in six-well plates. Cells were harvested 48 h after transfection and lysed with 300 µl lysis-buffer (25 mM Tris/HCl pH 7.5, 8 mM MgCl, 1 mM EDTA, 1% Triton X 100, 15% glycerin and freshly added 1 mM DTT) per well. Luciferase reporter activity was determined and corrected for βGAL expression.
Data deposition
The microarray and the ChIPseq data from this publication have been submitted to the GEO database as entry GSE41010.
Cell lines and transfection, Immunofluorescence analysis, Western Blot, Chromatin immunoprecipitation and ChIPseq analysis and bioinformatics analyses
RESULTS
The NuRD complex can be assembled on LacO repeats
To study possible MBD2- or MBD3-induced changes in chromatin compaction, we used the LacO/LacI system that uses 100s of LacO repeats integrated into single genomic loci (17). In many cases, genomic integration of the repeat cluster is found in heterochromatic regions. For such a situation, we used the F42B8 cell clone of U2OS cells, with a repeat cluster integrated close to the centromere (18). As a model for a euchromatic array, we used the RREB1 cell clone of CHO cells (19). In a first set of experiments, we wanted to know whether an MBD2 or MBD3 fusion with the DNA-binding domain of the Lac repressor LacI can be targeted to a LacO array, and whether such a bound protein recruits additional components of the NuRD complex. For this, we started with the heterochromatic array of the F42B8 cells, as the compact structure of such an array can be easily visualized and analysed by cytological methods. As a requirement for these experiments, we analysed the heterochromatic nature of the array to assure that it is lacking NuRD components. The array can be easily identified by expression of a GFP-LacI fusion (Supplementary Figure S1) and was found to be devoid of NuRD components. In contrast to the expression of GFP-LacI, both MBD fusions, GFP-LacI-MBD2b and GFP-LacI-MBD3, resulted in positive staining by immunofluorescence with antibodies against the NuRD components Mi2, HDAC1 and RbAp46 (Figure 1A). Thus, these NuRD factors were recruited by MBD2 or MBD3 targeted to the LacO array.
Figure 1.
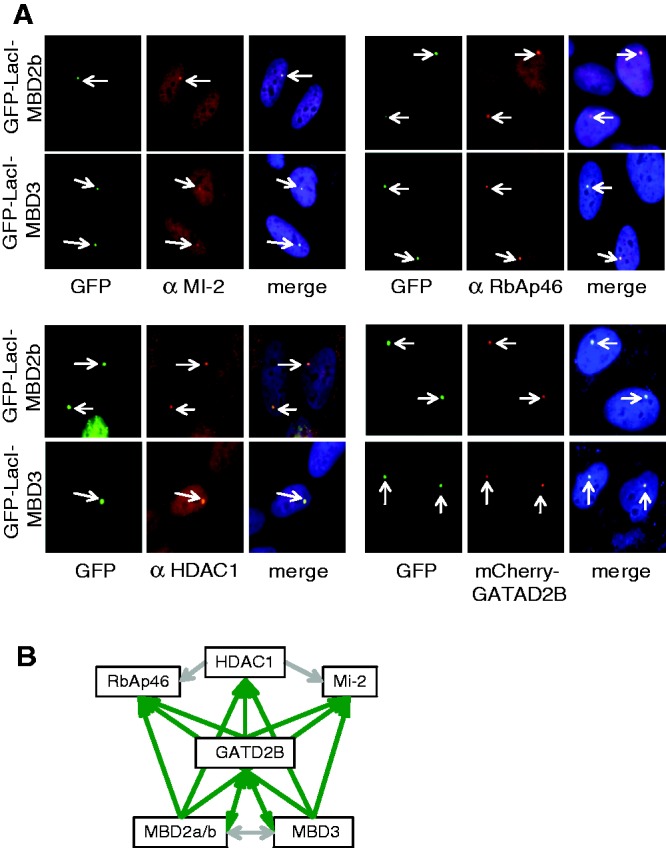
LacI-MBD2 or LacI-MBD3 assemble MBD2–NuRD or MBD3–NuRD on the LacO array. (A) F42B8 (U2OS) cells were transfected with GFP-LacI-MBD2b or GFP-LacI-MBD3. GFP or Cherry signals were directly determined, and indirect immunofluorescence was carried out with antibodies against Mi2 (αMi-2), RbAp46 (αRbAp46) and HDAC1 (αHDAC1). Merged colour images show DAPI staining in addition. (B) Schematic representation and summary of the assembly of the different NuRD components. Green arrows indicate recruitment of the components pointed out by the arrowhead, whereas the grey arrows indicate no assembly.
For MBD2, two isoforms are known, MBD2b and MBD2a, that contains an N-terminal extension. Within the assays described in this manuscript, there was no difference seen between using GFP-LacI-MBD2a or GFP-LacI-MBD2b (Supplementary Figure S2). Recruitment of GATAD2B was detected by co-expressing Cherry-GATAD2B (Figure 1Α). When using GATAD2B fused to GFP-LacI (GFP-LacI-GATAD2B), the NuRD components Mi2, HDAC1 and RbAp46 could be similarly recruited to the LacO array (Supplementary Figure S3). Furthermore, GFP-LacI-GATAD2B also recruited MBD2 and MBD3 to the array. This might potentially be a source for generating mixed complexes, i.e. MBD2 recruits GATAD2B, which in turn might recruit MBD3. To test for this potential problem, we co-expressed GFP-LacI-MBD2 with Cherry-MBD3 or GFP-LacI-MBD3 with Cherry-MBD2. These combinations clearly showed that binding of neither of the MBD factors resulted in recruitment of the paralogous factor to the array and that only homogeneous NuRD complexes were generated on the array (Supplementary Figure S4). In addition, we targeted HDAC1 to the LacO array by the use of GFP-LacI-HDAC1, but this did not result in assembly of the NuRD complex (Supplementary Figure S5). The results of these experiments are summarized in Figure 1B and demonstrate that the LacO array system is suitable to study NuRD. Furthermore, MBD2, MBD3 or GATAD2B can be used as starting component to assemble the NuRD complex. When targeting MBD2 to the DNA, only MBD2–NuRD is assembled, as is the case for MBD3, which assembles solely MBD3–NuRD.
In contrast to MBD3, MBD2 converts euchromatin into repressed chromatin
To study the repressive function of NuRD, we used the above LacO/LacI targeting system within a euchromatin background. This was provided by the RREB1 cell clone (19). NuRD complex assembly was similarly possible as with the heterochromatin array of the F42B8 cells above (Supplementary Figure S6). After transfection of GFP-LacI, the expanded array can be visualized. Upon expression of GFP-LacI-MBD2, the expanded array dramatically shrinks to a small spot similarly as seen for the heterochromatin array of the F42B8 cells. This array compression is observed when targeting MBD2a or MBD2b to the LacO array (Figure 2), but is not seen when GFP-LacI-MBD3 is expressed. Similarly, the array remains expanded when expressing GFP-LacI-HDAC1, a factor that is not able to assemble the NuRD complex on the LacO array (Figure 1B). If array binding, and therefore compaction, was indeed caused by the DNA-bound MBD2–NuRD complex, recruitment should be dependent on the LacI DNA-binding domain. Indeed, the deleted DNA-binding domain in construct GFP-ΔLacI-MBD2b resulted in a nuclear GFP staining not localized to the LacO array (Supplementary Figure S7). We wanted to test whether the compaction of the chromatin array is marked by the chromatin modification H3K9me3, which is specific for inactive and repressed chromatin. Therefore, we purified chromatin for immune-precipitation with antibodies specific for H3K9me3 or H3K9ac. The ratio of these precipitations is a mark for active or for repressed chromatin. In contrast to cells transfected with GFP-LacI, expression of GFP-LacI-MBD2a resulted in a strong increase of the repressive mark and a similarly strong decrease of the active mark. In contrast, expression of GFP-LacI-MBD3 induced a lower decrease in H3K9ac and a lower increase in H3K9me3 (Supplementary Figure S8). This low, but reproducible, effect of MBD3 in the absence of chromatin compaction may indicate that these modification changes are not sufficient to compact chromatin as is the case with MBD2.
Figure 2.
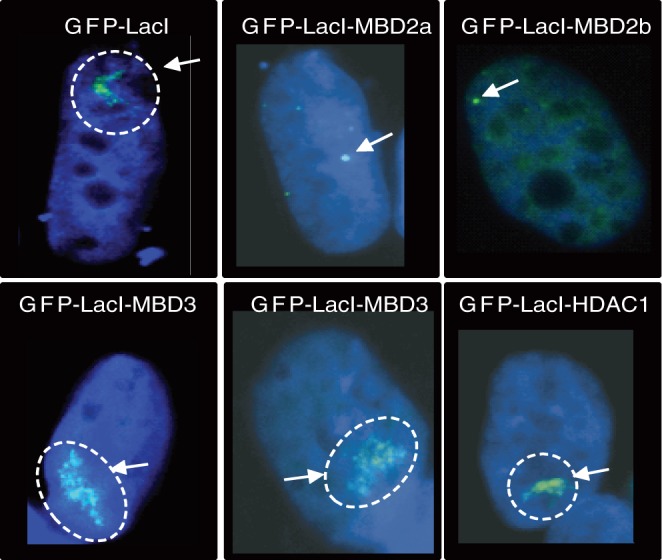
In contrast to MBD3 and to other NuRD components, LacI-MBD2a or LacI-MBD2b dramatically compact the euchromatic LacO array. RREB1 cells were transfected with GFP-LacI, GFP-LacI-MBD2a/b, GFP-LacI-MBD3 and GFP-LacI-HDAC1 as indicated. Samples were visualized for DNA (blue) and GFP (green). The arrows point to the compact array or to the expanded array (dotted circle).
To study the relationship between the chromatin status and gene activity, we generated HeLa cell clones containing an integrated luciferase reporter gene. The luciferase gene is controlled by seven copies of the LacO sequence, such that any LacI fusion protein (Figure 3A) can be tested for its regulatory capacity. We selected a single clone (A10), from several different cell clones based on the robust ground state of luciferase activity, which was expected to be further increased by an activator or to be reduced by a repressor. Indeed, transient control transfection with an expression plasmid coding for a GFP-LacI-VP16 fusion resulted in a 8-fold increase of luciferase activity (Figure 3C). Expression of GFP-LacI-MBD2a, GFP-LacI-MBD2b or GFP-LacI-MBD3 in clone A10 led to comparable amounts of the respective fusion protein (Figure 3B). Preparing these cells for ChIP analysis with an antibody directed against GFP allowed for the detection of the GFP-LacI-MBD fusions bound at the LacO repeats (Figure 3D). Luciferase gene activity was determined without transfection or after expression of GFP-LacI or of the MBD fusion factors (Figure 3C). Expression of either MBD2a or MBD2b led to a ∼10-fold reduction of luciferase activity, in contrast to the GFP-LacI control or to GFP-LacI-MBD3.
Figure 3.

In contrast to GFP-LacI-MBD3, GFP-LacI-MBD2a or GFP-LacI-MBD2b repress the euchromatic LacO promoter. (A) Illustration of the reporter-gene including seven LacI-binding sites (LacO-repeats). Reporter construct was used to generate stable reporter cell clones in HeLa cells. (B) Western Blot analysis of the indicated GFP-LacI-fusion proteins expressed in the stable A10 clone. GAPDH antibody was used as loading control. (C) The A10 clone was transiently transfected with either GFP-LacI, GFP-LacI-VP16, GFP-LacI-MBD2a, GFP-LacI-MBD2b or GFP-LacI-MBD3. Gene activity is expressed as fold change relative to the untransfected A10 clone. (D) Binding analysis by ChIP-qPCR was performed after transfection of the indicated constructs using a GFP antibody and expressed relative to untransfected cells. (E, F) GFP-LacI-MBD2a and GFP-LacI-MBD2b change the H3K9me3/H3K9ac ratio after binding to the reporter. ChIP-qPCR was performed with antibodies recognizing H3K9ac (E) and H3K9me3 (F) and expressed relative to input. Error bars indicate the standard error of three individual replicates.
Clearly, MBD2 mediates strong chromatin compaction and transcriptional repression, whereas MBD3 does not. Therefore, we determined the chromatin modification at the reporter gene with antibodies against H3K9me and against H3K9ac. The ratio of methylated K9 versus acetylated K9 is an indication for repressed chromatin. GFP-LacI-MBD2a and GFP-LacI-MBD2b expression resulted in an increased repression ratio in contrast to GFP-LacI-MBD3, which did not change the repression ratio as compared with the GFP-LacI control expression (Figure 3E and F).
Both MBD2 or MBD3 bind to similar sequence classes
Because of the functional differences of MBD2 and MBD3, we wanted to know whether both factors are targeted to factor-specific sites throughout the genome. Such an analysis has not yet been performed within a single cell type. The quality and specificity of available antibodies directed against these two proteins is a limiting factor in carrying out chromatin precipitation experiments. Therefore, we generated MBD versions fused to the V5 domain. This domain has been shown to be a universal and optimal tag for ChIPchip or ChIPseq analysis (20).
Expression of V5, V5-MBD2b or V5-MBD3 in HeLa cells did not alter cell proliferation. Western-blot detection of the tagged MBD factors revealed similar amounts of proteins expressed from the different constructs (Supplementary Figure S9A). Therefore, we generated chromatin from HeLa cells after expression of these three constructs and purified V5 bound DNA for ChIPseq. We identified regions of significant binding for MBD2 and MBD3 by comparing precipitations from cells transfected with V5-MBD2 and V5-MBD3 with those from cells transfected with the vector expressing the V5 tag only. We used PeakRanger to call peaks with a P-value <10–5 at an FDR <5%. Using this approach, we were able to detect ∼8200 MBD2- and 490 MBD3-binding regions. Despite this difference in the number of binding sites, both factors were found to bind to similar sequence classes. The peak distribution indicates highly specific binding to transcriptional start sites including upstream sequences (Figure 4A and B). Furthermore, exon sequences are highly enriched including transcriptional end sites. In contrast, intron sequences are underrepresented as compared with the total genomic sequence distribution (Figure 4A and B). In contrast to sequences associated with genes, intergenic sequences are almost depleted from either MBD2 or MBD3 binding (Figure 4B).
Figure 4.

MBD2 and MBD3 bind similar sequence classes. (A) MBD2 and MBD3 distribution of significantly bound regions in comparison with the distribution of genomic features (TSS, TSS upstream, TES, exon, intron, intergenic). (B) Peaks of MBD2 and MBD3 binding are enriched at exons and TSS, but not at intergenic regions and introns. Enrichments are calculated relative to the frequency of these features in the genome. (C) MBD2 and MBD3 bind to TSS. Plotted is the average profile of V5-MBD2, V5-MBD3 or V5 read coverage in a 6000 bp window around the TSS of all RefSeq genes. (D) MBD3 sites are often co-occupied by MBD2. Plots illustrate average V5 binding (left panel), V5-MBD2 (center panel) and V5-MBD3 binding (right panel) over MBD2 peaks (orange) and MBD3 peaks (blue).
Binding specificity was verified at positive and negative sites (Supplementary Figure S9B). To confirm that MBD2- or MBD3-binding sites are represented by the V5-tagged MBD proteins, we used the host HeLa cells without transfection and immune-precipitated chromatin with antibodies directed against MBD2 or MBD3. Although the specificity and efficiency of these antibodies was low, we could confirm MBD2 and MBD3 binding even without expression of the tagged protein (Supplementary Figure S10).
Binding to the transcriptional start sites was highly position specific with a sharp peak at the TSS (Figure 4C). Because of the lower number of MBD3-bound sites, the binding peak at the TSS was less pronounced. No specific enrichment of promoter DNA could be detected in the ChIPseq data from cells transfected with the empty V5 vector.
Because similar sequence classes are bound by both MBD factors, we tested for a possible overlap of binding sites. We determined the cumulative binding profiles for MBD2 and MBD3 across MBD2 and MBD3 peak regions. As expected MBD2 and MBD3 binding can be detected across the respective peak regions (Figure 4D). Interestingly, we are able to detect strong MBD2 binding even across MBD3 peak regions (Figure 4D, central panel; for genome-browser examples see Supplementary Figure S11), although the analysis of overlapping peaks suggested only moderate overlap between MBD2 and MBD3 peak ranges (Figure 5A). In contrast, we cannot detect much MBD3 binding across MBD2 peak regions (Figure 4D, right panel).
Figure 5.
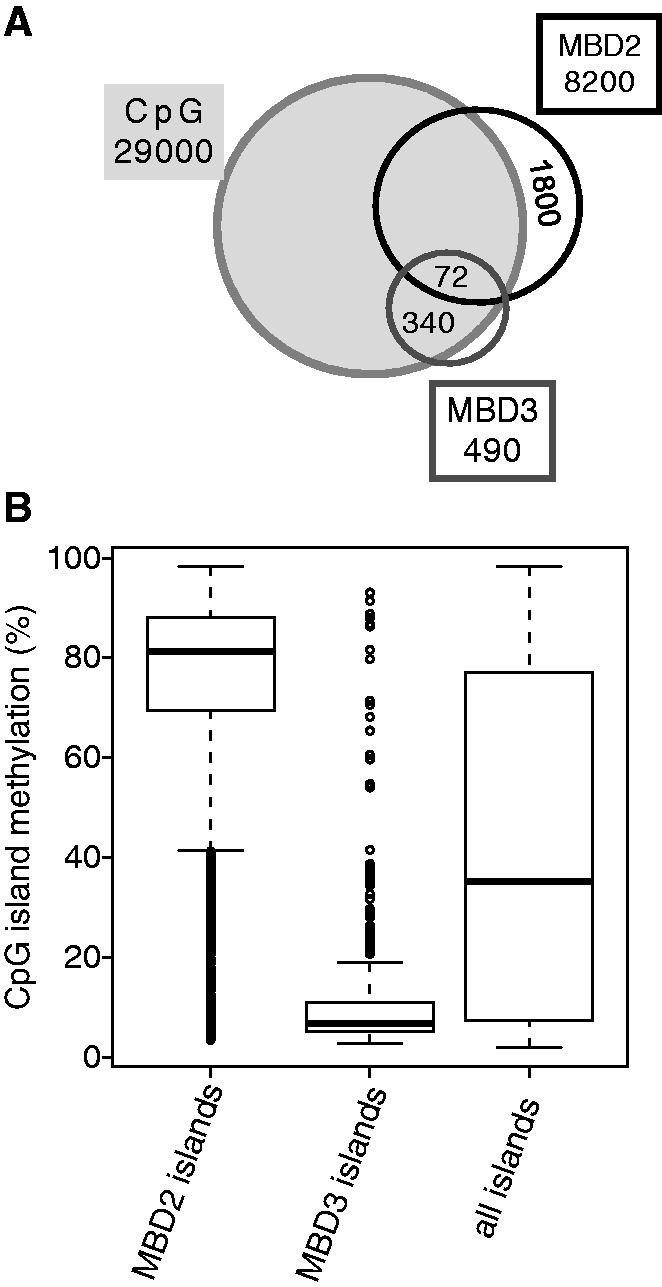
MBD2 sites are highly enriched for methylated DNA, whereas MBD3 sites are devoid of DNA methylation. (A) Venn diagram shows overlap of MBD2- and MBD3-binding regions with CpG islands. (B) Average relative (percentage) CpG methylation level within CpG islands associated with MBD2, MBD3 or of all CpG islands [island definition from (21)]. MBD2-associated islands show high methylation, whereas MBD3-associated islands are devoid of methylation as compared with the average island methylation [Wilcoxon signed-rank test: P = 0 (MBD2) and P = 3.5 × 10−57(MBD3)].
Thus, overall binding of MBD2 or MBD3 is targeted to similar sequence classes and MBD3 sites are often marked by robust MBD2 binding.
MBD2 sites are highly enriched for methylated DNA, whereas MBD3 sites are devoid of methylation
Despite the similarity of the MBD domains of MBD2 and MBD3, it has been shown that only MBD2 is able to bind to methylated CpG islands in vitro (4). The genome-wide methylation pattern of HeLa cells is known [GSE40699 (22)]. CpG dinucleotides are underrepresented in the genome and occur in two classes, dispersed throughout the genome and clustered within CpG islands (23). Both MBD2- and MBD3-binding sites are highly enriched for CpG islands (Figure 5A). Seventy-nine per cent of the MBD2 peaks are overlapping with CpG islands and in case of MBD3, 69% are overlapping. The MBD2 overlap with CpG islands involves a sub-fraction (22%) of the CpG islands only. Taking into account that a sub-fraction of CpG islands in HeLa cells is methylated, we determined binding of both MBD factors to methylated CpG islands. For MBD2, a high percentage of the MBD2-bound CpG islands were methylated, whereas for MBD3, <10% of the bound CpG islands were methylated (Figure 5B).
Thus, although both factors bind to similar sequence elements such as TSS and CpG islands, their binding differs dramatically in respect to CpG methylation.
MBD2 is associated with repressed genes and MBD3 with activated genes
Because we have seen the striking difference between MBD2 and MBD3 in mediating reporter-gene repression and chromatin compaction, we compared MBD2 and MBD3 promoter binding in respect to gene activity. Indeed, MBD2 is bound to promoters of genes with low transcriptional activity, whereas MBD3-bound promoters show a 4-fold higher gene activity, as determined from the mean values (Figure 6A). When comparing these differences with the activity of all genes in HeLa cells, MBD2 was found to be associated with genes with lower transcriptional activity as compared with all genes. In contrast, gene activity of MBD3-associated genes was higher than the mean activity of all genes (Figure 6A).
Figure 6.
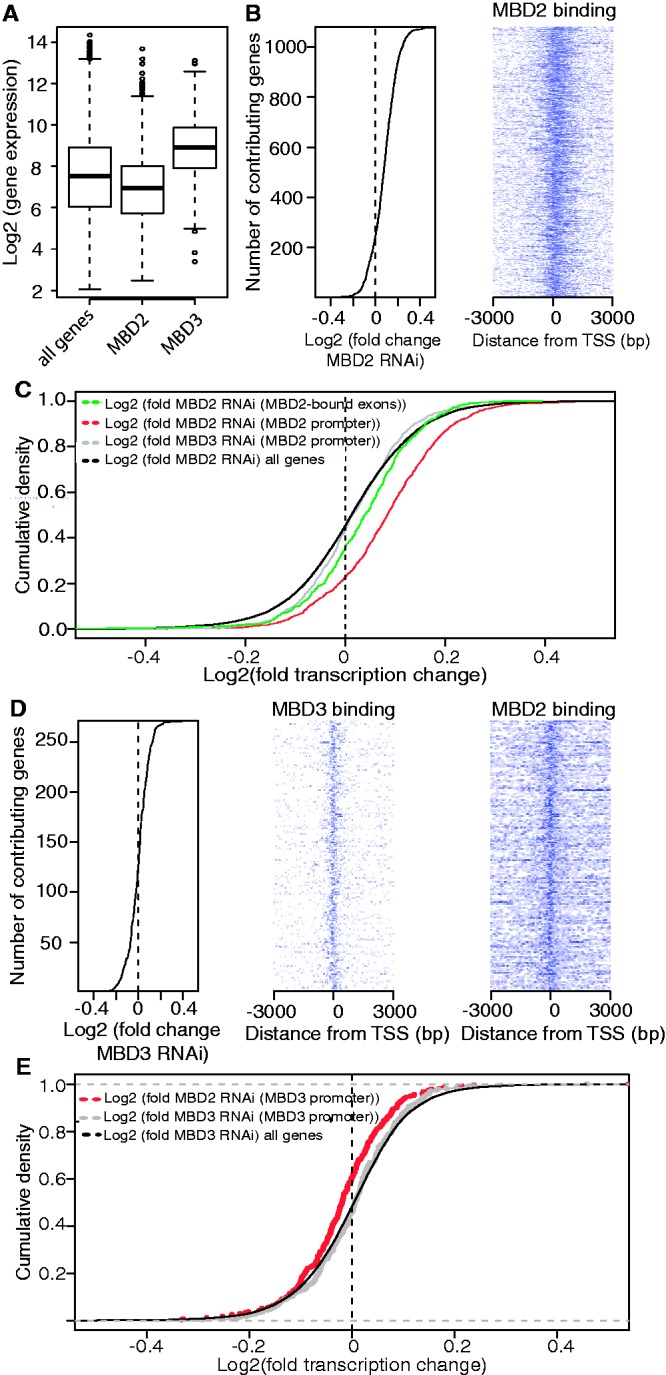
MBD2 binding to promoters is associated with gene repression and combined MBD2/MBD3 binding with gene activation. (A) MBD3-bound genes are significantly higher expressed than MBD2-bound genes. Gene expression in HeLa cells as measured by Affymetrix Gene 1.0 ST arrays [Wilcoxon signed rank test: P = 2 × 10−28 (MBD2) and P = 8 × 10−33(MBD3)]. (B) Analysis of the expression level of 1080 MBD2-bound genes sorted by fold change after MBD2 knock down (kd) in HeLa cells (left panel). Density map of ChIPseq data of MBD2 at these promoters shows MBD2 binding around the TSS (right panel). (C) MBD2 represses transcription on MBD2 targets: Cumulative density plot of gene expression changes after MBD2 kd (red) or MBD3 kd (grey) on MBD2-bound promoters, and after MBD2 kd on MBD2-bound exons (green) in HeLa cells in comparison with the expression of all genes after MBD2 kd (black) (Wilcoxon signed rank test: P = 7 × 10−76). (D) MBD3-knockdown results in gene repression. Analysis of the expression level of 271 MBD3-bound genes sorted by fold change after MBD3 kd in HeLa cells (left panel). Density map of ChIPseq data from MBD3-bound promoters shows MBD3 binding around the TSS (middle panel) as well as binding of MBD2 (right panel). (E) Co-localization of MBD2 and MBD3 at TSS mediates gene activation: Cumulative density plot depicts gene expression changes after MBD2 kd (red) or MBD3 kd (grey) on MBD3-bound promoters in HeLa cells in relation to the expression changes after MBD3 knockdown on all genes (black) (Wilcoxon signed rank test: P = 6 × 10−5).
To gain insights into the MBD2 or MBD3 function in the context of the endogenous gene repertoire, we depleted HeLa cells by RNAi against either MBD protein or as a negative control with control siRNA, determined the respective expression profiles on the Affymetrix GeneChip® Gene 1.0 ST Array platform and verified expression changes at individual genes (Supplementary Figures S12 and S13). In almost all cases, when well-studied repressors have been depleted, the expression profiles of target genes not only increased, but also a substantial number of genes decreased their expression. Similarly, depletion of well-characterized activators resulted in decreased as well as in increased gene activity (e.g. data compiled at http://www.ncbi.nlm.nih.gov/geo/). One of several reasons for this is an indirect effect on genes that do not harbour a binding site for the respective factor. Therefore, we focussed our view on 1080 genes with MBD2 binding within the promoter region (Figure 6B). The majority of these genes show a mild change in gene activity towards activation. In comparing MBD2 depletion with MBD3 depletion of MBD2-bound genes, an increase in gene activity of >75% is seen after MBD2 RNAi (red curve in Figure 6C shifted to the right). Because the expression changes are rather low, we wanted to determine the significance of this observation. Therefore, we determined the expression change of all genes, irrespective of a MBD2-binding site (Figure 6C) and found that 50% of these are either induced or repressed. This difference between MBD2-bound genes and all genes was significant, with a P value of 7 × 10–76. Expression changes after MBD3 depletion of MBD2-bound genes resulted in equal gene numbers repressed or induced. Similarly, we analysed genes with MBD2 binding to exon sequences (see below) and found that these genes responded with a slight shift towards activation upon MBD2 depletion (Figure 6C green line). Similar results were achieved, when we used the gene set enrichment analysis (GSEA; Supplementary Figure S14). Thus, again MBD2 functions primarily as a repressor at MBD2-bound genes.
Because MBD3 is found at non-methylated DNA (see above), we wanted to test for a possible functional difference of both MBD factors by analysing gene expression changes of 271 MBD3-bound genes upon MBD3 depletion. Small changes were found, resulting in both slightly increased or decreased activity of genes (Figure 6D). The binding analysis of MBD3-bound promoters shows a significant number of cases with MBD2 binding as well (heatmap in Figure 6D). In contrast to the whole genome binding analysis, where we applied a stringent peak-calling algorithm (compare Figure 5A), here within this small set of sites, we applied a different peak-calling algorithm (Macs) with less stringent settings. This revealed ∼140 of MBD3-bound promoters to be significantly associated with MBD2 peaks as well. Therefore, we asked for the effect of MBD2 depletion on the activity of these genes. For these, we found expression changes towards repression (red curve in Figure 6E shifted to the left). The difference in expression change after MBD2 depletion at MBD2 plus MBD3-bound genes and expression change of all genes after MBD3 depletion (black curve in Figure 6E) is significant (P < 6 × 10−5).
Therefore, we can conclude that MBD2 primarily causes gene repression, whereas MBD2 in the context of MBD3 promoter binding might be involved in gene activation.
In contrast to MBD2, MBD3 is associated with chromatin modification characteristic for active chromatin
To identify additional chromatin features MBD2 and MBD3 might be associated with, we compared the binding sites with known features of histone modification in HeLa cells. First, we identified that MBD3-bound sites are generally associated with open chromatin as indicated by the precise peaks in the FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) (24) and DNaseI profiles. In contrast, MBD2 sites are not in an open conformation.
Additionally, we found for MBD3 a correlation with active promoter marks H3K4me2, H3K4me3 and H3K9ac at MBD3-binding sites, which is in line with MBD3 binding to promoters of actively transcribed genes (Figure 6A) and to non-methylated CpG islands (Figure 5B), which often are found at active promoters. Indeed, MBD3 binding clearly correlated with these active promoter marks (Figure 7) with the characteristic ‘dip’ of the respective marks, since transcriptional start sites and other regulatory sites are depleted from nucleosomes. In general, sites occupied by both MBD factors (grey line in Figure 7) show similar features as the MBD3 sites. This binding is in stark contrast to MBD2, which does not show any correlation with these marks. A similar difference for the two MBD factors is seen with polymerase II binding at MBD3 sites, but not at MBD2-bound regions. Contrary, testing specifically for marks associated with silent chromatin, such as H3K9me3, we do not find a specific enrichment at MBD2 sites.
Figure 7.

In contrast to MBD2, MBD3 is associated with chromatin modifications characteristic for active genes. Plots show the average localization of the indicated chromatin modification around MBD2 (dashed line) or MBD3 (black) peaks or at sites occupied by both factors (grey). Whereas MBD2 shows no specific association with the analysed chromatin marks, MBD3 peaks are highly enriched for active chromatin marks (DNaseI, FAIRE, H3K4me3, H3K9Ac).
Because we have seen for both, MBD2 and MBD3, an enrichment over gene and exon sequences (Figure 4A and B), we wanted to compare the binding profiles with chromatin marks characteristic for the gene body and for transcriptional elongation, such as H3K36me3 and H3K79me2 (25–27). These showed a preference for MBD3 binding, but with a wide gap at the MBD3-binding site (Figure 7). This pattern is best explained by MBD3 binding to promoters, whereas the H3K36me3 and the H3K79me2 signals are known to mark the gene body with H3K79me2 peaking within the first kb of the transcribed region (28). Because these plots are compiling genes in both orientations, the increase for these two marks is seen on both sides of the MBD3-binding site.
We can conclude that chromatin modifications associated with MBD2 or MBD3 binding is in line with the LacO array studies, with reporter gene activity and with the response of endogenous gene activity upon depletion of either MBD factor.
MBD2 marks specifically exon sequences
Overall sequence class enrichment for MBD2 or MBD3 binding revealed that, among others, exon sequences were targeted (see Figure 4A and B). Therefore, we compiled the MBD2- and MBD3-binding sites at gene regions to test for enriched exon sequences in comparison with intron sequences. An instructive method is to plot binding sites over a meta-gene. Therefore we plotted the average MBD2/3 occupancy across 5′-upstream, first, internal and last exons as well as 3′-downstream regions according to their position relative to all RefSeq transcripts. (Figure 8A). With this analysis, a clear correlation for MBD2 with exon sequences was evident, whereas MBD3 was primarily detected at the promoter region and overlapping into the first exon. Control ChIPseq using the V5 tag resulted in only a marginal preference for the promoter region. The last exon, which is often not translated to a varying extend, depicts a decline of MBD2 binding from the 5′end towards the transcriptional end site. Such a finding is in line with results on DNA methylation that promoter regions of inactive genes may be methylated, whereas methylation of active genes is found in gene bodies (29,30). To verify the MBD2-binding pattern over the meta gene and to test for a dependency on transcriptional activity, we grouped the MBD2-binding sites into those binding to genes with low or with high transcriptional activity. Again, the preference for exons is evident with a stronger enrichment at the promoter and first exon at genes with low or no transcriptional activity, whereas the active genes show reduced binding to the promoter and first exon, but increased binding to internal exons (Figure 8B).
Figure 8.
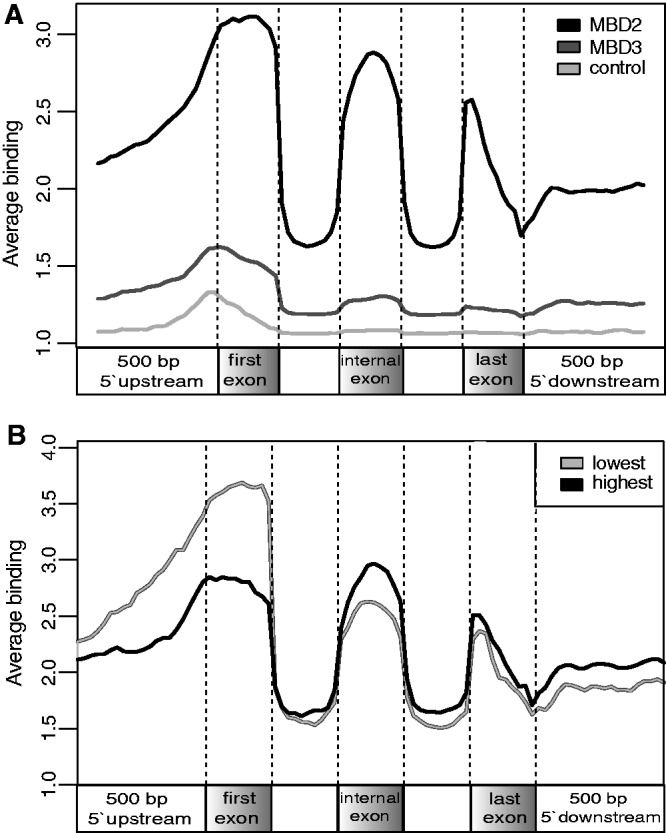
MBD2 specifically marks exon sequences. (A) MBD2 and MBD3 occupancy plotted as average of all sequencing reads across the following intervals: 500 bp upstream, first exon, all internal exons compiled as a single exon, last exon and 500 bp downstream according to their relative positions within the transcripts. First exon, internal exons and last exon were partitioned into 10 bins. 500 bp upstream and 500 bp downstream were partitioned into 20 bins. (B) MBD2 binding in respect to its association with low (inactive) and highly expressed genes (active). Highly expressed genes have lower MBD2 binding in the promoter in contrast to higher binding across internal exons.
Thus, we can conclude that MBD2 shows an exon specificity within active genes.
DISCUSSION
MBD2 and MBD3 have been analysed extensively for possible functional differences and similarities. Striking differences are embryonic lethality for MBD3-knockout mice (3) and differential expression of MBD3, but not MBD2, found in ES cells (16). Similarity between these two factors is given by their amino acid similarity (sequence is 70% identical), and indeed, earlier work suggested that both MBD factors are components of the same complex such that MBD2 recruits MBD3 complexes to methylated DNA (3). In contrast, complex purification revealed that both MBD proteins are incorporated into similar, but separable, complexes MBD2–NuRD and MBD3–NuRD (8). Furthermore, except for ES cells, many cell types seem to express both factors (http://genome.csdb.cn/cgi-bin/hgTrackUi?hgsid=1142799&c=chrX&g=gnfAtlas2). Despite the importance of both MBD factors to read and to modify epigenetic marks, a direct comparison of functional mechanisms and of target sites has been missing.
In general, the NuRD complex with its histone deacetylase activity is known as a factor that mediates gene repression [for recent reviews see (2,31,32)]. Nevertheless, when comparing MBD2 with MBD3 in their activities to effect reporter gene activity and chromatin modification, we could show that only MBD2 mediates repression and chromatin compaction. This is in line with the identification of a trans-repression domain within MBD2 (33).
The MBD has been defined by its ability to bind methylated CpG sequences. This was found for MBD2 in contrast to MBD3 (4,5). Because heterochromatic compaction of chromatin and DNA methylation correlate (34,35), the most direct explanation of such a correlation would be that the ‘reader’ of CpG methylation induces chromatin compaction. Binding of MBD2 to methylated DNA has been well established by in vitro experiments (4,5). In vivo, sequence-specific targeting of MBD2 to methylated CpGs has been tested (15). The authors focussed on 25 000 promoters in HeLa cells and found that MBD2 localized to the fraction of promoters characterized by DNA methylation. Here, we extended this analysis by testing the whole HeLa genome for MBD2 binding. In fact, we find MBD2 binding to be highly correlated with methylated CpG sequences, irrespective of their location within the gene body or within methylated promoter regions. Thus, MBD2 seems to be the ‘reader’ for many of the methylated CpGs. Therefore, it is interesting to note that MBD2 is also able to dramatically compact a widely expanded euchromatic LacO array such that MBD2 may also be the mediator of chromatin compaction at methylated DNA sequences.
In addition to methylated DNA binding and chromatin compaction, a functional effect on gene activity is observed. Previous studies (36) are in agreement with our observation that depletion of MBD2 results in changes of gene expression characterized by a release from repression. A more direct repressive role by MBD2 is evident from our binding site analysis that shows a preference for MBD2 to be associated with genes of low activity as compared with the mean activity of all genes. Therefore, we focussed on these genes bound by MBD2 and could find gene activation upon MBD2 depletion, supporting the repressive role of MBD2 bound to methylated CpGs.
For MBD3, a repressive function has been postulated and has also been documented in a few cases. For example, PML-RARa binds and recruits MBD3–NuRD to target genes, thereby facilitating Polycomb binding and H3K27 methylation (37). Recently, deacetylation of H3K27 by MBD3 could be shown, which subsequently causes H3K27 trimethylation (16). Nevertheless, when depleting mouse ES cells from MBD3, a majority of responsive genes are down-regulated (16), indicating that a potential activation function is associated with MBD3. This is in line with our HeLa cell analysis showing that genome-wide binding of MBD3 is associated with genes showing a significantly higher expression as compared with the mean value of the expression of all genes. This non-repressive activity of MBD3 is further supported by our finding that MBD3 neither causes chromatin compaction nor does it repress reporter gene activity. In general, we find MBD3 binding to non-methylated CpGs, i.e. active promoters, which often show MBD2 binding as well. This is the small fraction of MBD2 sites that are not methylated. In these cases, depletion of MBD3 activates as well represses gene activity, whereas depletion of MBD2 causes a small down-regulation of the associated genes. A dual role of a repressor complex in gene repression as well as in gene activation is often triggered by the neighbouring chromatin features. In case of MBD3, repression may require synergizing repressive factors, which may be absent in TSS regions bound by MBD3.
Overall, both MBD factors bind similar sequence classes, which might support earlier results on MBD2 recruiting MBD3 complexes (3), but the differences arguing for separate complexes are prevailing. First, there is an enrichment of promoter sequences, with MBD2 binding to methylated and MBD3 binding to unmethylated promoters. Previously, the analysis of MBD3 or of MBD3–NuRD revealed differing results concerning binding to promoters in ES cells (7,16,38,39). As discussed previously (40), distribution of the NuRD complex was identified in broad regions with a depletion at the TSS, or specifically binding at the TSS. Here, we show that within a single cell type, both MBD2 and MBD3 are found at the TSS tailing into the first exon.
Most striking is the binding to gene bodies and exon sequences. In general, we found MBD2 and MBD3 binding at exon sequences. However the absolute number of cases with exon binding by MBD2 was much higher and therefore we analysed these in detail. Primarily internal (translated) exons are enriched for MBD2 binding. The terminal exon with varying length of the open reading frame shows MBD2 binding enriched within the 5′-region. Previously, CpG methylation has been seen to be enriched in the exons of active genes (41–43). This enrichment is further specified by the fact that non-coding exons are hypomethylated, whereas internal and last exons are more frequently methylated (29,44). This methylation pattern reflects the MBD2 distribution within the gene bodies, with declining MBD2 binding towards the 3′-end of the last exon, which usually is not translated. Furthermore, we find a correlation of MBD2 binding to internal exons and gene activity. It has been suggested that this methylation pattern may foreshadow the content of mature mRNA (41). A functional connection between DNA methylation and splicing has been recently demonstrated at the CD45 gene. In this case, methylation inhibits binding of CTCF, which influences the splicing reaction (45). Here we are extending this idea that MBD2 binding may affect splicing, although formally, the reverse may be true as well in that the transcribing polymerase/splicing complex may determine CpG methylation and MBD2 binding.
Thus, what has been generally called the NuRD repressor complex turns out to be associated with a diverse set of functions, such as repression, activation and potentially connecting chromatin with splicing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–14, Supplementary Methods and Supplementary References [21,46–54]
FUNDING
Deutsche Forschungsgemeinschaft DFG [Re433/20 and IRTG 1384]. Funding for open access charge: Deutsche Forschungsgemeinschaft.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Helmut Dotzlaw and Reinhard Dammann for carefully reading the manuscript. Additionally, we would like to thank Nadine Müller, Sonja Sahner and Sylvia Thomas for technical assistance.
REFERENCES
- 1.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat. Rev. Cancer. 2011;11:588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hendrich B, Guy J, Ramsahoye B, Wilson VA, Bird A. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 2001;15:710–723. doi: 10.1101/gad.194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–1935. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saito M, Ishikawa F. The mCpG-binding domain of human MBD3 does not bind to mCpG but interacts with NuRD/Mi2 components HDAC1 and MTA2. J. Biol. Chem. 2002;277:35434–35439. doi: 10.1074/jbc.M203455200. [DOI] [PubMed] [Google Scholar]
- 7.Yildirim O, Li R, Hung J-H, Chen PB, Dong X, Ee L-S, Weng Z, Rando OJ, Fazzio TG. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147:1498–1510. doi: 10.1016/j.cell.2011.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WA, Cohen A, Lasonder E, Stunnenberg HG. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol. Cell. Biol. 2006;26:843–851. doi: 10.1128/MCB.26.3.843-851.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brackertz M, Boeke J, Zhang R, Renkawitz R. Two highly related p66 proteins comprise a new family of potent transcriptional repressors interacting with MBD2 and MBD3. J. Biol. Chem. 2002;277:40958–40966. doi: 10.1074/jbc.M207467200. [DOI] [PubMed] [Google Scholar]
- 10.Feng Q, Cao R, Xia L, Erdjument-Bromage H, Tempst P, Zhang Y. Identification and functional characterization of the p66/p68 components of the MeCP1 complex. Mol. Cell. Biol. 2002;22:536–546. doi: 10.1128/MCB.22.2.536-546.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gnanapragasam MN, Scarsdale JN, Amaya ML, Webb HD, Desai MA, Walavalkar NM, Wang SZ, Zu Zhu S, Ginder GD, Williams DC. p66Alpha-MBD2 coiled-coil interaction and recruitment of Mi-2 are critical for globin gene silencing by the MBD2-NuRD complex. Proc. Natl Acad. Sci. USA. 2011;108:7487–7492. doi: 10.1073/pnas.1015341108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie W, Ling T, Zhou Y, Feng W, Zhu Q, Stunnenberg HG, Grummt I, Tao W. The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc. Natl Acad. Sci. USA. 2012;109:8161–8166. doi: 10.1073/pnas.1201262109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Rechem C, Boulay G, Pinte S, Stankovic-Valentin N, Guérardel C, Leprince D. Differential regulation of HIC1 target genes by CtBP and NuRD, via an acetylation/SUMOylation switch, in quiescent versus proliferating cells. Mol. Cell. Biol. 2010;30:4045–4059. doi: 10.1128/MCB.00582-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spruijt CG, Bartels SJJ, Brinkman AB, Tjeertes JV, Poser I, Stunnenberg HG, Vermeulen M. CDK2AP1/DOC-1 is a bona fide subunit of the Mi-2/NuRD complex. Mol. BioSyst. 2010;6:1700–1706. doi: 10.1039/c004108d. [DOI] [PubMed] [Google Scholar]
- 15.Chatagnon A, Perriaud L, Nazaret N, Croze S, Benhattar J, Lachuer J, Dante R. Preferential binding of the methyl-CpG binding domain protein 2 at methylated transcriptional start site regions. Epigenetics. 2011;6:1295–1307. doi: 10.4161/epi.6.11.17875. [DOI] [PubMed] [Google Scholar]
- 16.Reynolds N, Salmon-Divon M, Dvinge H, Hynes-Allen A, Balasooriya G, Leaford D, Behrens A, Bertone P, Hendrich B. NuRD-mediated deacetylation of H3K27 facilitates recruitment of polycomb repressive complex 2 to direct gene repression. EMBO J. 2012;31:593–605. doi: 10.1038/emboj.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinett CC, Straight A, Li G, Willhelm C, Sudlow G, Murray A, Belmont AS. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol. 1996;135:1685–1700. doi: 10.1083/jcb.135.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jegou T, Chung I, Heuvelman G, Wachsmuth M, Gorisch SM, Greulich-Bode KM, Boukamp P, Lichter P, Rippe K. Dynamics of telomeres and promyelocytic leukemia nuclear bodies in a telomerase-negative human cell line. Mol. Biol. Cell. 2009;20:2070–2082. doi: 10.1091/mbc.E08-02-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verschure PJ, van der Kraan I, de Leeuw W, van der Vlag J, Carpenter AE, Belmont AS, van Driel R. In vivo HP1 targeting causes large-scale chromatin condensation and enhanced histone lysine methylation. Mol. Cell. Biol. 2005;25:4552–4564. doi: 10.1128/MCB.25.11.4552-4564.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolodziej KE, Pourfarzad F, de Boer E, Krpic S, Grosveld F, Strouboulis J. Optimal use of tandem biotin and V5 tags in ChIP assays. BMC Mol. Biol. 2009;10:6. doi: 10.1186/1471-2199-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, Bird AP. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010;6:e1001134. doi: 10.1371/journal.pgen.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 24.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17:877–885. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hon G, Wang W, Ren B. Discovery and annotation of functional chromatin signatures in the human genome. PLoS Comput. Biol. 2009;5:e1000566. doi: 10.1371/journal.pcbi.1000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoang SA, Xu X, Bekiranov S. Quantification of histone modification ChIP-seq enrichment for data mining and machine learning applications. BMC Res. Notes. 2011;4:288. doi: 10.1186/1756-0500-4-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi JK. Contrasting chromatin organization of CpG islands and exons in the human genome. Genome Biol. 2010;11:R70. doi: 10.1186/gb-2010-11-7-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayakawa T, Nakayama J-I. Physiological roles of class I HDAC complex and histone demethylase. J. Biomed. Biotechnol. 2011;2011:129383. doi: 10.1155/2011/129383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li DQ, Pakala SB, Nair SS, Eswaran J, Kumar R. Metastasis-associated protein 1/nucleosome remodeling and histone deacetylase complex in cancer. Cancer Res. 2012;72:387–394. doi: 10.1158/0008-5472.CAN-11-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boeke J, Ammerpohl O, Kegel S, Moehren U, Renkawitz R. The minimal repression domain of MBD2b overlaps with the methyl-CpG-binding domain and binds directly to Sin3A. J. Biol. Chem. 2000;275:34963–34967. doi: 10.1074/jbc.M005929200. [DOI] [PubMed] [Google Scholar]
- 34.Hiragami-Hamada K, Xie SQ, Saveliev A, Uribe-Lewis S, Pombo A, Festenstein R. The molecular basis for stability of heterochromatin-mediated silencing in mammals. Epigenetics Chromatin. 2009;2:14. doi: 10.1186/1756-8935-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stancheva I. Caught in conspiracy: cooperation between DNA methylation and histone H3K9 methylation in the establishment and maintenance of heterochromatin. Biochem. Cell Biol. 2005;83:385–395. doi: 10.1139/o05-043. [DOI] [PubMed] [Google Scholar]
- 36.Lopez-Serra L, Ballestar E, Ropero S, Setien F, Billard LM, Fraga MF, Lopez-Nieva P, Alaminos M, Guerrero D, Dante R, et al. Unmasking of epigenetically silenced candidate tumor suppressor genes by removal of methyl-CpG-binding domain proteins. Oncogene. 2008;27:3556–3566. doi: 10.1038/sj.onc.1211022. [DOI] [PubMed] [Google Scholar]
- 37.Morey L, Brenner C, Fazi F, Villa R, Gutierrez A, Buschbeck M, Nervi C, Minucci S, Fuks F, Di Croce L. MBD3, a component of the NuRD complex, facilitates chromatin alteration and deposition of epigenetic marks. Mol. Cell. Biol. 2008;28:5912–5923. doi: 10.1128/MCB.00467-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reynolds N, Latos P, Hynes-Allen A, Loos R, Leaford D, O'Shaughnessy A, Mosaku O, Signolet J, Brennecke P, Kalkan T, et al. NuRD suppresses pluripotency gene expression to promote transcriptional heterogeneity and lineage commitment. Cell Stem Cell. 2012;10:583–594. doi: 10.1016/j.stem.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482:221–225. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu G, Wade PA. NuRD and pluripotency: a complex balancing act. Cell Stem Cell. 2012;10:497–503. doi: 10.1016/j.stem.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hodges E, Smith AD, Kendall J, Xuan Z, Ravi K, Rooks M, Zhang MQ, Ye K, Bhattacharjee A, Brizuela L, et al. High definition profiling of mammalian DNA methylation by array capture and single molecule bisulfite sequencing. Genome Res. 2009;19:1593–1605. doi: 10.1101/gr.095190.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malousi A, Kouidou S. DNA hypermethylation of alternatively spliced and repeat sequences in humans. Mol. Genet. Genomics. 2012;287:631–642. doi: 10.1007/s00438-012-0703-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verschure PJ, Van Der Kraan I, Enserink JM, Mone MJ, Manders EM, Van Driel R. Large-scale chromatin organization and the localization of proteins involved in gene expression in human cells. J. Histochem. Cytochem. 2002;50:1303–1312. doi: 10.1177/002215540205001003. [DOI] [PubMed] [Google Scholar]
- 47.Zhang R, Burke LJ, Rasko JE, Lobanenkov V, Renkawitz R. Dynamic association of the mammalian insulator protein CTCF with centrosomes and the midbody. Exp. Cell Res. 2004;294:86–93. doi: 10.1016/j.yexcr.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 48.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng X, Grossman R, Stein L. PeakRanger: a cloud-enabled peak caller for ChIP-seq data. BMC Bioinformatics. 2011;12:139. doi: 10.1186/1471-2105-12-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 52.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- 53.Qi HH, Sarkissian M, Hu GQ, Wang Z, Bhattacharjee A, Gordon DB, Gonzales M, Lan F, Ongusaha PP, Huarte M, et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature. 2010;466:503–507. doi: 10.1038/nature09261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin H, Liu T, Manrai AK, Liu XS. CEAS: cis-regulatory element annotation system. Bioinformatics. 2009;25:2605–2606. doi: 10.1093/bioinformatics/btp479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.