

Abstract
Changes in the expression of γ-aminobutyric acid type A (GABAA) receptors can either drive or mediate homeostatic alterations in neuronal excitability. A homeostatic relationship between α5 subunit-containing GABAA (α5GABAA) receptors that generate a tonic inhibitory conductance, and HCN channels that generate a hyperpolarization-activated cation current (Ih) was recently described for cortical neurons, where a reduction in Ih was accompanied by a reciprocal increase in the expression of α5GABAA receptors resulting in the preservation of dendritosomatic synaptic function. Here, we report that in mice that lack the α5 subunit gene (Gabra5−/−), cultured embryonic hippocampal pyramidal neurons and ex vivo CA1 hippocampal neurons unexpectedly exhibited a decrease in Ih current density (by 40% and 28%, respectively), compared with neurons from wild-type (WT) mice. The resting membrane potential and membrane hyperpolarization induced by blockade of Ih with ZD-7288 were similar in cultured WT and Gabra5−/− neurons. In contrast, membrane hyperpolarization measured after a train of action potentials was lower in Gabra5−/− neurons than in WT neurons. Also, membrane impedance measured in response to low frequency stimulation was greater in cultured Gabra5−/− neurons. Finally, the expression of HCN1 protein that generates Ih was reduced by 41% in the hippocampus of Gabra5−/− mice. These data indicate that loss of a tonic GABAergic inhibitory conductance was followed by a compensatory reduction in Ih. The results further suggest that the maintenance of resting membrane potential is preferentially maintained in mature and immature hippocampal neurons through the homeostatic co-regulation of structurally and biophysically distinct cation and anion channels.
Introduction
Proper functioning of the central nervous system depends on the delicate control of neuronal excitability through a balance of excitation and inhibition. The homeostatic regulation of ion channels that regulate membrane conductance contributes to the maintenance of this balance [1], [2]. Pathological brain states can result when this balance is disrupted, such as the development of seizures following the loss of neuronal inhibition [3], [4]. Ample evidence suggests that homeostatic mechanisms exist to compensate for the loss of neuronal inhibition to maintain normal brain function [5], [6].
The neurotransmitter γ-aminobutyric acid (GABA) largely mediates inhibitory neurotransmission in the mammalian brain [7]. Activation of synaptically-localized type A GABA (GABAA) receptors results in rapid transient inhibition of postsynaptic neurons whereas activation of extrasynaptic GABAA receptors by low concentrations of ambient GABA generates a tonic inhibitory conductance [8]. A tonic GABAergic conductance in the hippocampus is predominantly generated by GABAA receptors that contain either the α5 subunit (α5GABAA) or δ subunit (δGABAA) [9], [10]. Tonic GABAergic inhibition can exert powerful regulatory constraints on neuronal firing, excitability, and plasticity of excitatory synapses of hippocampal pyramidal neurons [11]–[13].
Loss of tonic inhibition can induce compensatory changes in the expression of other ion channels that maintain normal neuronal function. For example, in cerebellar granule cells of α6GABAA receptor-null mutant mice, the loss of tonic inhibition mediated by putative extrasynaptic δGABAA receptors was accompanied by a homeostatic increase in the expression of two-pore domain K+ TASK-1 channels that generate a tonic inhibitory K+ current [14]. This increase in TASK-1 channel expression maintained neuronal excitability at levels observed in wild-type (WT) neurons.
Genetic deletion of voltage-dependent ion channels can also induce homeostatic changes in tonic GABAergic inhibition [15]. In particular, the genetic deletion of the hyperpolarization-activated cyclic nucleotide-gated type 1 (HCN1) channel which generates a hyperpolarization-activated cation current (Ih) increased the expression of α5GABAA receptors in cortical pyramidal neurons [15]. HCN channels are encoded by four genes (HCN1–HCN4), and are activated at hyperpolarized membrane potentials. HCN channels are permeable to both Na+ and K+ ions and mediate an inward current [16]. These non-inactivating ion channels exert complex effects on neuronal function by providing a tonic depolarizing current which contributes to resting membrane potential and opposes deviations away from the prevailing membrane potential. In hippocampal and neocortical pyramidal neurons, these biophysical properties of Ih, together with a preferential distribution of the channels in distal dendrites limits the influence of excitatory synaptic input on membrane potential [17].
Pyramidal neurons of the hippocampus and cortex predominantly express the type-1 isoform of HCN (HCN1), and deletion of HCN1 strongly decreases Ih in these neurons [18], [19]. Surprisingly, the summation of evoked excitatory post-synaptic potentials (EPSPs) in cortical neurons was unchanged following genetic deletion of HCN1 [15]. A homeostatic upregulation of α5GABAA receptors in the cortex maintained the sublinear somatic summation of EPSPs following deletion of HCN1 [15]. As such, the increase in tonic inhibition compensated for the loss of Ih and constrained dendritosomatic efficacy. Notably, there was no upregulation of α5GABAA receptors in hippocampal pyramidal neurons of HCN1−/− mice, perhaps due to a saturation of α5GABAA receptor expression in these neurons [15].
α5GABAA receptors and HCN1 channels have several common biophysical and functional properties that suggest they may mutually co-regulate neuronal excitability. For example, both channels can remain persistently activated following a hyperpolarization of the membrane to regulate resting membrane potential and conductance [11], [16], [20]. Additionally, HCN1 channels are expressed in high levels in the distal dendrites of hippocampal pyramidal neurons [21] where α5GABAA receptors are also clustered [22]. Tonic inhibition and Ih both regulate the induction of long-term synaptic plasticity of hippocampal pyramidal neurons and limit sublinear EPSP summation in neocortical pyramidal neurons [15]. Finally, both α5GABAA receptors and HCN1 channels constrain hippocampus-dependent memory performance [13], [19].
The functional commonalities between α5GABAA receptors and HCN1 channels suggest that the potential reciprocal homeostatic co-regulation of these proteins is plausible. However, it is unknown whether the expression of α5GABAA receptors regulates Ih. In this study, we tested the hypothesis that a reduction in the expression of α5GABAA receptors causes a reciprocal upregulation of Ih in hippocampal pyramidal neurons. Unexpectedly, we found the opposite, where a reduction in the expression of α5GABAA receptors was associated with a reduction of Ih that contributes to homeostatic maintenance of resting membrane potential in these cells.
Methods
Electrophysiology
Hippocampal cell culture
The experiments reported here were approved by the Animal Care Committee of the University of Toronto. All experiments were conducted with hippocampal tissue harvested from WT Gabra5+/+ or α5GABAA null mutant mice (Gabra5−/−) mice. Generation of the Gabra5−/− mice has been described previously [23]. Briefly, all mice were of mixed genetic background (50:50 C57BL/6 and 129SvEv), and WT and Gabra5−/− mice were generated by crossing heterozygous Gabra5+/− mice. Cultures of hippocampal neurons were prepared as previously described [11] from Gabra5−/− and WT littermates on postnatal day 1. Cells were maintained in culture for 14 to 21 days before experimentation.
Hippocampal brain slices
Slices were prepared from WT and Gabra5−/− mice that ranged in age from postnatal day 17–21. After administration of isoflurane anesthesia, the mice were decapitated and their brains quickly removed and placed in ice-cold, oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid (aCSF; containing in mM: NaCl 124, KCl 3, MgCl2 1.3, CaCl2 2.6, NaH2PO4 1.25, NaHCO3 26, d-glucose 10) with osmolarity adjusted to 300–310 mOsm. Brain slices (350 µm) containing coronal sections of the hippocampus were prepared with a VT1200 tissue slicer (Leica, IL, USA).
Data Acquisition
Data were acquired with a Multiclamp 700B amplifier (Molecular Devices Corporation, Sunnyvale, CA, USA) controlled with pClamp 9.0 software (Molecular Devices Corporation) via a Digidata 1322 interface (Molecular Devices Corporation). Membrane current and voltage were filtered at 2 kHz and sampled at 10 kHz for all electrophysiological experiments. Membrane capacitance was measured with the membrane test protocol in pClamp 9.0. Access resistance was monitored periodically throughout the experiments by a brief 10-mV or 10-pA hyperpolarizing step during voltage-clamp and current-clamp experiments, respectively. Cells were eliminated from further analysis if the access resistance changed by more than 20% over the recording period. Liquid junction potential and pipette capacitance were corrected using the pClamp 9.0 software before the whole-cell configuration was established.
Patch pipettes, pulled from thin-walled borosilicate glass capillary tubes, had open-tip resistances of 4 to 6 MΩ when filled with an intracellular solution that contained (in mM) 145 K+ gluconate, 5 Na+ gluconate, 2 KCl, 10 HEPES, 11 EGTA, 4 Mg2+ATP, and 1 CaCl2 with an osmolarity of 300 to 320 mOsm and the pH adjusted to 7.3 with KOH. Extracellular solutions for all experiments contained (in mM) 140 NaCl, 1.3 CaCl2, 2.0 KCl, 25 HEPES, and 33 glucose; the osmolarity was adjusted to 290 to 300 mOsm with sucrose, and the pH was adjusted to 7.4 with 10 N NaOH. The extracellular solution was applied directly to neurons at a rate of 1 ml/min by a computer-controlled, multi-barreled perfusion system (SF-77B; Warner Instruments, Hamden, CT, USA). Whole-cell current was recorded with the holding potential clamped at −60 mV except where indicated otherwise.
Experiments in cultured pyramidal neurons were performed as previously described [11]. For experiments in hippocampal slices, whole-cell recordings were obtained from the pyramidal cell layer using a blind-patch technique. Neurons with small membrane capacitances suggestive of non-pyramidal neurons in this preparation (<60 pF) were excluded from study (3 WT, 1 Gabra5 −/− neuron). The composition of the intracellular solution and the recording procedures were identical to those described for the recordings from cultured neurons.
In all experiments, the ionotropic glutamate antagonists 6-cyano-7-nitroquinoxaline-2,3-dione (10 µM) and 2-amino-4-phosphonovaleric acid (40 µM) were added to the extracellular solution. In experiments designed to measure Ih and membrane impedance, the Na+ channel blocker tetrodotoxin (0.3 µM; Alomone Labs, Jerusalem, Israel) was added to the extracellular solution. Aqueous stock solutions of all drugs were prepared with distilled water. All drugs and chemicals were purchased from Sigma-Aldrich (Oakville, Ontario, Canada) except where indicated otherwise.
Measurement of Ih
Ih was activated by changing the holding potential from −60 mV through a range of test potentials (from −120 mV to −30 mV) in 10-mV steps. Each test potential was maintained for 500 ms. The net Ih conductance was measured as the difference between the steady-state current at the end of the test potential and the minimum current measured within 100 ms of the start of the test potential (Fig 1A). The Ih tail current was measured as the peak amplitude of the residual current measured at the end of each test potential immediately after the return the holding potential to −60 mV. The membrane potential that evoked half-maximal activation (V50) of Ih was determined by fitting the tail current activation data to a Boltzmann sigmoidal function using Graphpad 4 (Graphpad, San Diego, CA, USA). The kinetics of Ih activation, measured at holding potentials between −120 mV and −70 mV, were determined by fitting onset of the current with a single exponential curve using Clampfit 10 (Molecular Devices Corporation) with the equation: 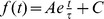 . The net Ih was measured at the end of the test holding potential, and the Ih conductance was estimated by fitting the net Ih measured between −120 mV and −90 mV with a linear regression line.
. The net Ih was measured at the end of the test holding potential, and the Ih conductance was estimated by fitting the net Ih measured between −120 mV and −90 mV with a linear regression line.
Figure 1. Reduced Ih in cultured Gabra5−/− neurons.

A) Schematic illustrating the method of Ih measurement B) Ih was activated in cultured hippocampal pyramidal neurons of wild-type (WT) and Gabra5−/− neurons by changing the membrane potential from −120 mV to −30 mV in 10-mV increments. C) Estimation of Ih conductance from the linear portion of the current-voltage curve generated by hyperpolarizing the resting membrane potential revealed a 43% reduction of Ih conductance in Gabra5−/− neurons. D) Quantification of the Ih tail currents that remained after membrane potential was returned to −60 mV revealed significantly lower Ih density in Gabra5−/− neurons (n = 16) than in WT neurons (n = 9). Neither the kinetics of Ih activation (E) nor sensitivity to Ba2+ (0.5 mM; n = 5) or Cs+ (0.5 mM; n = 4) (F) were changed in Gabra5−/− neurons, which suggested no change in the subtypes of HCN channels generating Ih. G) Enhancing or reducing the tonic current in WT neurons with 1 µM GABA (n = 6) or 1 µM picrotoxin (PTX; n = 6), respectively, did not change Ih measured at −120 mV, demonstrating that the lower level of Ih in Gabra5−/− neurons is independent of changes in tonic inhibition.
Measurement of after-hyperpolarization
An after-hyperpolarization of the membrane was induced by stimulating neurons with a train of action potentials in current-clamp mode. A depolarizing current sufficient to stimulate action potential firing at a frequency of 5 Hz for 2 s was applied and after-hyperpolarization was measured as the area under the curve, relative to resting membrane potential, of the membrane potential over the period of hyperpolarization following the train of action potentials. The decay time constant (τ) of the after-hyperpolarization was measured with Clampfit 10 by fitting the decay with a standard single exponential curve.
Determination of membrane impedance
The neuronal frequency-dependent membrane impedance was studied using the impedance (Z) amplitude profile (ZAP) as described previously [24]. In brief, in whole-cell current-clamp mode, neurons were injected with a sinusoidal current of constant amplitude and linearly increasing frequency (0–40 Hz over 30 s). The amplitude of the ZAP current was adjusted to maintain a peak depolarization of the membrane potential of approximately 10 mV positive to resting potential. The frequency-dependent membrane impedance was determined by transforming the membrane voltage and input current recordings with a fast Fourier transform over the range of frequencies from 0.5 to 40 Hz with Clampfit 10 and dividing the transformed voltage by the current. The peak resonance frequency was determined as the input frequency at which membrane resistance was the greatest.
Western blot measurements
Hippocampal tissue was collected from adult (16 weeks old) WT and Gabra5−/− mice. Hippocampal tissue was dissected from whole brains in ice cold phosphate-buffered saline (pH 7.4) and homogenized with a Dounce homogenizer (Wheaton, NJ, USA). The homogenization buffer contained (in mM): 10 Tris-HCl, 5 NaF, 1 Na3VO4, 1 EDTA, 1 EGTA, 320 sucrose, protease inhibitor EDTA free tablet (Roche Diagnostics, Germany) at pH 7.4. The homogenate was centrifuged at 900 g for 10 min at 4°C, and the supernatant was spun again at 10,000 g for 20 min. The final supernatant was isolated in lysis buffer containing (in mM): 20 Tris-HCl, 150 NaCl, 5 EDTA, 10 NaF, 2 Na3VO4, 10 sodium pyrophosphate, 1% (v/v) Triton X-100, and 0.1% (w/v) sodium dodecyl sulfate (SDS), EDTA-free protease inhibitor tablet (Roche Diagnostics, Germany). The supernatant was homogenized using a probe sonicator (Cole Parmer Instruments, IL, USA) and protein concentration was determined using bicinchoninic acid assay (BCA) (Thermo Scientific, IL, USA).
Hippocampal protein (15 µg) was loaded on 10% Bis-Tris gels and transferred onto nitrocellulose membranes (Pall Life Sciences, NY, USA) followed by SDS-PAGE. The membranes were rinsed in TBS-Tween that contained 50 mM Tris-HCl, 150 mM NaCl, and 0.05% (v/v) Tween 20 and then incubated in 5% (w/v) milk in TBS-Tween at room temperature for 1 hr. Primary and secondary antibodies were diluted in 3% (w/v) bovine serum albumin in TBS-Tween. The membranes were incubated with 1∶1000 anti-HCN1 antibody (clone N70/28; NeuroMab, UC Davis NeuroMab facility, CA, USA) overnight at 4°C, washed with TBS-Tween, and incubated in 1∶1000 anti-mouse antibody (Cell Signaling, MA, USA) at room temperature for 1 hr. The membranes were treated with enhanced chemiluminesence western blotting substrate (Thermo Scientific, IL, USA) for protein band visualization. HCN1 primary and secondary antibodies were stripped from the membranes by incubating in stripping buffer (Thermo Scientific, IL, USA) at room temperature for 20 min, followed by 4 washes in TBS-Tween. To allow the normalization of HCN1 blot densities, β-actin blots were then performed using the western blotting procedure described above with 1∶1000 anti-β-actin antibody (Millipore, MA, USA), followed by 1∶1000 anti-rabbit antibody (Cell Signaling, MA, USA).
All membranes were exposed and quantified using the Kodak Image Station 2000R (Kodak, USA). Because HCN1 is known to exist in a glycosylated (108 kDa) and unglycosylated (100 kDa) form, both of which are recognized by the Anti-HCN1 antibody used (clone N70/28; NeuroMab), the densities of both bands were pooled for analysis as described elsewhere [25]. The density of HCN1 bands were normalized to β-actin, a prototypical loading control.
Statistical analyses
Statistical analyses were performed using Graphpad Prism 4. Membrane impedance and Ih tail current and activation kinetics were analyzed with two-way repeated-measures ANOVA followed by a Bonferroni post hoc test. The remaining comparisons were performed with one-way ANOVA or Student t-tests, as appropriate. Any p value less than 0.05 was considered significant. All data are shown as mean ± standard error of the mean.
Results
The biophysical properties of cultured hippocampal neurons from WT and Gabra5−/− mice were studied using standard whole-cell patch clamp techniques. WT and Gabra5−/− neurons had similar membrane capacitances (WT: 33.5 pF±2.0 pF, n = 16; Gabra5−/−: 35.0 pF±1.6 pF, n = 14; p>0.05) and resting membrane potentials (WT: −67.5 mV±0.8 mV, n = 16; Gabra5−/−: −67.9 mV±0.7 mV, n = 14; p>0.05), as shown previously [11]. However, Gabra5−/− neurons had higher input resistances than WT neurons (WT: 231 MΩ±9 MΩ, n = 16; Gabra5−/−: 313 MΩ±11 MΩ, n = 14; p<0.0001), owing in part to the loss of tonic inhibition as described in a previous report [11].
Reduced Ih in cultured Gabra5−/− neurons
Next, the amplitude of the Ih current was measured in WT and Gabra5−/− neurons (Fig 1A) The net Ih was measured as the time-dependent inward current activated by the voltage step (Fig 1B). The Ih conductance was estimated from the near linear current-voltage relationship of Ih measured between −120 mV and −90 mV (Fig 1C). From these analyses, the total Ih conductance was estimated to be 43% smaller in Gabra5−/− neurons compared with WT neurons (WT: 6.0 nS±0.2 nS, n = 9; Gabra5−/−: 3.4 nS±0.1 nS, n = 16; p<0.0001). The maximum amplitude of the tail current measured following the hyperpolarizing voltage steps was smaller in Gabra5−/− neurons (n = 16) than in WT neurons (n = 9; Fig 1D; voltage × genotype: F9,198 = 4.09; p<0.0001), consistent with a reduced Ih in these neurons. The HCN channel blocker ZD-7288 caused a complete block of Ih in both WT and Gabra5−/− neurons (data not shown).
The reduced Ih in Gabra5−/− neurons may result from the substitution of HCN1 with another HCN isoform. The subtype of HCN channels determines its sensitivity to cAMP and voltage-dependent activation and kinetics [16]. Thus, a substitution of HCN subtype is predicted to be accompanied by changes in the activation kinetics and voltage-dependent activation of Ih. However, we observed that the time course of current activation (τ Ih) was similar between WT and Gabra5−/− neurons (Fig 1E) (genotype × voltage: F5,99 = 0.05, p>0.05). In addition, the voltage-sensitivity of Ih, measured as the half-maximal activation voltage (V50) of the tail currents (Fig 1C), was similar between WT and Gabra5−/− mice (WT: −91.5 mV±5.0 mV, n = 9; Gabra5−/−: −93.3 mV±7.3 mV, n = 16, p>0.05). These results suggest that the lower Ih in Gabra5−/− neurons is not likely due to a change in the subpopulation of HCN channels that generate Ih.
A pharmacological characteristic of Ih generated by HCN channels is an insensitivity to low concentrations of extracellular barium and potent inhibition induced by low concentrations of extracellular cesium [26]. To confirm that the reduction in Ih in Gabra5−/− neurons resulted from a decrease in HCN-generated current; we applied a low concentrations of either BaCl2 (0.5 mM) or CsCl (0.5 mM). Consistent with HCN pharmacology, BaCl2 (0.5 mM) did not block Ih in WT (n = 5) or Gabra5−/− (n = 5) neurons, but CsCl (0.5 mM) caused near complete inhibition of Ih in both WT (n = 4) and Gabra5−/−−/− (n = 4) neurons, when Ih was activated at −120 mV (Fig 1F).
We next sought to determine whether the acute enhancement or inhibition of α5GABAA receptor-mediated current changed Ih, similar to the reduction of Ih observed following genetic deletion of α5GABAA receptors. The tonic current was either enhanced by applying 1 µM GABA (n = 6) or inhibited by applying 1 µM picrotoxin (n = 6), as described previously [11] then Ih was activated in WT neurons by hyperpolarizing the membrane potential to −120 mV. Neither enhancement or inhibition of the tonic current changed the amplitude of Ih (Fig 1G; one-way ANOVA F2,18 = 0.08, p>0.05).
Ih can exert a powerful regulatory effect on the resting membrane potential of neurons [16]. Further, the dynamic voltage-dependent activity of depolarizing Ih opposes any changes in membrane potential away from the resting membrane potential. We next sought to determine whether the lower Ih in Gabra5−/− neurons would exert less control over resting membrane potential than in WT neurons. Application of the HCN antagonist ZD-7288 (20 µM) induced a similar hyperpolarization of the resting membrane potential of approximately 5.5 mV in both WT and Gabra5−/− neurons (WT + ZD-7288: −72.8 mV±2.0 mV, n = 8; Gabra5−/− + ZD-7288: −73.6 mV±1.8 mV, n = 7, p>0.05). These results suggest that the baseline level of Ih depolarized the resting membrane potential to a similar degree in both WT and Gabra5−/− neurons.
Reduced after-hyperpolarization in cultured Gabra5−/− neurons
Following a train of action potentials, the membrane potential is often hyperpolarized below the resting potential, in part due to deactivation of Ih [27]. This after-hyperpolarization is a key determinant of spike frequency adaptation in hippocampal neurons [28], [29]. We next studied the potential functional consequences of the reduced Ih conductance on neuronal after-hyperpolarization. Neurons were depolarized to fire action potentials at a rate of 5–6 Hz for 2 s, and the after-hyperpolarization was measured as the area of the subsequent membrane hyperpolarization (i.e., after the action potential train) (Fig 2A). The amount of excitatory current required to produce similar frequencies of action potentials was lower in Gabra5−/− neurons than in WT neurons (WT: 7.67 pA/pF±1.00 pA/pF, n = 12; Gabra5−/−: 4.81 pA/pF±0.64 pA/pF, n = 9; p = 0.038) as reported previously [11]. The resting membrane potential was similar between WT and Gabra5−/− neurons (WT: −68.0 mV±1.4 mV, n = 12; Gabra5−/−: −68.8 mV±1.2 mV, n = 9; p>0.05). Application of ZD-7288 (20 µM) abolished the after-hyperpolarization in both WT and Gabra5−/− neurons (Fig 2B), which confirmed the important role of Ih in the after-hyperpolarization. The after-hyperpolarization was smaller in Gabra5−/− than in WT neurons (Fig 3B) (WT: −3528 mV·s±540 mV·s, n = 12; Gabra5−/−: −1883 mV·s±369 mV·s, n = 9; p = 0.022). This effect was largely due to a reduction in peak after-hyperpolarization potential seen in Gabra5−/− neurons compared to WT (Fig 2C) (WT: −5.1 mV±0.8 mV, n = 12; Gabra5−/−: −2.9 mV±0.5 mV, n = 9; p = 0.040). However, the τ of the after-hyperpolarization did not differ between WT and Gabra5−/− neurons (Fig 2D) (WT: 538 ms±86 ms, n = 12; Gabra5−/−: 501 ms±119 ms, n = 9; p>0.05), consistent with similar Ih kinetics in these neurons.
Figure 2. Reduced after-hyperpolarization in cultured Gabra5−/− neurons.
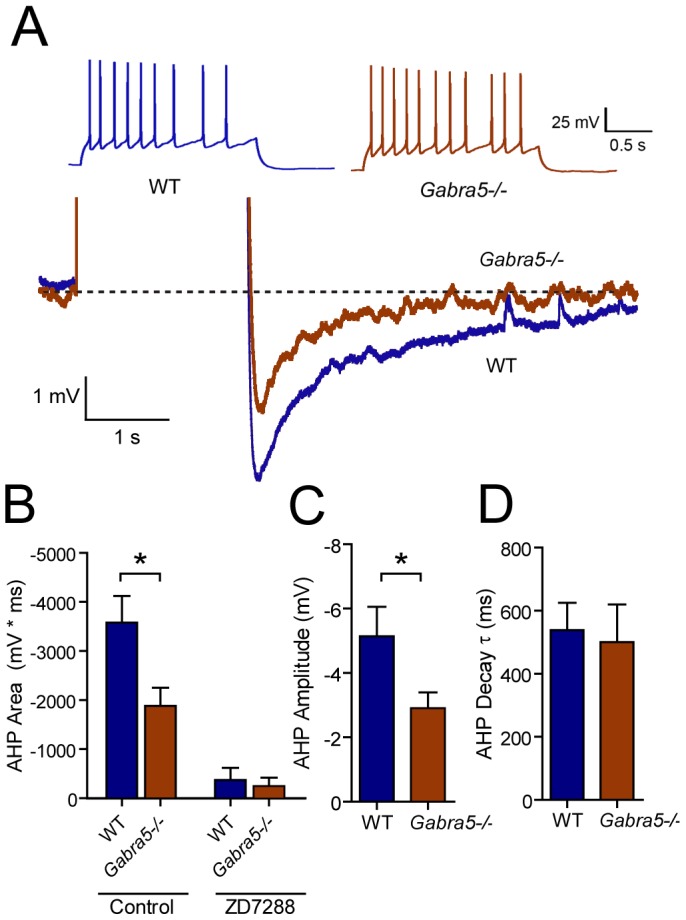
A) Action potentials were elicited in WT and Gabra5−/− neurons at a frequency of 5 Hz (examples shown in upper traces). The membrane hyperpolarization that occurred following depolarization was measured relative to resting membrane potential to reveal a reduced after-hyperpolarization in Gabra5−/− neurons (example traces enlarged to emphasize after-hyperpolarization are shown in lower traces). B) The area of after-hyperpolarization was smaller in Gabra5−/− neurons (n = 9) than in WT neurons (n = 12). Application of the HCN antagonist ZD-7288 (20 µM) blocked the after-hyperpolarization in neurons of both genotypes (n = 5), confirming the contribution of Ih to after-hyperpolarization. C) The peak after-hyperpolarization, measured relative to the resting membrane potential, was also smaller in Gabra5−/− neurons compared to WT. D) The decay kinetics of after-hyperpolarization were similar between WT and Gabra5−/− neurons.
Figure 3. Increased membrane impedance in response to low-frequency input in cultured Gabra5−/− neurons.

A) The membrane impedance properties of WT and Gabra5−/− neurons were determined by quantifying membrane resistance during the injection of a sinusoidal current ranging in frequency from 0 to 40 Hz. Example traces show larger changes in membrane potential in the Gabra5−/− neuron at low frequencies, indicative of an increased membrane impedance. B) The membrane impedance of Gabra5−/− neurons (n = 10) was greater than that of WT neurons (n = 7) in response to low-frequency input from 1 to 4 Hz (p1.0 Hz<0.01, p1.5 Hz<0.001, p2.0 Hz<0.001, p2.4 Hz<0.01, p2.9 Hz<0.05, p3.4 Hz>0.05, p3.9 Hz<0.01). The inset shows the membrane impedance ratio of Gabra5−/− to WT neurons. C) Blockade of Ih in WT neurons with ZD-7288 (n = 4) increases membrane impedance to input from 1 to 6 Hz (p1.0 Hz<0.001, p1.5 Hz<0.001, p2.0 Hz<0.001, p2.4 Hz<0.001, p2.9 Hz<0.001, p3.4 Hz<0.001, p3.9 Hz<0.001, p4.4 Hz<0.001, p4.9 Hz<0.001, p5.4 Hz<0.05, p5.9 Hz<0.05). D) Blockade of Ih in Gabra5−/− neurons with ZD-7288 (n = 5) caused a modest but significant increase in membrane impedance. Post hoc analysis did not reveal significant differences within any specific frequency range. E) No differences were observed in the impedance of Gabra5−/− and WT neurons in the presence of ZD-7288. Asterisks indicating significant differences within specific frequency ranges have been omitted for clarity.
Increased low-frequency membrane impedance in Gabra5−/− neurons
Previous studies have demonstrated that Ih contributes to the frequency-dependent membrane impedance of neurons [19], [24]. Specifically, Ih generated by HCN1 in hippocampal pyramidal neurons selectively attenuates changes in membrane potential resulting from low-frequency input (< 5Hz), which in turn reduces the subthreshold membrane resonance properties of neurons in response to input in this frequency range [19], [24]. We examined the membrane impedance properties of cultured WT (n = 7) and Gabra5−/− (n = 10) neurons by injecting an oscillating current of linearly increasing frequency and then measuring the impedance (Fig 3A). Gabra5−/− neurons had a higher frequency-dependent impedance than WT neurons in response to stimulation in the frequency range of 0 to 4 Hz (Fig 3B) (genotype × frequency: F80,1215 = 1.39; p = 0.016). Post hoc analysis revealed a significantly greater membrane impedance in Gabra5−/− than in WT neurons over most of the frequency range from 1 to 4 Hz (Fig 4B).
Figure 4. Reduced Ih and HCN1 expression in hippocampus of Gabra5−/− mice.

A) Representative traces of Ih in CA1 pyramidal neurons in hippocampal slices obtained from postnatal WT and Gabra5−/− mice. Ih was activated and measured by changing the membrane potential from −120 mV to −30 mV in 10-mV increments. B) Estimation of Ih conductance from the linear portion of the current-voltage curve revealed a 28% reduction of Ih in Gabra5−/− neurons. C) A modest but significant reduction in Ih tail current was also observed in Gabra5−/− neurons. Post hoc analysis did not reveal significant differences at any specific test potential. D) The expression of HCN1 protein and β-actin in hippocampal tissue from adult WT and Gabra5−/− mice. E) After normalization to β-actin, the expression of HCN1 was reduced in hippocampal tissue from Gabra5−/− mice by 41% relative to WT mice, paralleling the decrease in Ih current.
To ascertain whether this difference in membrane impedance between WT and Gabra5−/− resulted from the lower Ih, we tested for changes in membrane impedance following the application of ZD-7288 (20 µM). In WT neurons (n = 4), blockade of Ih by ZD-7288 resulted in a robust frequency-dependent increase in membrane impedance (Fig 3C) (genotype × frequency: F80,729 = 3.35; p<0.0001). Post hoc analysis of this interaction revealed a significant increase in membrane impedance in the frequency range 1 to 6 Hz (Fig 3C). In contrast, ZD-7288 caused only a modest frequency-dependent increase in membrane impedance in Gabra5−/− neurons (n = 5), which was not significantly different from control at any specific frequency (Fig 3D) (main effect of drug: F1,1053 = 32.05, p<0.0001). Notably, the membrane impedance of WT and Gabra5−/− neurons was similar following application of ZD-7288 (Fig 3E) (main effect of genotype: F1,567 = 0.006; p>0.05). Finally, the peak resonant frequency of the neurons was similar in both genotypes and drug conditions (WT: 1.15 Hz±0.03 Hz; Gabra5−/−: 1.20 Hz±0.03 Hz; WT + ZD-7288: 1.13 Hz±0.03 Hz; Gabra5−/− + ZD-7288: 1.18±0.06. Two way ANOVA genotype: F1,22 = 1.67, p<0.05; drug: F1,22 = 0.268, p>0.05). The similarities between the membrane impedance of WT and Gabra5−/− neurons in the presence of ZD-7288 suggest that the tonic GABAergic inhibitory conductance does not significantly contribute to membrane impedance properties of pyramidal neurons.
Reduced Ih and HCN1 in Gabra5−/− hippocampal neurons in brain slices
We next sought to determine whether the reduced Ih observed in cultured Gabra5−/− hippocampal neurons was also present in neurons of the hippocampal CA1 pyramidal layer recorded in brain slices (Fig 4A). Similar to cultured neurons, we observed an increased membrane resistance in Gabra5−/− neurons compared to WT (Gabra5−/−: 212 MΩ±14 MΩ, n = 13; WT: 158 MΩ±19 MΩ, n = 12; p = 0.027). Ih current density was again reduced in Gabra5−/− neurons (n = 12) compared to WT (n = 8) (Fig 4B) (voltage × genotype: F8,144 = 9.64; p<0.0001). Relative to WT neurons, the total Ih conductance was estimated to be 28% lower in Gabra5−/− neurons (WT: 4.5 nS±0.3 nS, n = 8; Gabra5−/−: 3.2 nS±0.4 nS, n = 12; p = 0.030). Ih tail current was also reduced in Gabra5−/− neurons compared to WT (voltage × genotype: F8,144 = 3.03; p = 0.004), although post-hoc analysis did not reveal a significant reduction at any specific potential (Fig 4C). The difference in Ih current density was not attributable to differences in cell size (WT: 166 pF±23 pF; Gabra5−/−: 196 pF±14 pF; p = 0.30). Additionally, the V50 of Ih was similar between WT and Gabra5−/− mice (WT: −84.2 mV±3.4 mV, n = 8; Gabra5−/−: −84.6 mV±3.0 mV, n = 12; p>0.05). These data suggest that the reduction of Ih in Gabra5−/− neurons is robust and occurs at different stages of development and in different neuronal environments.
Protein levels of HCN1 are reduced in Gabra5−/− neurons
One likely explanation for the reduction of Ih in Gabra5−/− neurons, in the absence of changes in Ih kinetics, is a decrease in the expression of HCN1 protein. This hypothesis was tested by measuring levels of HCN1 protein in hippocampal tissue samples from adult WT (n = 6) and Gabra5−/− (n = 6) mice (Fig 4D). HCN1 was selected for measurement since it is the most highly expressed isoform in the hippocampus CA1 [21]. Densitometric analysis showed that compared to WT mice, total protein expression of HCN1 in the hippocampus of Gabra5−/− mice was decreased by 40.8%±9.1% (Fig 4E) (one-sample t-test, p = 0.002). Thus, the magnitude of the reduction of HCN1 protein in Gabra5−/− hippocampal neurons closely paralleled the reduction of Ih.
Discussion
Here, we tested the hypothesis that reduced expression of α5GABAA receptors would be accompanied by a reciprocal increase in Ih [15]. Unexpectedly, we observed a reduction in Ih in Gabra5−/− hippocampal neurons compared to WT neurons, as indicated by the lower hyperpolarization-activated current, lower after-hyperpolarization, and greater low-frequency membrane impedance. The reduction in Ih was observed in both cultured neurons and in hippocampal pyramidal neurons. We observed no change in Ih activation kinetics in Gabra5−/− neurons, suggesting that changes in HCN channel isoform did not contribute to the reduced Ih in Gabra5−/− neurons. Finally, we observed a decrease in the protein levels of HCN1 in Gabra5−/− hippocampus that paralleled the reduction of Ih observed in Gabra5−/− neurons.
Reduced Ih maintains normal resting membrane potential in Gabra5−/− neurons
The resting membrane potential was not different in Gabra5−/− neurons, despite the fact that the tonic inhibitory conductance generated by α5GABAA receptors was absent in Gabra5−/− neurons [11]. These data raise the possibility that a decrease in Ih, which normally provides a tonic depolarizing current, serves to homeostatically maintain the same resting membrane potential in Gabra5−/− and WT neurons. It is notable that the reduced Ih current associated with deletion of the α5GABAA receptor was observed in both cultured hippocampal pyramidal neurons and in CA1 hippocampal neurons. This finding suggests that there exists a robust relationship between α5GABAA receptor and HCN1 channel expression that persists in very different neuronal environments and at different developmental stages.
The lack of change in resting membrane potential contrasted with the differences between WT and Gabra5−/− mice in after-hyperpolarization and membrane impedance. The after-hyperpolarization was reduced in Gabra5−/− neurons. Since ZD-7288 blocked the after-hyperpolarization in both WT and Gabra5−/− neurons, the after-hyperpolarization measured here was predominantly generated through the voltage-dependent deactivation of Ih during depolarization. Despite the differences in peak after-hyperpolarization, activation of Ih terminated the after-hyperpolarization similarly in WT and Gabra5−/− neurons. Because of the role Ih plays in regulating the firing of action potentials [28], a reduced after-hyperpolarization may disturb the firing frequency of Gabra5−/− neurons. Nonetheless, the reduced Ih in Gabra5−/− neurons appears to maintain membrane potential even at the expense of a reduced after-hyperpolarization and the potential consequences on firing activity.
A reduction in Ih also increased the frequency-dependent membrane impedance in Gabra5−/− neurons. These findings are consistent with the established role of Ih in reducing membrane impedance to low-frequency, fluctuating input [19], [24]. Similar to after-hyperpolarization, we found that membrane impedance was not greatly influenced by tonic α5GABAA receptor activity, since WT and Gabra5−/− neurons exhibit similar membrane impedances when Ih was blocked by ZD-7288. Overall our data suggest that the reduced Ih in Gabra5−/− hippocampal neurons homeostatically maintains resting membrane potential, with consequential changes in other neuronal properties and behaviours that are regulated by Ih, such as after-hyperpolarization and membrane impedance. Whether the reduced Ih also restores normal synaptic integration in Gabra5−/− neurons [15] remains to be determined.
Homeostasis of neuronal excitability following reduction of tonic GABAergic inhibition
Deletion of the GABAA receptors that contribute to tonic GABAergic inhibition causes changes in other conductances that regulate neuronal excitability. For example, the genetic deletion of α6GABAA receptors, which mediate a tonic current in cerebellar granule cells, causes the upregulation of the two-pore-domain leak K+ channel, TASK-1 [14]. The converse relationship has also been found: genetic deletion of Kv4.2 K+ channels was associated with an increased tonic inhibitory current in hippocampal pyramidal neurons [30]. In both of these examples, the loss of one inhibitory current was offset by an increase in another inhibitory current to maintain normal neuronal excitability. We showed that the genetic deletion of α5GABAA receptors that generate tonic outward currents in hippocampal neurons [9] was associated with a decrease in Ih that provides tonic inward current. As such, the normal relative levels of outward and inward current could be maintained, as reflected in the lack of difference in resting membrane potential between WT and Gabra5−/− neurons.
It is notable that in previous studies, an upregulation of α5GABAA receptors was not observed in hippocampal pyramidal neurons of HCN1−/− mice [15]. The expression of α5GABAA receptors in the hippocampus is among the highest in the mammalian brain [31]. The high basal level of expression of α5GABAA receptors may reduce or eliminate the capacity for further upregulation of these receptors [15]. Alternatively, HCN1 channels and α5GABAA receptors may serve different functional roles in hippocampal pyramidal neurons and may be homeostatically co-regulated in a manner different from that observed in cortical neurons. The cortex and hippocampus are distinct neuronal environments that may exert unique homeostatic pressures, such that either resting membrane potential or EPSP summation is preferentially preserved through compensatory mechanisms [15]. Thus, the mechanisms of compensation may be diverse and likely vary depending on the primary contribution of the ionic currents to neuronal function and the prevailing activity patterns of the neurons [32], [33]. Finally, tonic inhibitory currents are subject to regulation by endogenous hormones, such as neuroactive steroids and insulin [34], [35]. It would be of interest to ascertain whether the endogenous regulation of tonic inhibition also induces changes in Ih.
Lastly, HCN1 channels expressed in hippocampal CA1 pyramidal neurons play an important role in the regulation of hippocampus-dependent memory [19]. Specifically, deletion of HCN1 in forebrain neurons enhanced short- and long-term memory in mice [19]. Similarly, Gabra5−/− mice display better hippocampus-dependent memory performance [13], [23]. Thus, it is possible that reduced Ih contributes to the enhanced memory performance of Gabra5−/−mice. Additionally, Gabra5−/−mice exhibit a reduced sensitivity to memory impairment by etomidate, which potently enhances the activity of α5GABAA receptors [36], [37]. HCN channels are similarly inhibited by anesthetics including propofol and isoflurane [18], and a reduction of Ih may also contribute to the reduced sensitivity of Gabra5−/− mice to the amnestic effects of anesthetics. Overall, the results of this study suggest a co-regulation of α5GABAA receptors that generate a tonic GABAergic conductance and HCN1 channels that generate Ih in hippocampal pyramidal neurons. It will be of future interest to determine whether alterations in Ih contribute to the behavioural phenotype of Gabra5−/− mice.
Funding Statement
This study was funded by a grant from the National Institutes of Health (GM66181) to DAB, a grant from the Canadian Institutes of Health Research (MOP 38028, MOP 79428) and a Canada Research Chair to BAO. RPB is supported by Fonds de Recherche du Québec – Santé, RPB and AAZ by the Natural Sciences and Engineering Council, and JY is supported by the Ontario Students Opportunity Trust Fund. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Turrigiano G (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135: 422–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schulz D (2006) Plasticity and stability in neuronal output via changes in intrinsic excitability: it's what's inside that counts. J Exp Biol 209: 4821–4828. [DOI] [PubMed] [Google Scholar]
- 3. Peng Z, Hauer B, Mihalek RM, Homanics GE, Sieghart W, et al. (2002) GABA(A) receptor changes in delta subunit-deficient mice: altered expression of alpha4 and gamma2 subunits in the forebrain. J Comp Neurol 446: 179–197. [DOI] [PubMed] [Google Scholar]
- 4. Naylor D, Liu H, Wasterlain C (2005) Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci 25: 7724–7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marder E, Goaillard J-M (2006) Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci 7: 563–637. [DOI] [PubMed] [Google Scholar]
- 6. Destexhe A, Marder E (2004) Plasticity in single neuron and circuit computations. Nature 431: 789–884. [DOI] [PubMed] [Google Scholar]
- 7. Rudolph U, Crestani F, Mohler H (2001) GABA(A) receptor subtypes: dissecting their pharmacological functions. Trends Pharmacol Sci 22: 188–194. [DOI] [PubMed] [Google Scholar]
- 8. Semyanov A, Walker MC, Kullmann DM, Silver RA (2004) Tonically active GABA A receptors: modulating gain and maintaining the tone. Trend Neurosci 27: 262–269. [DOI] [PubMed] [Google Scholar]
- 9. Caraiscos VB, Elliott EM, You T, Cheng VY, Belelli D, et al. (2004) Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by a5 subunit-containing gamma-aminobutyric acid type A receptors. Proc Nat Acad Sci 101: 3662–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Glykys J, Mann EO, Mody I (2008) Which GABA(A) receptor subunits are necessary for tonic inhibition in the hippocampus? J Neurosci 28: 1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bonin RP, Martin LJ, MacDonald JF, Orser BA (2007) Alpha5GABAA receptors regulate the intrinsic excitability of mouse hippocampal pyramidal neurons. J Neurophysiol 98: 2244–2254. [DOI] [PubMed] [Google Scholar]
- 12. Glykys J, Mody I (2006) Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABA A receptor alpha5 subunit-deficient mice. J Neurophysiol 95: 2796–2807. [DOI] [PubMed] [Google Scholar]
- 13. Martin LJ, Zurek AA, MacDonald JF, Roder JC, Jackson MF, et al. (2010) Alpha5GABAA receptor activity sets the threshold for long-term potentiation and constrains hippocampus-dependent memory. J Neurosci 30: 5269–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brickley SG, Revilla V, Cull-Candy SG, Wisden W, Farrant M (2001) Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature 409: 88–92. [DOI] [PubMed] [Google Scholar]
- 15. Chen X, Shu S, Schwartz LC, Sun C, Kapur J, et al. (2010) Homeostatic regulation of synaptic excitability: tonic GABA(A) receptor currents replace I(h) in cortical pyramidal neurons of HCN1 knock-out mice. J Neurosci 30: 2611–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wahl-Schott C, Biel M (2009) HCN channels: structure, cellular regulation and physiological function. Cell Mol Life Sci 66: 470–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Magee J (1998) Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci 18: 7613–7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen X, Shu S, Kennedy DP, Willcox SC, Bayliss DA (2009) Subunit-specific effects of isoflurane on neuronal Ih in HCN1 knockout mice. J Neurophysiol 101: 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nolan M, Malleret G, Dudman J, Buhl D, Santoro B, et al. (2004) A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 119: 719–751. [DOI] [PubMed] [Google Scholar]
- 20. Robinson R, Siegelbaum S (2003) Hyperpolarization-activated cation currents: from molecules to physiological function. Ann Rev Physiol 65: 453–533. [DOI] [PubMed] [Google Scholar]
- 21. Magee J, Cook E (2000) Somatic EPSP amplitude is independent of synapse location in hippocampal pyramidal neurons. Nat Neurosci 3: 895–1798. [DOI] [PubMed] [Google Scholar]
- 22. Vargas-Caballero M, Martin L, Salter M, Orser B, Paulsen O (2010) alpha5 Subunit-containing GABA(A) receptors mediate a slowly decaying inhibitory synaptic current in CA1 pyramidal neurons following Schaffer collateral activation. Neuropharmacology 58: 668–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, et al. (2002) Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J Neurosci 22: 5572–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu H, Vervaeke K (2002) Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J Physiol 545: 783–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stradleigh TW, Ogata G, Partida GJ, Oi H, Greenberg KP, et al. (2011) Colocalization of hyperpolarization-activated, cyclic nucleotide-gated channel subunits in rat retinal ganglion cells. J Comp Neurol 519: 2546–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fu XW, Brezden BL, Wu SH (1997) Hyperpolarization-Activated Inward Current in Neurons of the Rat's Dorsal Nucleus of the Lateral Lemniscus In Vitro. J Neurophysiol 78: 2235–2245. [DOI] [PubMed] [Google Scholar]
- 27.Storm JF (1990) Potassium currents in hippocampal pyramidal cells. In: J. Storm-Mathisen JZ, Ottersen OP, editors. Progress in Brain Research: Elsevier. pp. 161–187. [DOI] [PubMed] [Google Scholar]
- 28. Lancaster B, Nicoll RA (1987) Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol 389: 187–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Disterhoft JF, Wu WW, Ohno M (2004) Biophysical alterations of hippocampal pyramidal neurons in learning, ageing and Alzheimer's disease. Age Res Rev 3: 383–406. [DOI] [PubMed] [Google Scholar]
- 30. Andrásfalvy BK, Makara JK, Johnston D, Magee JC (2008) Altered synaptic and non-synaptic properties of CA1 pyramidal neurons in Kv4.2 knockout mice. J Physiol 586: 3881–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wisden W, Laurie DJ, Monyer H, Seeburg PH (1992) The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J Neurosci 12: 1040–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jung S, Warner L, Pitsch J, Becker A, Poolos N (2011) Rapid loss of dendritic HCN channel expression in hippocampal pyramidal neurons following status epilepticus. J Neurosci 31: 14291–14296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gibson J, Bartley A, Huber K (2006) Role for the subthreshold currents ILeak and IH in the homeostatic control of excitability in neocortical somatostatin-positive inhibitory neurons. J Neurophysiol 96: 420–452. [DOI] [PubMed] [Google Scholar]
- 34. Belelli D, Lambert JJ (2005) Neurosteroids: endogenous regulators of the GABA(A) receptor. Nat Rev Neurosci 6: 565–576. [DOI] [PubMed] [Google Scholar]
- 35. Caraiscos VB, Bonin RP, Newell JG, Czerwinska E, Macdonald JF, et al. (2007) Insulin increases the potency of glycine at ionotropic glycine receptors. Mol Pharmacol 71: 1277–1287. [DOI] [PubMed] [Google Scholar]
- 36. Saab B, Maclean A, Kanisek M, Zurek A, Martin L, et al. (2010) Short-term memory impairment after isoflurane in mice is prevented by the α5 γ-aminobutyric acid type A receptor inverse agonist L-655,708. Anesthesiology 113: 1061–1132. [DOI] [PubMed] [Google Scholar]
- 37. Zurek A, Bridgwater E, Orser B (2012) Inhibition of α5 γ-aminobutyric acid type A receptors restores recognition memory after general anesthesia. Anesth Analg 114: 845–900. [DOI] [PubMed] [Google Scholar]