

Abstract
Objective
Menkes disease is a lethal neurodegenerative disorder of infancy caused by mutations in a copper-transporting ATPase gene, ATP7A. Among its multiple cellular tasks, ATP7A transfers copper to dopamine-beta-hydroxylase (DBH) within the lumen of the Golgi network or secretory granules, catalyzing the conversion of dopamine to norepinephrine. In a well-established mouse model of Menkes disease, mottled-brindled, we tested whether systemic administration of L-threo-dihydroxyphenylserine (L-DOPS), a drug used successfully to treat autosomal recessive norepinephrine deficiency, would improve brain neurochemical abnormalities and neuropathology.
Methods
At 8, 10, and 12 days of age, wild type and mo-br mice received intraperi-toneal injections of 200μg/g body weight of L-DOPS, or mock solution. Five hours after the final injection, the mice were euthanized and brains removed. We measured catecholamine metabolites affected by DBH via high-performance liquid chromatography with electrochemical detection, and assessed brain histopathology.
Results
Compared to mock-treated controls, mo-br mice that received intraperitoneal L-DOPS showed significant increases in brain norepinephrine (P<0.001) and its deaminated metabolite, dihydroxyphenylglycol (DHPG, P<0.05). The ratio of a non-beta-hydroxylated metabolite in the catecholamine biosynthetic pathway, dihydroxyphenylacetic acid, to the beta-hydroxylated metabolite, dihydroxyphenylglycol, improved equivalently to results obtained previously with brain-directed ATP7A gene therapy (P<0.01). However, L-DOPS treatment did not arrest global brain pathology or improve somatic growth, as gene therapy had.
Interpretation
We conclude that 1) L-DOPS crosses the blood-brain barrier in mo-br mice and corrects brain neurochemical abnormalities, 2) norepinephrine deficiency is not the cause of neurodegeneration in mo-br mice, and 3) L-DOPS treatment may ameliorate noradrenergic hypofunction in Menkes disease.
Introduction
Menkes disease is an X-linked recessive disorder of copper metabolism described 50 years ago1 caused by mutations in ATP7A (EC 3.6.3.4), a copper-transporting ATPase critical for absorbing copper from the diet and providing it for copper-dependent enzymes, such as dopamine-β-hydroxylase (DBH).2 Plasma and cerebrospinal fluid neurochemical levels are highly sensitive and specific for diagnosis of this illness.3,4 While the major clinical features of Menkes disease are delayed growth and neurodevelopment, seizures5,6 and death by 3 years of age, daily copper injections are sufficient to rescue the neurodegenerative aspects of this disease and prolong survival in some patients.7 However Menkes disease survivors as well as patients with its milder allelic variant, occipital horn syndrome, often exhibit persistent signs of DBH deficiency, including low norepinephrine levels, orthostatic hypotension, hypothermia, and chronic diarrhea.8–10 These findings are similar to the autonomic problems encountered in patients with norepinephrine deficiency caused by mutations in the DBH gene, an autosomal recessive trait.11–13
The drug L-threo-dihydroxyphenylserine (L-DOPS) is converted to norepinephrine through the action of aromatic-L-amino acid decarboxylase (ALAAD, EC 4.1.1.28), and has been useful in treatment of this latter condition, both in human subjects,14,15 and a mouse DBH knockout.16,17 Since ALAAD is not copper-dependent, Menkes disease survivors and occipital horn syndrome patients should convert L-DOPS into NE and secure improved noradrenergic function.10
In the current study, we formally evaluated this hypothesis, using an animal model of Menkes disease, the mottled-brindled (mo-br) mouse.18 The mo-br mutation is a 6 base pair deletion in exon 11 of the mouse ATP7A homolog, Atp7a. This results in deletion of two highly conserved amino acid residues, 799 (alanine) and 800 (leucine) in Atp7a. Since we recently documented improved brain neurochemical ratios in this model by an alternative approach, ATP7A gene therapy,18 the current study also afforded a comparative assessment.
Materials and Methods
L-DOPS formulation and administration
L-threo-3,4-dihydroxyphenylserine (L-DOPS) was obtained via a Material Transfer Agreement between Chelsea Therapeutics, Inc., Charlotte, NC and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (SG Kaler, MTA33023). Since L-DOPS is poorly soluble in water except at low pH, 0.2 M HCl and 1 M NaOH were prepared from concentrated stocks. Titration of the NaOH solution into a sample of the HCl solution was performed to determine the precise volume needed to adjust the HCl solution to pH 7. L-DOPS was freshly dissolved each day (with ~30 minutes of vortexing) in a suitable volume of HCl such that the concentration following neutralization would be 20 mg/ml. Immediately prior to injection, an aliquot of L-DOPS solution was mixed with the appropriate volume of neutralizing NaOH. The neutralized L-DOPS solution in a dose of 200 μg/g body weight was injected via intraperitoneal injection using a syringe fitted with a 28 gauge needle. If precipitate was observed during the injections, the solution was discarded and a new mixture was prepared. Control mice received a comparable volume of neutralized HCl, based on body weight.
Animal Care
All animal experimental procedures were approved by the NICHD Animal Studies Committee. C57BL/6-Atp7amo-br breeding pairs were obtained from Jackson Laboratories (Bar Harbor, ME). Each breeding pair consisted of a female heterozygous for the Atp7amo-br allele and a wild-type male. After the first generation, breeding pairs consisted mainly of sib crosses. For experiments, litters were culled to 4–5 pups. Shortly after birth, toe biopsies were performed for genotyping and subsequent identification as previously described.18 At 8, 10, and 12 days of age, wild type and mutant mice received either 200 μg of L-DOPS per gram body weight by intraperitoneal injection, a regimen used previously by others,19 or a commensurate volume of carrier solution.
Five hours after the final injection, a post-treatment interval considered optimal,17 the mice were euthanized by decapitation. The brains were removed promptly, weighed, and divided sagittally into two halves. One half was fixed in 10% neutral buffered formalin and submitted for paraffin embedding (Histoserve, Germantown, MD) and histopathological staining (hemotoxylin and eosin). The other half was divided into anterior, middle and posterior sections, frozen on dry ice, and stored at −70°C until neurochemical assay. The age of animals at sacrifice (12 days) and processing of mouse brains were the same as in our earlier study of this mouse model.18
Catecholamine metabolite analyses
Individual anterior and posterior brain samples were weighed and homogenized in 5–10 volumes of 0.4 N perchloric acid containing 0.1% ethylene diamine tetra-acetic acid. The homogenates were centrifuged and supernatant frozen at −70 °C until assay. Concentrations (pg/mg protein) of dopamine (DA), norepinephrine (NE), 3,4-dihydroxyphenylalanine (DOPA), 3,4-dihydroxyphenylacetic acid (DOPAC), 3,4-dihydroxyphenylglycol (DHPG), and dihydroxyphenylacetaldehyde (DOPAL) were determined by high-performance liquid chromatography with electrochemical detection, as previously described.18
Results
Brain catecholamine metabolite levels
In anterior brain segments, DA was unchanged in mo-br compared to wild type, whereas DOPAC was markedly elevated in mo-br mice, relative to normal controls (P<0.001, Fig. 2A). Levels were not significantly affected by treatment with L-DOPS. In contrast, NE (P<0.001) and DHPG (P<0.001) levels were markedly lower in mo-br mice, and L-DOPS treatment significantly increased the levels of these neurotransmitters in the mo-br mice (P<0.001 and P<0.05, respectively, Fig. 2A). The peak for DOPA was obscured by a co-migrating peak in five anterior brain samples (three mo-br and two wild type); DOPA was thus omitted from the analysis.
Figure 2.
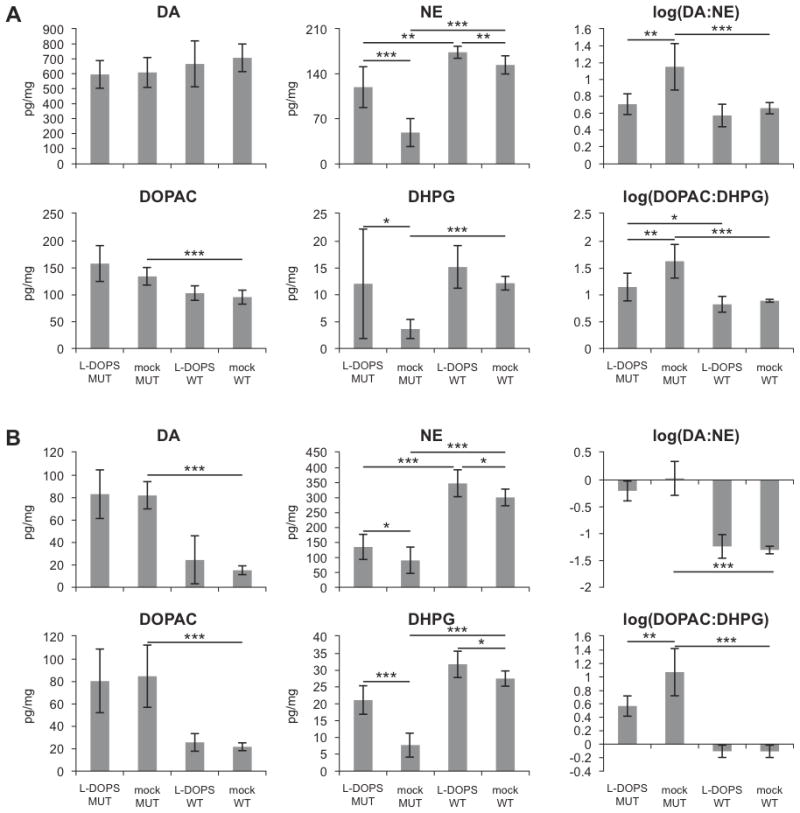
L-DOPS treatment increases NE and DHPG concentrations in the brain. Concentrations of DA, NE, DOPAC, and DHPG were measured in the anterior (A) and posterior (B) brain sections. Data are mean ± standard deviation, with n=6 except for DHPG (see Materials and Methods). *p<0.05, **p<0.01, ***p<0.001.
In the posterior brain, DA and DOPAC were both markedly elevated in mo-br mice relative to wild type mice (P<0.001, Fig. 2B), and L-DOPS treatment did not affect their concentrations (Fig. 2B). NE and DHPG levels were each markedly decreased in mo-br mice relative to controls (P<0.001, Fig. 2B), and L-DOPS treatment increased the concentration of these molecules in both mutant and normal individuals (P<0.001 in mutant; P<0.05 in wild type, Fig. 2B). As with anterior brain, we were unable to quantitate brain DOPA levels due to a co-migrating peak that obscured this catechol in numerous samples.
The ratio of a non-beta-hydroxylated metabolite in the catecholamine biosynthetic pathway, DOPAC, to a beta-hydroxylated metabolite, DHPG, improved in both anterior and posterior brain, equivalently to results obtained previously with brain-directed ATP7A gene addition (P<0.01).18
The concentration of DOPAL was also measured and found to be significantly increased by L-DOPS treatment in both brain regions of mo-br mice, and in the posterior region of wild-type mice (Fig. 3).
Figure 3.
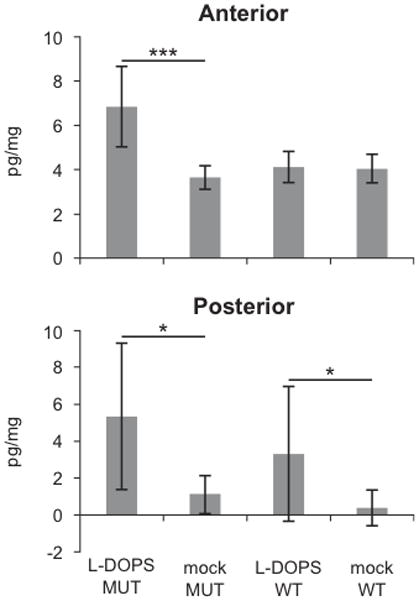
DOPAL levels increase in the brain following L-DOPS treatment. DOPAL was increased in the posterior brain in wild-type (WT) and in mo-br animals treated with L-DOPS, and in the anterior brain in mo-br (MUT) animals. Data are mean ± standard deviation, n=6, *p<0.05, ***p<0.001.
Brain Pathology
Mo-br mice at 12 days of age showed abnormal neurons with pyknotic nuclei throughout the hippocampus and cerebral cortex regardless of treatment with L-DOPS (Fig. 4A,C). In comparison, wild-type mice that received L-DOPS or mock treatment showed no obvious neuropathology (Fig. 4B,D).
Figure 4.
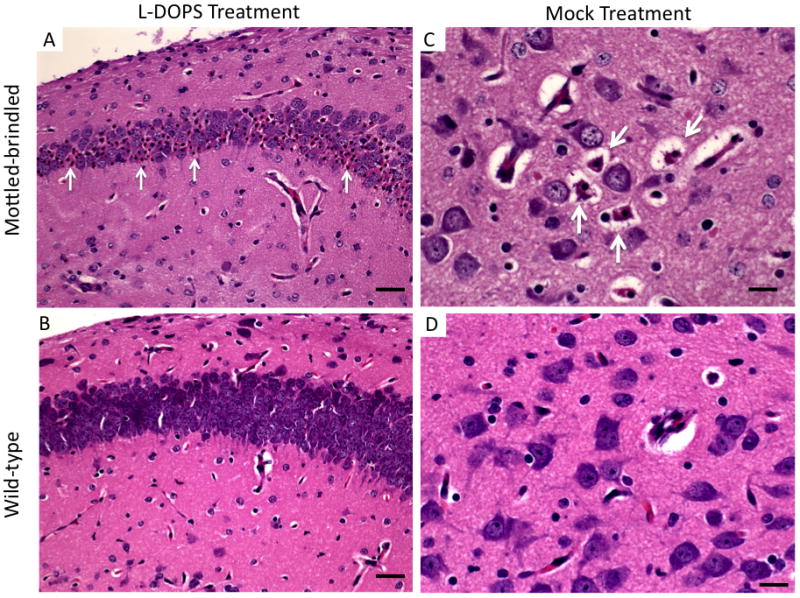
L-DOPS does not prevent brain pathology in mo-br mice. A. Numerous abnormal hippocampal neurons with pyknotic nuclei (arrows) in 12 day mo-br brain. B. Normal appearance of the wild-type hippocampus. C. Variable degrees of neuron shrinkage in 12 day mo-br cerebral cortex is evident (arrows). D. Wild-type cerebral cortical neurons show no appreciable pathology. Mo-br mouse brains had equivalently abnormal histopathology with or without L-DOPS treatment. Hematoxylin & eosin stain. Scale bars: 350 μm (A,B) and 200 μm (C,D)
Growth rate of mice
Body weights were measured at 8, 10, and 12 days of age before injections with L-DOPS or mock carrier. Untreated mo-br mice grew at a slower rate than untreated wild type mice (Fig. 5). Treatment with L-DOPS had no discernable growth effects in either mutant or wild type mice.
Figure 5.

Weight gain in L-DOPS-treated and untreated mice. Animal weights were measured on each injection day (ages eight, ten, and twelve days). Weight gain was not adversely affected by L-DOPS treatment for either mo-br (MUT, treated n=7, untreated n=6) or wild type (WT, treated n=7, untreated n=5) mice. Data are mean ± SEM.
Discussion
Among its multiple cellular tasks, ATP7A transfers copper to dopamine-beta-hydroxylase (DBH) within the lumen of the trans-Golgi network or secretory granules where DBH mediates the conversion of dopamine to norepinephrine. Other ATP7A functions include removal of copper from cells across the plasma membrane in response to increases in its concentration, pumping copper into melanosomes for metallation of tyrosinase, contribution to axonal and synaptic development, and involvement in neuronal activation.2 Mutations in ATP7A, or its murine homolog, Atp7a, impair all these processes, with the defect in copper transfer to DBH resulting in distinctively abnormal neurochemical patterns, including markedly elevated ratios of non-beta-hydroxylated: beta-hydroxylated metabolites in the catecholamine biosynthetic pathway.3,4,7,8–10,18,20,21
In our previous study of the mo-br mouse,18 we found that brain-directed adeno-associated virus-mediated transfer of ATP7A plus copper significantly lowered the brain ratios of dihydroxyphenylacetic acid: dihydroxyphenylglycol (DOPAC: DHPG), reflecting enhanced dopamine-β-hydroxylase activity. In fact, this improvement reflected the only statistically significant change among several copper enzyme activities that correlated with rescue of mo-br. A plausible explanation for these findings is that ATP7A is specifically required for copper delivery to the trans-Golgi network, the compartment in which DBH apoenzyme is processed and refined to its mature, functional, active form. In contrast, the other cuproenzymes examined (Cu/Zn superoxide dismutase and cytochrome c oxidase) may be influenced by intracellular copper availability but are not directly dependent on ATP7A for receipt of the metal.
Coupled with the high prenatal lethality associated with DBH gene knockout in mice,16 the neurochemical effects of ATP7A gene transfer suggested that correction of brain norepinephrine deficiency by small molecule therapeutic L-DOPS might also mediate rescue of the mo-br mouse model. However, the present study demonstrates unequivocally that L-DOPS does not prevent growth retardation or neurodegeneration in mo-br, despite correction of the brain neurochemical abnormality. Brain catechol levels were measured at the same age and in the same fashion in our gene therapy and L-DOPS studies. However, the timing and routes of administration were necessarily distinct. The former study involved a single intracerebroventricular (ICV) injection of recombinant adeno-associated virus, serotype 5 (AAV5) carrying a compact version of the human ATP7A complementary DNA (cDNA) on postnatal day 2, followed by 50ng ICV copper on postnatal day 3.18 No L-DOPS was used. In the current study, only L-DOPS was administered by intraperitoneal injection on postnatal days 8, 10, and 12, and brains were harvested 5h after the third dose, parameters based on the work and findings of others.17,18 While we cannot formally exclude effects on brain catechol levels and neuropathology based on the timing of treatment and administration route, we speculate that the agents used in these interventions and their respective mechanisms of action represent more critical factors in the cumulative results.
We measured neurochemical levels in two regions of the mouse brain: the anterior third, which includes cerebral cortex, ventral striatum, caudate putamen, and anterior olfactory nucleus, and the posterior third, which includes cerebellum, medulla, and pons. The findings were similar in both regions (Fig. 2). As expected, NE and DHPG levels were reduced in mo-br mice relative to wild type, while DA and DOPAC were elevated, reflecting the DBH deficiency associated with Menkes disease. One exception was DA in the anterior brain, which showed no difference in comparison to wild type.
These changes in neurochemical levels also resulted in statistically significant differences in the DA:NE and DOPAC:DHPG ratios between mo-br and wild type mice, similar to results obtained in CSF obtained from untreated Menkes disease patients.3,20,21 One notable difference we observed was a significantly lower DHPG level in mo-br brain relative to wild type controls, a finding not observed in CSF of Menkes disease patients.3 This may indicate a difference in central DBH activity between humans and mice with impaired copper-ATPase-mediated transport.
DA appeared to predominate in the anterior portion of the brain, most likely due to the density of dopaminergic neurons in the caudate putamen and ventral striatum. In contrast, NE dominated in the posterior brain region, likely related to noradrenergic neurons located in the pons (including the locus ceruleus) and the relative lack of dopaminergic neurons in this region.
In agreement with studies of DBH knockout mice,16,17 treatment of mo-br mice with L-DOPS significantly increased NE and DHPG levels in the anterior and posterior brain regions. These findings lend additional evidence that L-DOPS crosses the mammalian blood-brain barrier.
DOPAL is a highly reactive intermediate metabolite of DA that is rapidly converted to DOPAC (Fig. 1). DOPAL has toxic effects on neurons in vitro and in vivo, especially dopaminergic neurons in the substantia nigra.23–25 It has been hypothesized that DOPAL may contribute to neuronal death in Parkinson’s disease.26 We detected this catecholaldehyde in untreated, as well as treated mice, although treatment increased brain DOPAL levels by 2 to 8 fold. Trace contamination of the L-DOPS formulation with DOPAL is one possible explanation for this result.22 Alternatively, treatment with L-DOPS may enhance MAO-mediated catabolism of DA, which would increase DOPAL production (Fig. 1). Of note, the highest concentration of DOPAL we observed, 6.8 pg/mg wet tissue in the posterior brain samples of mo-br mice, was more than 58-fold lower than in the substantia nigra from a 67 year old male (397 pg/mg wet wt) reported by others.27
Figure 1.
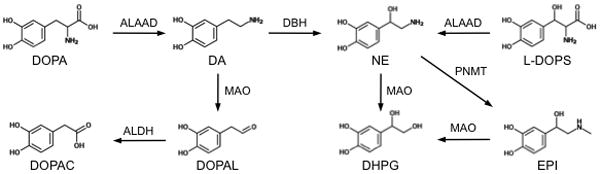
Synthesis of dopamine (DA) and norepinephrine (NE) and their metabolites. Normally, NE is synthesized from DA by dopamine-β-hydroxylase (DBH). Loss of DBH activity results in reduced NE concentrations. DBH deficiency can be bypassed by the administration of L-threo-dihydroxyphenylserine (L-DOPS), which is converted by aromatic-L-aminoacid decarboxylase (ALAAD) directly into NE. EPI – epinephrine, DHPG – 3,4-dihydroxyphenylglycol, DOPAL - 3,4-dihydroxyphenylacetaldehyde, DOPAC – 3,4-dihydroxyphenylacetic acid, DOPA – L-3,4-dihydroxyphenylalanine, MAO – monoamine oxidase, ALDH – aldehyde dehydrogenase, PNMT – phenolethanolamine-N-methyltransferase.
In summary, we show here that L-DOPS can be converted to NE in the central nervous system of an animal model of Menkes disease. Taken together with anecdotal successes of L-DOPS in medical management of Menkes disease survivors10 and individuals with DBH deficiency,11,12,14,15 our preclinical results in an animal model suggest that L-DOPS may have wider clinical application. Alternative treatments such as pharmacological chaperones, recently proposed for subjects with norepinephrine deficiency due to misfolded mutant DBH proteins,13 are not relevant for situations in which defective copper transport represents the underlying molecular basis. While L-DOPS alone does not correct the fundamental defect in copper transport responsible for neurodegeneration in Menkes disease, it may ameliorate noradrenergic hypofunction and be useful, in combination with other treatment. Long-surviving Menkes disease patients with favorable neurodevelopmental outcomes due to very early copper treatment, and subjects with mild Menkes disease phenotypes, including occipital horn syndrome, may especially benefit from this medical intervention.
Acknowledgments
This work was supported the intramural research programs of NICHD and NINDS. We thank Tracey Rouault and Irv Kopin for critical review of the manuscript, and Marie Reine Haddad for assistance with mouse brain dissection.
Footnotes
Potential Conflicts of Interest: Nothing to report.
References
- 1.Menkes JH, Alter M, Steigleder GK, et al. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics. 1962;29:764–779. [PubMed] [Google Scholar]
- 2.Kaler SG. The neurology of ATP7A copper transporter disease: emerging concepts and future trends. Nat Rev Neurol. 2011;7:15–29. doi: 10.1038/nrneurol.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaler SG, Goldstein DS, Holmes C, et al. Plasma and cerebrospinal fluid neurochemical pattern in Menkes disease. Ann Neurol. 1993;33:171–175. doi: 10.1002/ana.410330206. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein DS, Holmes CS, Kaler SG. Relative efficiencies of plasma catechol levels and ratios for neonatal diagnosis of Menkes disease. Neurochem Res. 2009;34:1464–1468. doi: 10.1007/s11064-009-9933-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White SR, Reese K, Sato S, Kaler SG. Spectrum of EEG findings in Menkes disease. Electroenceph Clin Neurophysiol. 1991;87:57–61. doi: 10.1016/0013-4694(93)90175-u. [DOI] [PubMed] [Google Scholar]
- 6.Kaler SG, Liew CJ, Donsante A, et al. Molecular correlates of epilepsy in early diagnosed and treated Menkes disease. J Inher Metab Dis. 2010;33:583–589. doi: 10.1007/s10545-010-9118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaler SG, Holmes CS, Goldstein DS, Tang J, Godwin SC, Donsante A, Liew CJ, Sato S, Patronas N. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358(6):605–14. doi: 10.1056/NEJMoa070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaler SG, Gallo LK, Proud VK, et al. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat Genet. 1994;8:195–202. doi: 10.1038/ng1094-195. [DOI] [PubMed] [Google Scholar]
- 9.Tang J, Donsante A, Desai V, et al. Clinical outcomes in Menkes disease patients with a copper-responsive ATP7A mutation, G727R. Mol Genet Metab. 2008;95:174–181. doi: 10.1016/j.ymgme.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christodoulou J, Danks DM, Sarkar B, et al. Early treatment of Menkes disease with parenteral copper-histidine: long-term follow-up of four treated patients. Am J Med Genet. 1998;76(2):154–164. [PubMed] [Google Scholar]
- 11.Senard JM, Rouet P. Dopamine-beta-hydroxylase deficiency. Orphanet J Rare Dis. 2006;1:7. doi: 10.1186/1750-1172-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robertson D, Haile V, Perry SE, et al. Dopamine beta-hydroxylase deficiency. A genetic disorder of cardiovascular regulation. Hypertension. 1991;18(1):1–8. doi: 10.1161/01.hyp.18.1.1. [DOI] [PubMed] [Google Scholar]
- 13.Kim C, Leung A, Huh Y, et al. Norepinephrine deficiency is caused by combined abnormal mRNA processing and defective protein trafficking of dopamine β-hydroxylase. J Biol Chem. 2011;286(11):9196–9204. doi: 10.1074/jbc.M110.192351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biaggioni I, Robertson D. Endogenous restoration of noradrenaline by precursor therapy in dopamine-beta-hydroxylase deficiency. Lancet. 1987;2(8569):1170–2. doi: 10.1016/s0140-6736(87)91317-1. [DOI] [PubMed] [Google Scholar]
- 15.Man in’t Veld AJ, Boomsma F, van den Meiracker AH, Schalekamp MA. Effect of unnatural noradrenaline precursor on sympathetic control and orthostatic hypotension in dopamine-beta-hydroxylase deficiency. Lancet. 1987;2(8569):1172–5. doi: 10.1016/s0140-6736(87)91318-3. [DOI] [PubMed] [Google Scholar]
- 16.Thomas SA, Matsumoto AM, Palmiter RD. Noradrenaline is essential for mouse fetal development. Nature. 1995;374(6523):643–646. doi: 10.1038/374643a0. [DOI] [PubMed] [Google Scholar]
- 17.Thomas SA, Marck BT, Palmiter RD, Matsumoto AM. Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine beta-hydroxylase. J Neurochem. 1998;70(6):2468–76. doi: 10.1046/j.1471-4159.1998.70062468.x. [DOI] [PubMed] [Google Scholar]
- 18.Donsante A, Yi L, Zerfas PM, et al. ATP7A gene addition to the choroid plexus results in long-term rescue of the lethal copper transport defect in a Menkes disease mouse model. Mol Ther. 2011;19(12):2114–23. doi: 10.1038/mt.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalinin S, Polak PE, Lin SX, et al. The noradrenaline precursor L-DOPS reduces pathology in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2012;33(8):1651–63. doi: 10.1016/j.neurobiolaging.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donsante A, Johnson P, Jansen LA, Kaler SG. Somatic mosaicism in Menkes disease suggests choroid plexus-mediated copper transport to the developing brain. Am J Med Genet A. 2010;152A(10):2529–34. doi: 10.1002/ajmg.a.33632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haddad MR, Macri C, Holmes CS, et al. In utero copper treatment for Menkes disease associated with a severe ATP7A mutation. Mol Genet Metab. 2012 May 18; doi: 10.1016/j.ymgme.2012.05.008. http://dx.doi.org/10.1016/j.ymgme.2012.05.008. [DOI] [PMC free article] [PubMed]
- 22.Holmes C, Whittaker N, Heredia-Moya J, Goldstein DS. Contamination of the norepinephrine prodrug L-DOPS by dihydroxyphenylacetaldehyde. Clin Chem. 2010;56(5):832–838. doi: 10.1373/clinchem.2009.139709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamensdorf I, Eisenhofer G, Harvey-White J, et al. 3,4-Dihydroxyphenyl-acetaldehyde potentiates the toxic effects of metabolic stress in PC12 cells. Brain Res. 2000;868(2):191–201. doi: 10.1016/s0006-8993(00)02309-x. [DOI] [PubMed] [Google Scholar]
- 24.Lamensdorf I, Eisenhofer G, Harvey-White J, et al. Metabolic stress in PC12 cells induces the formation of the endogenous dopaminergic neurotoxin, 3,4-dihydroxyphenyl-acetaldehyde. J Neurosci Res. 2000;60:552–558. doi: 10.1002/(SICI)1097-4547(20000515)60:4<552::AID-JNR14>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 25.Marchitti SA, Deitrich RA, Vasiliou V. Neurotoxicity and metabolism of the catechol-amine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenyl-glycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev. 2007;59:125–150. doi: 10.1124/pr.59.2.1. [DOI] [PubMed] [Google Scholar]
- 26.Mattammal MB, Haring JH, Chung HD, et al. An endogenous dopaminergic neurotoxin: implication for Parkinson’s disease. Neurodegeneration. 1995;4:271–81. doi: 10.1016/1055-8330(95)90016-0. [DOI] [PubMed] [Google Scholar]
- 27.Burke WJ, Chung HD, Li SW. Quantitation of 3,4-dihydroxyphenylacetaldehyde and 3, 4-dihydroxyphenylglycolaldehyde, the monoamine oxidase metabolites of dopamine and noradrenaline, in human tissues by microcolumn high-performance liquid chromatography. Anal Biochem. 1999;273:111–116. doi: 10.1006/abio.1999.4196. [DOI] [PubMed] [Google Scholar]