

Background: α-Synuclein is known to undergo exchange between membrane and cytosolic compartments.
Results: α-Synuclein interacts with GTP-bound Rab3a on synaptic vesicles, and its dissociation is mediated by GDI/Hsp90.
Conclusion: α-Synuclein's membrane association and dissociation cycle is linked to synaptic activity by the Rab3a recycling machinery.
Significance: Significance: Impairments to α-synuclein interactions with vesicles and with the Rab3a recycling machinery may affect neurodegeneration.
Keywords: GTPase, Membrane Proteins, Neurodegeneration, Protein Complexes, Rab Proteins, Subcellular Fractionation, Synaptosomes, Synuclein
Abstract
α-Synuclein is an abundant presynaptic protein and a primary component of Lewy bodies in Parkinson disease. Although its pathogenic role remains unclear, in healthy nerve terminals α-synuclein undergoes a cycle of membrane binding and dissociation. An α-synuclein binding assay was used to screen for vesicle proteins involved in α-synuclein membrane interactions and showed that antibodies directed to the Ras-related GTPase Rab3a and its chaperone RabGDI abrogated α-synuclein membrane binding. Biochemical analyses, including density gradient sedimentation and co-immunoprecipitation, suggested that α-synuclein interacts with membrane-associated GTP-bound Rab3a but not to cytosolic GDP-Rab3a. Accumulation of membrane-bound α-synuclein was induced by the expression of a GTPase-deficient Rab3a mutant, by a dominant-negative GDP dissociation inhibitor mutant unable to recycle Rab3a off membranes, and by Hsp90 inhibitors, radicicol and geldanamycin, which are known to inhibit Rab3a dissociation from membranes. Thus, all treatments that inhibited Rab3a recycling also increased α-synuclein sequestration on intracellular membranes. Our results suggest that membrane-bound GTP-Rab3a stabilizes α-synuclein on synaptic vesicles and that the GDP dissociation inhibitor·Hsp90 complex that controls Rab3a membrane dissociation also regulates α-synuclein dissociation during synaptic activity.
Introduction
α-Synuclein (α-syn)6 is a key factor in the pathogenesis of Parkinson disease (PD). Missense mutations in its amino-terminal domain and gene multiplication cause autosomal dominant forms of PD (1). In addition, accumulation of fibrillar α-syn is a primary pathological characteristic in brains of individuals affected with PD and a wider group of neurodegenerative disorders named synucleinopathies. Several lines of evidence suggest that the physiological function of α-syn involves a nonessential regulatory role in neurotransmitter secretion. Animals with α-syn gene deletions are viable but display subtle increases in transmitter release and some rearrangements of the reserve synaptic vesicle pool (2, 3). α-syn overexpression is associated with a modest inhibition of secretion (4–6). These effects are consistent with studies showing that α-syn is concentrated at presynapses, associated with both cytosolic and membrane compartments (7–10), and it may be linked to the organization and stability of multimeric SNARE fusion machinery (11).
Although it lacks a conventional transmembrane domain, the amino-terminal half of α-syn can assume an amphipathic α-helical conformation upon interaction with lipid bilayers. The alignment of hydrophobic and hydrophilic amino acids in opposing directions enables partial insertion of α-syn into lipid bilayers that is further stabilized by laterally oriented lysines that interact with the negatively charged lipid head groups (12–15). The presence of PD-linked familial mutations (A30P, A46K, and A53T) within α-syn's lipid binding domain has raised speculation that α-syn's association with subcellular membranes, particularly synaptic vesicles, may relate to α-syn's role in disease.
Studies that have examined α-syn distribution and mobility at presynaptic terminals indicate that α-syn is highly mobile and its membrane association is linked to neuronal activity. Experiments with neurons expressing GFP-tagged α-syn show that depolarization increases α-syn mobility and diffusion away from nerve terminals followed by a slower recovery to the resting state during repolarization (10). Using a cell-free assay composed of permeabilized synaptosomes, we reported that cytosolic proteins accelerate α-syn dissociation from membranes (9). In addition, α-syn binding to permeabilized synaptosomes is stabilized by chemical cross-linking suggesting that protein assemblies on synaptic membranes regulate α-syn membrane binding in addition to its α-helical structure (16).
Therefore, to gain further insight into the molecular regulation of α-syn compartmentalization, we screened various antibodies to synaptic vesicle proteins for their ability to block α-syn binding. We surmised that such interference might underlie a functional relationship. This study presents several lines of evidence that α-syn cycling on and off synaptic vesicles is coupled to exocytosis in partnership with the Rab3a recycling machinery that includes Rab-specific GDP dissociation inhibitor (GDI) and the 90-kDa heat shock protein (Hsp90).
EXPERIMENTAL PROCEDURES
Transgenic Mice
Mice expressing human wild-type, A30P, or A53T α-syn were created as described previously (9) and crossed with SNCA null mice (2) from The Jackson Laboratory to create transgenic lines lacking endogenous murine α-syn. All animal experiments were performed according to guidelines established in the Canadian Guide for the Care and Use of Laboratory Animals.
Synaptosome Preparation
Synaptosomes were prepared as described previously (17, 18). Briefly, the murine brains minus cerebella were homogenized with 10 strokes of a Teflon pestle at 500 rpm in ice-cold buffer A (320 mm sucrose, 1 mm EGTA, and 5 mm HEPES, pH 7.4). The homogenate was centrifuged at 1000 × g for 10 min. Next, the supernatant was spun for 10 min at 13,300 × g, and the resulting pellet (P2) was resuspended in buffer A. The P2 fraction was loaded onto a discontinuous Ficoll gradient (13, 9, and 5% in buffer A) and centrifuged for 35 min at 35,000 × g. Intact synaptosomes at the 9–13% interface were resuspended in buffer B (140 mm NaCl, 5 mm KCl, 20 mm HEPES, 5 mm NaHCO3, 1.2 mm Na2HPO4, 1 mm MgCl2, and 10 mm glucose). To generate synaptosomal membranes, intact synaptosomes centrifuged at 24,000 × g for 10 min and the resulting pellet were resuspended for 10 min in hypotonic buffer C (10 mm HEPES, 18 mm KOAc, pH 7.2), centrifuged at 24,000 × g for 10 min, and resuspended in buffer D (25 mm HEPES, 125 mm KOAc, and 2.5 mm MgCl2) for use in binding or dissociation experiments.
Cytosol Preparation
Mouse brains were thoroughly homogenized in 250 μl/brain volume of buffer A (85 mm sucrose, 100 mm KOAc, 1 mm MgOAc, and 20 mm HEPES, pH 7.4). The homogenate was centrifuged for 10 min at 15,000 × g and the supernatant spun for 1 h at 100,000 × g. The supernatant was subsequently dialyzed for 4 h in buffer B (145 mm KOAc and 25 mm HEPES, pH 7.2) and then centrifuged for 25 min at 28,000 × g. Protein concentration was determined by BCA protein assay (Pierce) and aliquots stored at −80 °C.
Glycerol Gradient Analyses
Synaptosomes were cross-linked with 4% paraformaldehyde for 15 min at room temperature, centrifuged at 24,000 × g for 10 min, and then rinsed with buffer B (from synaptosome preparation). Samples were then resuspended into buffer D (from synaptosome preparation) containing 1% Triton X-100. After a 10-min spin at 24,000 × g, supernatants were loaded on the top of 5–40% (v/v) glycerol gradient. The gradients were centrifuged at 35,000 × g for 15 h and separated into 1-ml fractions for analysis by Western blotting.
α-syn Binding
Synaptosomal membranes prepared from α-syn-deficient mice were incubated for 10 min at 37 °C with 1.5 mg/ml α-syn-deficient cytosol supplemented with 3 μg of recombinant WT, A30P, or A53T α-syn in the presence or absence of specific antibodies. Membranes were then centrifuged at 24,000 × g for 10 min, and supernatants were saved for Western blotting analyses. Pellets were rinsed twice with buffer D, centrifuged at 24,000 × g for 10 min, and then resuspended in 1% SDS buffer. α-syn binding was quantified by Western blotting (16).
α-syn Dissociation
Synaptosomal membranes prepared from transgenic WT, A30P, or A53T α-syn mice were incubated for 10 min at 37 °C with 1.5 mg/ml α-syn-deficient cytosol in the presence or absence of Hsp90 inhibitors. Samples were then centrifuged at 24,000 × g for 10 min. α-syn dissociation into the supernatants was assessed by Western blotting (9).
Immunoprecipitation
Membrane-bound or cytosolic fractions from ∼1.5 mg of murine synaptosomes were resuspended in Tris-lysis buffer with 1% CHAPS and incubated for 1 h with a monoclonal anti-α-syn antibody (syn-1; BD Biosciences) or preimmune mouse serum. Protein G-agarose beads (Sigma) were then added to each sample overnight. Following three washes with Tris-lysis buffer, the resulting bead-bound proteins were removed through incubation with 50 μl of 2× Sample Buffer (20% v/v glycerol, 0.1 m Tris, pH 6.8, 4% SDS, 0.008% bromphenol blue, and 2.5% β-mercaptoethanol) at 95 °C for 5 min.
Cell Culture and Rab3a/GDI Expression
SH-SY5Y neuroblastoma cells (ATCC) were maintained Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and 0.5% penicillin/streptomycin (Wisent). High potassium stimulation was achieved by replacing the media with 55 mm high potassium buffer (80 mm NaCl, 55 mm KCl, 5 mm NaHCO3, 1.2 mm Na2HPO4, 1 mm MgCl2, 10 mm glucose, 2.5 mm CaCl2, and 20 mm NaHEPES, pH 7.2) for 10 min at 37 °C prior to harvesting. In some experiments, CaCl2 was reduced or replaced with EGTA (1 mm); radicicol (50 μm) or geldanamycin (20 μm) was included. Care was taken so that the DMSO used to dissolve radicicol or geldanamycin did not exceed a final concentration of 0.2%.
For fractionation of membrane and cytosolic proteins, cells were hypotonically lysed in swelling buffer (10 mm HEPES and 18 mm KOAc, pH 7.2) and centrifuged at 20,000 × g for 5 min and separated into supernatant and pellet. Supernatant was centrifuged again at 100,000 × g for 15 min to remove any contaminating membranes, and the resulting supernatant was kept as the cytosolic fraction.
His6 epitope-tagged Rab3a and GDI mutant constructs were generously provided by William Balch (Scripps Research Institute). Rab3a sequences were subcloned into pcDNA3.1 vector (Invitrogen) for transient transfection using a Nucleofector II (Amaxa). GDI constructs were inserted into pWPI lentiviral vectors for expression. Virus production was done in HEK293T cell by co-transfection with 3 μg of envelope plasmid pMD2.G (Addgene plasmid 12259), 5 μg of packaging plasmid pPAX2 (Addgene plasmid 12260), and 8 μg of either pWPI/−/Neo, pWPI/hGDIWT/Neo, pWPI/hGDIR240A/Neo, or pWPI/hGDIR218E/Neo using Lipofectamine 2000 (Invitrogen). The lentivirus was collected 48 h post-transfection, cleared by centrifugation (3000 rpm for 15 min), and filtered through 0.45-μm pore size cellulose acetate filters. SH-SY5Y cells were then infected with the virus for 24 h, rinsed in DMEM to remove excess viral particles, and then cultured in DMEM with 10% FBS. 48 h post-infection, the infected SH-SY5Y cells were selected with G418 (50 μg/ml) for 2 weeks. Rab3a knockdown was done using four separate siRNAs against Rab3a A-D (J-009668-07, J-009668-08, J-009668-09, and J-009668-10, respectively, 0.5 nmol each) and one scrambled control (nontargeting control 1)(ON-TARGETplus from Dharmacon).
Expression and Purification of Recombinant α-syn
Human WT α-syn cDNAs were subcloned into the plasmid pET-28a (Novagen), using NcoI and HindIII restriction sites. α-syn was overexpressed in Escherichia coli BL21 (DE3) via an isopropyl-1-thio-3/4-d-galactopyranoside-inducible T7 promoter. The bacterial pellet was resuspended in phosphate-buffered saline (PBS) containing 1 mm phenylmethylsulfonyl fluoride (PMSF). The bacterial suspension was then sonicated for 30 s several times, boiled for 15 min, and ultracentrifuged at 150,000 × g for 30 min. The supernatant containing the heat-stable α-syn was dialyzed against 50 mm Tris, pH 8.3, loaded onto a Q-Sepharose column (GE Healthcare), and eluted with a 0–500 mm NaCl step gradient. The resulting α-syn fractions were desalted and concentrated on a Centricon-10 (Millipore) in 5 mm phosphate buffer, pH 7.3.
Western Blotting
Proteins were boiled briefly in loading buffer (glycerol 10% v/v; 0.05 m Tris, pH 6.8, SDS 2%, bromphenol blue, and 2.5% v/v β-mercaptoethanol) and separated by electrophoresis using 12% Tris-glycine polyacrylamide gels. Proteins were transferred to nitrocellulose (Life Sciences) and probed by Western blotting using the following antibodies: α-syn (monoclonals, 211 from Neomarkers and Syn-1 from BD Transduction Laboratories, both used at 1:1000); a rabbit polyclonal raised to a 24-mer α-syn-specific peptide (LWS1, 1:1000); synaptophysin (monoclonal antibody at 1:10,000, Biodesign International); synphilin-1 (goat polyclonal anti-carboxyl-terminal at 1:2000, Research Diagnostics, Inc.); Rab3a (polyclonal 3335; Abcam); GDI (monoclonal 81.2; Synaptic Systems); Hsp90 (polyclonal 44721; Abcam); tetra-His (Qiagen); syntaxin (monoclonal HPC-1; Sigma); and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (monoclonal 6C5; Meridian Life Science). Bound HRP-conjugated anti-mouse or anti-rabbit IgG (Sigma) was revealed by chemiluminescence using ECL Plus (GE Healthcare) and quantified with a Storm 860 fluorescent imager and ImageQuant software (Molecular Dynamics). Statistical comparisons were done with GraphPad InStat software using Student's t test for comparisons between two groups or ANOVA (Bonferroni test) for multiple comparisons.
RESULTS
α-syn Membrane Binding Is Inhibited by Rab3a Antibodies
As a preliminary screen to identify putative α-syn membrane binding partners, we took advantage of our in vitro α-syn binding assay (16) to assess whether antibodies to various synaptic vesicle proteins could modify the extent of α-syn membrane binding. Hypotonically lysed brain synaptosomes derived from α-syn-deficient mice were incubated with α-syn-deficient cytosol and E. coli-expressed recombinant α-syn. Under these conditions, α-syn was observed to bind to synaptosomal membranes. As expected, inclusion of a polyclonal α-syn antibody eliminated virtually all of the α-syn binding, whereas neither preimmune serum nor anti-synaptophysin antibodies had any effect (Fig. 1A). Interestingly, Rab3a and synphilin-1 antibodies also significantly reduced α-syn binding to synaptosomal membranes. Although synphilin-1 is a known α-syn interacting protein (19), the interaction between α-syn and Rab3a has not been reported. Our results suggested that Rab3a either plays a role in α-syn membrane association or is in close proximity to α-syn-binding sites. To distinguish between these possibilities, we determined whether α-syn and Rab3a form a stable complex. Rab3a was detected in anti-α-syn immunoprecipitates from mouse brains expressing wild-type or PD-linked mutant α-syn but not from α-syn-deficient mice (Fig. 1B).
FIGURE 1.
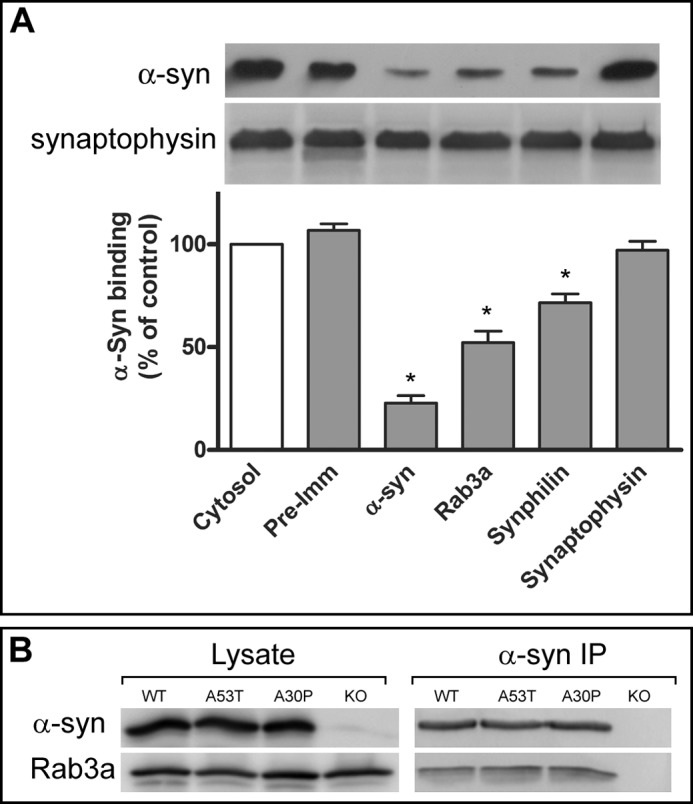
Blockade of α-syn membrane binding by specific antibodies. A, synaptic membranes from α-syn-deficient mice were incubated with 1.5 mg/ml α-syn-deficient brain cytosol and 3 μg of recombinant wild-type human α-syn in the absence (Cytosol) or presence of either pre-immune serum (Pre-Imm) or specific antibodies to presynaptic proteins (α-syn, Rab3a, Synphilin-1, and Synaptophysin). The top panel shows representative Western blots of membrane-bound α-syn and endogenous synaptophysin, and the lower panel shows α-syn binding as a percent of binding in the presence of cytosol alone (mean ± S.E., n = 4). B, α-syn was immunoprecipitated (IP) from detergent-solubilized lysates of synaptosomes from transgenic WT, A30P, A53T, and α-syn knock-out mice using LWS1 anti-α-syn polyclonal antibody. Western blots confirmed the co-immunoprecipitation of Rab3a from α-syn-expressing but not α-syn knock-out animals. *, p < 0.01.
Rab3a and α-syn Complex Are Associated with Synaptic Membranes
α-Syn and Rab3a are both abundant synaptic vesicle proteins known to cycle on and off synaptic vesicles (10, 17). Whereas the mechanisms regulating α-syn membrane binding and dissociation are not understood, the protein complex that mediates Rab3a cycling is composed of Rab-specific GDP-dissociation inhibitor (GDI) and Hsp90 (20, 21). To address whether α-syn might be functionally linked to the Rab3a recycling machinery, we first assessed whether they co-migrate on glycerol density gradients, which can differentiate large protein assemblies according to size (22).
Elution profiles of α-syn and Rab3a from detergent-solubilized synaptosomes showed co-migration in two major peaks as follows: an abundant low molecular weight fraction eluting with 29–66-kDa markers and a high molecular weight fraction associated with 200–443-kDa markers (Fig. 2A). In contrast, GDI and Hsp90 were localized to the middle of the gradient, overlapping with 66–200-kDa markers. A similar distribution was also observed in synaptosomal lysates from A30P and A53T α-syn mice (data not shown).
FIGURE 2.

Rab3a and α-syn co-elute in high molecular weight gradient fractions. Synaptosomes isolated from Tg α-syn WT mice were chemically cross-linked and detergent-solubilized for fractionation on a 5–40% glycerol gradient. A, Western blots probed for α-syn, Rab3a, GDI, and Hsp90 reveal the presence of α-syn and Rab3a in high molecular weight fractions. B, upper panel shows immunoblots of membrane (Membr), and cytosolic (Cytosol) fractions that were separated prior to loading onto glycerol gradients show that high molecular weight α-syn was preferentially associated with membrane-derived fractions, with only trace amounts in cytosol. Lower panels show representative immunoblots of protein markers for membrane (synaptophysin) and cytosolic (GAPDH) fractions. C, high molecular weight α-syn was not detected if recombinant WT α-syn was added to α-syn-deficient brain cytosol (Cytosol α-syn) but was if recombinant WT α-syn was pre-bound to synaptosomal membranes prior to gradient separation (Membr α-syn).
Experiments comparing elution profiles of synaptosomal membranes versus cytosol suggested that the α-syn in the high molecular weight fractions is preferentially associated with the membrane compartment. High molecular weight α-syn species were present predominantly in the membrane-derived fractions, whereas the low molecular weight α-syn was present in both membrane and cytosolic fractions (Fig. 2B). Furthermore, the distribution of recombinant α-syn added to α-syn-deficient cytosol prior to gradient separation was limited to the low molecular weight gradient fractions, whereas recombinant α-syn pre-bound to α-syn-deficient membranes eluted in the high molecular weight fractions (Fig. 2C). Thus, inclusion of recombinant α-syn to separate cytosol and membrane fractions demonstrated very distinct elution profiles, indicating that α-syn in the high molecular weight fractions was membrane-associated.
To explore the interaction between Rab3a and α-syn, we immunoprecipitated endogenous murine α-syn from either membrane or cytosolic fractions of nontransgenic murine synaptosomes. Despite the abundance of membrane and cytosolic Rab3a, endogenous Rab3a was co-immunoprecipitated with α-syn only in the membrane fractions and not in cytosolic fractions or in the control immunoprecipitation with preimmune serum (Fig. 3A). Neither GDI nor Hsp90 was co-immunoprecipitated with α-syn. Membrane Rab3a was also co-immunoprecipitated from Tg mice expressing human WT and mutant A30P α-syn (Fig. 3, B and C) but not from α-syn-deficient animals (Fig. 3D).
FIGURE 3.
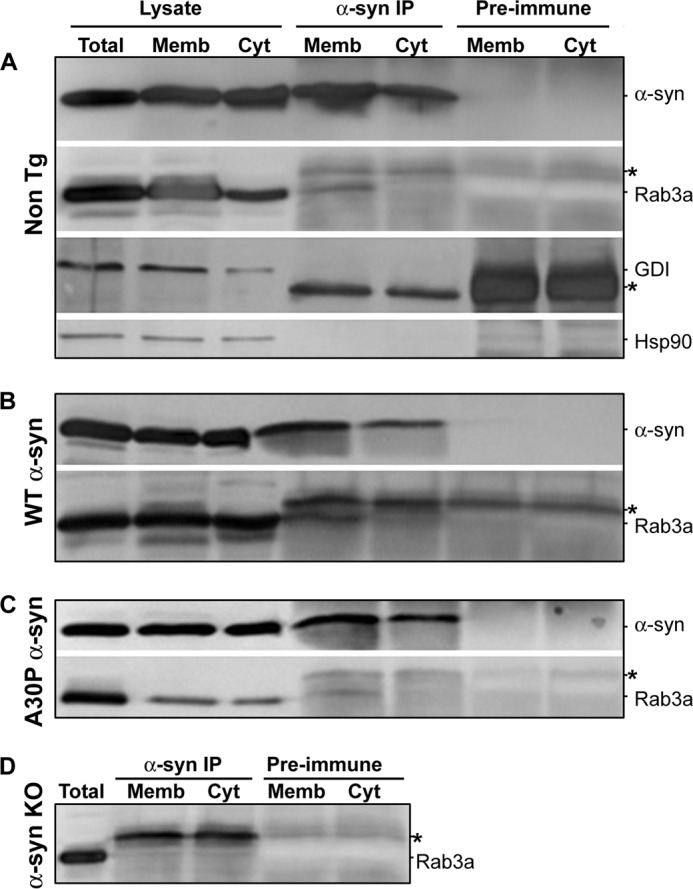
α-syn and Rab3a interaction occurs on presynaptic membranes. Lysates from synaptosomes expressing nontransgenic (Non Tg) (A), human wild type (WT) (B), A30P α-syn (C), or no α-syn (α-syn KO) (D) were separated into total, membrane-bound, and cytosolic proteins (Total, Memb, and Cyt) through hypotonic lysis. Membrane-associated and cytosolic fractions were immunoprecipitated (IP) either using anti-α-syn antibody (α-syn IP) or mouse pre-immune serum (Pre-immune). Western blots were probed for α-syn, Rab3a, Hsp90, and GDI. The blots shown are representative of three independent experiments. *, immunoglobulin heavy or light chain.
The results above suggest that α-syn and Rab3a are constituents of a membrane-bound complex and that antibodies to Rab3a can impede the acquisition of soluble α-syn. To determine whether the α-syn/Rab3a interaction can occur prior to membrane association or directly on membranes, we asked if pre-exposing membranes or cytosol separately to antibody could block α-syn membrane binding. Synaptosomal membranes were preincubated with antibodies to either Rab3a, GDI, or Hsp90, washed to remove excess antibody, and then examined for their capacity to bind recombinant WT or mutant α-syn. Pre-exposure with antibodies to Rab3a or GDI, but not Hsp90, significantly inhibited α-syn binding (Fig. 4A, one-way ANOVA, p < 0,0001). In contrast, there was no effect on α-syn membrane binding when the Rab3a, GDI, or Hsp90 antibodies were instead preincubated with cytosol and removed with protein G-coupled beads (Fig. 4B). These results imply that pre-existing membrane-bound Rab3a and GDI both play a role in the recruitment of α-syn to membranes. Our inability to detect GDI in α-syn immunoprecipitates or overlapping gradient fractions could reflect either a weak or transient interaction between GDI and α-syn or that GDI is in sufficiently close proximity to Rab3a that GDI antibodies sterically hinder α-syn recruitment.
FIGURE 4.

Reduced α-syn binding to membranes treated with Rab3a and GDI antibodies. A, α-syn-deficient synaptosomal membranes were preincubated with either preimmune serum (IgG) or with antibodies recognizing Rab3a, GDI, or Hsp90. Membranes were washed to remove excess unbound antibody and incubated with α-syn-deficient brain cytosol supplemented with recombinant WT, A30P, or A53T α-syn. Recovery of bound α-syn was reduced by membrane exposure to antibodies recognizing Rab3a and GDI but not Hsp90 (*, one-way ANOVA, p < 0.001, n = 3). B, α-syn-deficient cytosol was preincubated with preimmune serum (IgG) or antibodies to Rab3a, GDI, or Hsp90. All immunoglobulin was removed after 1 h using protein G-coupled beads. The exposure of cytosol to preimmune serum (IgG) or to specific antibodies had no significant effect on α-syn binding (one-way ANOVA, p > 0.05, n = 4).
Conformation-specific Rab3a and GDI Mutants Increase Membrane α-syn
The membrane-bound relationship between α-syn and Rab3a/GDI raised the possibility that the guanine nucleotide-dependent Rab3a conformation, which regulates Rab3a's attachment to synaptic vesicles, may also influence α-syn membrane binding. Therefore, we assessed the impact of conformation-specific mutants of Rab3a and GDI on α-syn distribution. First, we expressed two His6-tagged Rab3a mutants in either a predominantly GTP- or GDP-bound state in SH-SY5Y cells. The Q81L Rab3a has impaired GTP hydrolysis and is confined to its GTP-bound configuration, whereas the T36N Rab3a cannot accommodate the γ-phosphate in its GTP binding pocket and remains either GDP-bound or empty (23–26). There was a modest tendency to increase membrane-bound α-syn following 10 min of high potassium stimulation in mock-transfected and WT Rab3a cells (Fig. 5A). However, cells that expressed the constitutively GTP-bound Q81L Rab3a showed a significant increase in membrane-bound α-syn upon stimulation. The altered α-syn membrane binding did not appear linked to any change in Rab3a membrane binding as the amount of membrane-bound WT Rab3a and Q81L Rab3a did not differ between rest and stimulation (Fig. 5B). In contrast to Q81L Rab3a, the constitutively GDP-bound T36N Rab3a mutant had lower membrane distribution and did not alter α-syn localization, consistent with our immunoprecipitation data that α-syn preferentially interacts with membrane-bound GTP-Rab3a.
FIGURE 5.
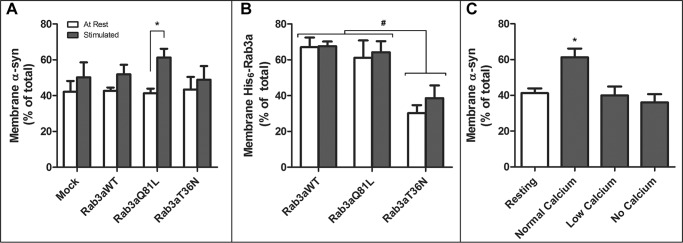
GTP-bound Rab3a mutant increases membrane α-syn following stimulation. SH-SY5Y cells transfected with an empty vector (Mock), WT Rab3a (Rab3aWT), GTP-bound Rab3a (Rab3aQ81L), or GDP-bound Rab3a (Rab3aT36N) were given no stimulation (At Rest) or stimulated for 10 min with a 55 mm potassium buffer (Stimulated) before being fractionated into membrane-bound and cytosolic proteins. Membrane-bound α-syn (A) or His6-tagged Rab3a (B) is shown as a percent of total (mean ± S.E., n = 5). C, Q81L Rab3a-transfected cells were left without (Resting) or with stimulation with a 55 mm high potassium buffer for 10 min containing either 2.5 mm (Normal Calcium) or 0.1 mm (Low Calcium) extracellular Ca2+ or with buffer containing 1 mm EGTA (No Calcium). *, significant change in membrane-bound α-syn between resting and stimulated conditions (p < 0.005, one-way ANOVA with Bonferroni multiple comparison post-test, n = 5). #, significant change in membrane-bound Rab3a levels between Rab3aWT and Rab3aT36N at both resting and stimulated states (p < 0.002, one-way ANOVA with Bonferroni post-test).
Activation of the Rab3a GTPase and the subsequent GDI-mediated recycling of GDP-Rab3a off synaptic vesicles are coupled to Ca2+ influx during exocytosis (27). Therefore, we examined whether depolarization alone was sufficient to increase membrane-bound α-syn in Q81L Rab3a-expressing cells or whether extracellular calcium was also required. When high potassium stimulation was done in combination with either low (0.1 mm) or no (with 1 mm EGTA) extracellular calcium, both conditions abrogated the increased membrane α-syn induced by Q81L Rab3a (Fig. 5C) indicating that normal Rab3a GTPase activity and Ca2+ influx are both necessary for normal α-syn distribution.
Next, we used two distinct loss-of-function GDI mutants that are deficient in either Rab3a or Hsp90 binding. The R240A mutation in the conserved Rab binding domain impairs the ability of GDI to extract Rab3a off vesicles (28), but it does not affect the GDI interaction with Hsp90. Conversely, the R218E mutation in its mobile effector loop disables the GDI·Hsp90 complex and its recycling off of membranes but allows binding to Rab3a (20, 29). SH-SY5Y cells expressing of WT, R240A, or R218E GDI showed normal α-syn distributions under resting conditions (data not shown), but the mutants had a differential effect on α-syn localization following 10 min of stimulation with high K+. Whereas the R240A GDI did not modify α-syn distribution compared with mock (empty vector) or WT GDI, cells expressing R218E GDI had a significant increase in membrane-bound α-syn (Fig. 6). These results suggested two conclusions. First, conformation-specific mutants of GDI and Rab3a shown previously to assert dominant-negative effects on Rab3a recycling also elevated α-syn membrane distribution. Second, a functional association between GDI and Hsp90 was involved in α-syn membrane dissociation.
FIGURE 6.
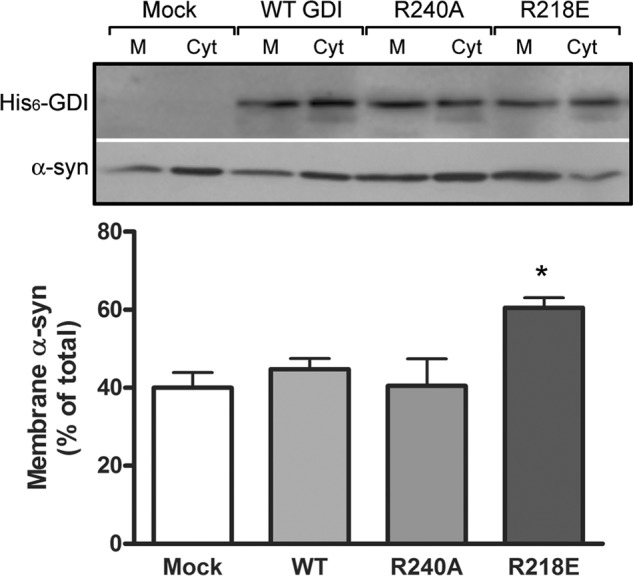
Effects of GDI mutants on α-syn membrane binding. SH-SY5Y cells were infected with lentivirus expressing empty vector (Mock), His6-tagged WT or mutant GDI (R240A and R218E) and stimulated for 10 min with a 55 mm high potassium buffer before fractionation into membrane-bound and cytosolic proteins. Upper panel shows representative Western blots of membrane (M) and cytosolic (Cyt) fractions probed for His6-GDI and α-syn. The lower panel shows quantification of mean ± S.E. from four independent experiments of membrane-associated α-syn, respectively (*, p < 0.05, one-way ANOVA with Bonferroni post-test).
Inhibition of Hsp90 Activity Impairs α-syn Dissociation from Synaptic Membranes
If the GDI interaction with Hsp90 is a precondition for α-syn cycling, we predicted that blockade of Hsp90 ATPase would influence α-syn accumulation much like the Q81L Rab3a and R218E GDI mutants. Therefore, we treated SH-SY5Y cells for up to 2 h with or without radicicol or geldanamycin, two structurally unrelated Hsp90 inhibitors, which impair Hsp90 function and block Rab recycling (20, 21). Following 10 min of high K+ stimulation, cells exposed to either Hsp90 inhibitor showed a reduction in membrane-bound GDI and an increase in the corresponding proportions of membrane-bound α-syn and Rab3a as compared with cells incubated without inhibitor (represented as time = 0) (Fig. 7).
FIGURE 7.
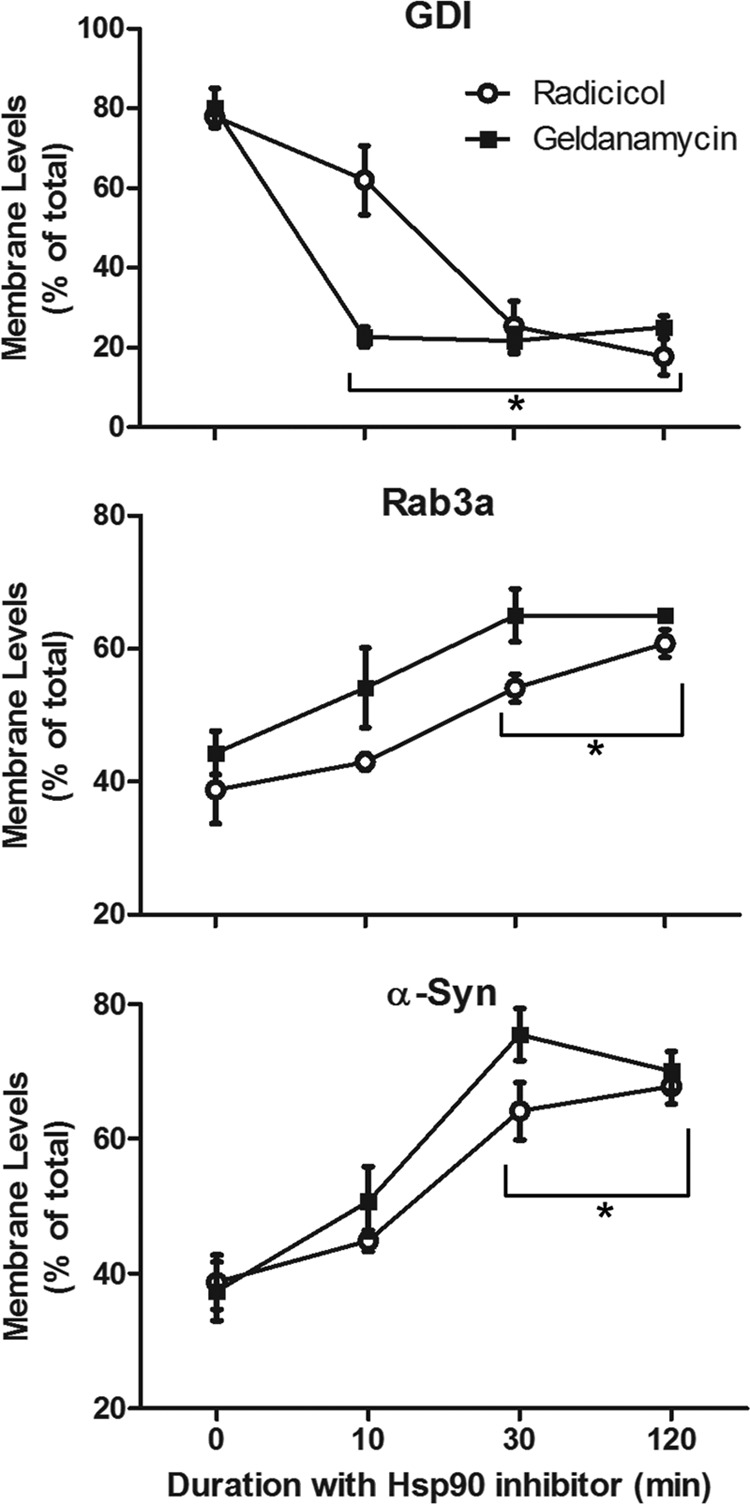
Inhibition of HSP90 activity increases membrane-bound α-syn. SH-SY5Y neuroblastoma cells were treated with 50 μm radicicol (open circles) or 20 μm geldanamycin (closed squares) for 0–120 min prior to a 10-min stimulation with 55 mm potassium buffer. Cells were immediately separated into membrane and cytosolic fractions, which were probed for GDI, Rab3a, and α-syn. Quantification of membrane protein levels is shown as a percent of total expression (mean ± S.E.). *, significant change in membrane-bound protein as compared with the t = 0 drug treatment control (p < 0.005, one-way ANOVA with Bonferroni post-test, n = 5).
Because our α-syn binding experiments (see Fig. 4) argued against a role for Hsp90 in α-syn membrane binding, we further dissected the Hsp90 function by using a cell-free assay designed to measure α-syn membrane dissociation. We previously showed this membrane dissociation to be triggered by unknown cytosolic proteins (9). Hypotonically lysed synaptosomal membranes from Tg mouse brains expressing WT α-syn were incubated for 10 min with α-syn-deficient cytosol, and α-syn dissociation from membranes into the cytosolic fraction was determined. Pre-exposure of both membranes and cytosol to Hsp90 inhibitors prior to co-incubation caused a significant reduction in α-syn dissociation (Fig. 8). We also selectively disabled either membrane or cytosolic Hsp90 by independently pretreating either fraction with inhibitors and found that inhibition of the cytosolic fraction was considerably more effective than inhibition of the membrane fraction alone. We found no detectable synaptophysin dissociated from synaptic membrane fractions in either the radicicol- or geldanamycin-treated assays, suggesting that there was no nonspecific protein dissociation due to membrane damage. Collectively, these results suggest that recruitment of cytosolic Hsp90 plays a role in mediating α-synuclein dissociation off membranes.
FIGURE 8.
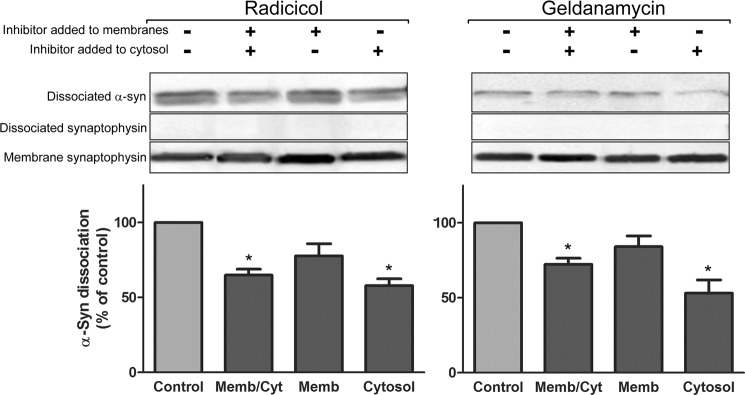
Inhibition of cytosolic Hsp90 activity reduces α-syn membrane dissociation. Dissociation of α-syn from presynaptic membranes was assessed in the absence or presence of 50 μm radicicol (left panel) or 20 μm geldanamycin (right panel). Synaptosomal membranes from Tg WT α-syn mice or α-syn-deficient cytosol were separately incubated with or without Hsp90 inhibitors for 10 min at 37 °C and then incubated together for another 10 min at 37 °C. Upper panels show representative immunoblots measuring the dissociation of membrane α-syn, and the lack of dissociation of integral membrane vesicle marker synaptophysin, into cytosol from experiments where inhibitor was added either to both membrane and cytosol (Memb/Cyt), only membrane (Memb), or only cytosol (Cyt). Lower panels show quantification of α-syn dissociation. Bars represent mean ± S.E. from seven to eight independent experiments and normalized to untreated controls. *, p < 0.05, one-way ANOVA with Bonferroni post-test.
Rab3a Distribution Is Not Affected by α-syn Expression, but Rab3a Knockdown Causes α-syn Levels to Increase
We tested whether α-syn expression or mutations affected the expression or localization of Rab3a. Rab3a expression was compared between in synaptosomes from α-syn-deficient mice and nontransgenic mice, as well as those expressing human WT, A30P, or A53T α-syn. The total level (Fig. 9A) and distribution (Fig. 9B) of Rab3a protein did not vary with α-syn expression or its PD-linked mutant. Approximately 70–80% of total Rab3a in murine synaptosomes was membrane-bound. There were also no changes observed in GDI and Hsp90 expression or distribution (data not shown).
FIGURE 9.

Reciprocal consequences of modifying α-syn and Rab3a expression levels. Synaptosomes from α-syn-deficient (KO), nontransgenic (Non-TG), overexpressing human wild-type or mutant α-syn (WT, A30P, and A53T) mice analyzed for total α-syn and Rab3a (A), and membrane-associated Rab3a (B). C, left panel shows relative Rab3a expression in SH-SY5Y cells that were either transfected with nontargeting siRNA (Cntrl) or four siRNA against Rab3a (KD). The right panel compares the Rab3a knockdown in individual experiments to α-syn expression. Total lysates from cells transfected with all four siRNAs were run on Western blots and probed for Rab3a, α-synuclein, and GAPDH (inset). Quantified α-syn expression levels in cells transfected with siRNA against Rab3a, relative to the levels in the nontargeting siRNA-transfected cells, were plotted against the level of Rab3a knockdown (all normalized to GAPDH signal). An inverse correlation between α-synuclein and Rab3a protein levels at rest can be seen using a least squares linear regression model (r2 = 0.5609). D, distribution of α-syn in membrane (Membr) and cytosolic (Soluble) fractions following 10 min of stimulation with 55 mm potassium of SH-SY5Y cells transfected with control (Scrambled) or four Rab3a siRNA. Bars represent mean ± S.E. from four independent experiments.
Conversely, to evaluate the effects of Rab3a expression on α-syn levels and membrane binding, SH-SY5Y neuroblastoma cells were transfected with each of four commercially available siRNAs against Rab3a. Differences in siRNA efficacy after 24 h generated variable decreases in Rab3a levels (Fig. 9C, left panel). Comparison of the relative protein levels of α-syn and Rab3a in these siRNA-transfected cells against those of control cells revealed an inverse correlation between α-syn and Rab3a protein levels (Fig. 9C, right panel). A 90% reduction in Rab3a protein level doubled α-syn expression (Fig. 9C, inset), although this did not alter the overall α-syn distribution compared with control cells with normal Rab3a content (Fig. 9D). These results, although concordant with numerous studies showing α-syn binding to synthetic membranes can proceed without additional cofactors, suggest that global α-syn levels are subject to feedback regulation to compensate for the loss of a functional complex.
DISCUSSION
Soluble unstructured α-syn binds to membranes by assuming an amphipathic α-helix that orients nonpolar residues into lipid bilayers and polar residues facing the aqueous cytoplasm (12–15). This accepted static model, however, does not account for regulated α-syn exchange between cytosolic and membrane compartments. Although purified recombinant α-syn readily associates with synthetic membranes without additional factors (30–34), there is compelling evidence that α-syn distribution is highly dynamic and regulated by membrane and cytoplasmic components as follows. (i) Dissociation of α-syn from synaptosome membranes is significantly augmented by cytosolic proteins (9, 35). (ii) Purified A30P α-syn has impaired binding to synthetic lipids (36–39) but shows normal compartmentalization in vivo (9). (iii) Cross-linking stabilizes α-syn on membranes suggesting proximity to other vesicular proteins (16). (iv) Neuronal activity promotes membrane dissociation of presynaptic α-syn and subsequent re-association (10).
Our screen for presynaptic α-syn interactions revealed that anti-Rab3a antibodies strongly inhibited α-syn binding to synaptosomal membranes. Several studies have linked α-syn with various Rab proteins, including interaction of α-syn and Rab3a in disease tissue (40–42), disruption of endoplasmic reticulum-Golgi trafficking by α-syn rescued by Rab1 or Rab3a (43, 44), and uptake and secretion of extracellular α-syn involving Rab1A and Hsp90 (45). However, the α-syn and Rab3a relationship in healthy neurons is not understood. We verified the interaction between α-syn and Rab3a by co-elution of both proteins in dense fractions of velocity gradients and by co-immunoprecipitation of α-syn and Rab3a from membrane fractions. Despite substantial levels of cytosolic α-syn and Rab3a, we failed to detect cytosolic α-syn·Rab3a complexes. Because neuronal activity is known to transiently augment the freely diffusible pools of both proteins by dissociation from synaptic vesicles (10, 17), the presence of an exclusively membrane-associated complex suggested a functional link between α-syn and Rab3a.
Rab proteins regulate membrane fusion events by switching between GTP- and GDP-bound states (46), and the machinery for chaperoning Rab3a vesicle association and dissociation is well characterized. Depolarization-induced Ca2+ influx activates GTP cleavage by membrane-bound Rab3a, followed by the retrieval of GDP-Rab3a by a GDI·Hsp90 complex (20, 47). The α-syn·Rab3a complex raised the possibility that the guanine nucleotide-dependent Rab3a conformation may also influence α-syn distribution. The coupling of α-syn binding and dissociation to the Rab3a GTP/GDP cycle was confirmed by two Rab3a mutants locked in either GTP- or GDP-bound conformation (23–26). Depolarization of cells expressing GTP-bound Q81L Rab3a increased membrane α-syn in a Ca2+-dependent manner, whereas GDP-bound T36N Rab3a, with weak membrane interaction, did not affect α-syn. The simplest interpretation is that α-syn is preferentially sequestered or stabilized on membranes by GTP-bound Rab3a awaiting GTP hydrolysis.
Rab3a lacks a transmembrane domain and is anchored to vesicles by covalent carboxyl-terminal prenylation (48–50). Consequently, Rab3a retrieval by GDI involves both a Rab binding domain and an adjacent hydrophobic pocket to accommodate its prenyl groups (51). The GDI/Rab3a interaction is also controlled by a separate Hsp90 binding domain called the GDI mobile effector loop. Although neither GDI nor Hsp90 were detected in α-syn immunoprecipitates, pre-exposure of membranes to GDI antibodies reduced α-syn binding as effectively as Rab3a antibodies. This may reflect a transient or weak interaction or that close apposition of GDI to Rab3a generates steric hindrance when anti-GDI immunoglobulin is bound. Interestingly, GDI mutants selectively deficient in either Rab3a or Hsp90 binding differentially affected α-syn localization during depolarization. The Rab binding domain mutant, R240A GDI, with defective Rab3a retrieval (28) did not affect α-syn distribution, suggesting that it may not compete with endogenous GDI in SH-SY5Y cells. In contrast, R218E GDI, which cannot bind Hsp90 (52), significantly increased membrane α-syn. Thus, an impaired GDI/Hsp90 interaction was sufficient to increase membrane α-syn, implicating this Rab3a recycling machinery as a regulatory link between α-syn and synaptic vesicles.
Hsp90 ATPase activity is essential for the conformational change in GDI necessary to shield the Rab3a hydrophobic prenyl moieties from aqueous cytoplasm prior to Rab3a extraction (20, 21). Two inhibitors of Hsp90 ATPase previously shown to block Rab recycling, radicicol and geldanamycin, also increased the membrane-bound pools of Rab3a and α-syn while reducing that of GDI. Furthermore, selective inhibition of cytosolic Hsp90 in a cell-free assay was more effective at blocking α-syn dissociation. This result implicates a preferential role for cytosolic Hsp90 in α-syn membrane dissociation, whereas membrane Rab3a and GDI appear to stabilize α-syn binding on vesicles. Therefore, activation of Rab3a GTPase by neuronal depolarization triggers the separation of α-syn from Rab3a (with the aid of the GDI·Hsp90 recycling complex), as well from the vesicle membrane, resulting in the mobilization of α-syn into cytosol (Fig. 10).
FIGURE 10.

Model of α-syn membrane binding and regulation by Rab3a machinery and synaptic activity. A, soluble α-syn binds to vesicle membranes in close proximity to Rab3a and GDI and forms a membrane-associated complex with GTP-bound Rab3a. B, depolarization and Ca2+ influx trigger Rab3a GTPase. Concomitantly, recruitment of cytosolic Hsp90 to vesicles and activation of its ATPase allow GDI to bind GDP-bound Rab3a. C, disruption of α-syn/Rab3a interaction by GDI·Hsp90 chaperone complex promotes α-syn dissociation from vesicles.
It is informative to consider other components of the Rab3a recycling machinery for potential relationships with α-syn. Immunoprecipitation of a cross-linked synaptosomal membrane GDI complex also pulled down another chaperone, cysteine string protein (CSP), in addition to Hsp90 (20). Interestingly, CSPα deficiency is known to cause progressive and lethal neurodegeneration that is exacerbated by α-syn gene deletion and rescued by α-syn overexpression, suggesting overlapping but probably independent functions for α-syn and CSPα (53, 54). Indeed, both proteins appear to modulate SNARE complex assembly (55–58). Although triple knock-out mice lacking all three synucleins (α, β, and γ) are viable, they too display age-dependent sensorimotor deficits and increasing mortality at 1 year of age. Thus, the loss of all synuclein function produces a form of degeneration similar to the CSPα-deficient phenotype albeit with later onset.
Additional evidence for α-syn's role in secretion comes from both knock-out and overexpression models. Single knock-out α-syn-deficient mice have normal life spans despite mildly elevated transmitter release and impairments in reserve pool maintenance during repetitive stimuli (2, 3). Conversely, overexpression of α-syn inhibits vesicle trafficking and exocytosis from non-neuronal and neuroendocrine cells (59–62). In murine brain neurons, it was shown to interfere with normal vesicular trafficking (4–6, 63). These knockdown and overexpression studies collectively imply that α-syn levels are closely titrated at presynapses to modulate exocytosis. Indeed, in our hands, transient knockdown of Rab3a levels was inversely matched by increased α-syn expression, perhaps to compensate for the loss of functional membrane assemblies. Surprisingly, despite the lower Rab3a levels, the elevated α-syn maintained an apparently normal membrane/cytosol, suggesting that α-syn membrane levels are impacted by impairments in Rab3a cycling but not by Rab3a levels per se. Alternatively, other Rab molecules (e.g. Rab1 or Rab3b/c/d) may compensate for reduced Rab3a.
If vesicle-bound α-syn regulates SNARE assembly as proposed (11), the functional consequence of α-syn membrane dissociation upon Rab3a GTPase activation may be to destabilize SNARE complexes and increase the probability of vesicle fusion. In accord, our study provides the underlying mechanism by which Rab1 and Rab3a can rescue the observed inhibition of exocytic pathways induced by α-syn overexpression (43, 44). In such a model, overexpression of α-syn reduces the likelihood of vesicle fusion by stabilizing SNARE complexes. This imbalance is rectified by Rab3a (or Rab1 for endoplasmic reticulum to Golgi traffic in non-neuronal cells) overexpression that enables the GDI/Hsp90 Rab recycling machinery to facilitate α-syn dissociation from membrane and relieve its stabilizing influence on SNARE complexes.
In conclusion, our results show that α-syn and GTP-bound Rab3a form a complex on presynaptic membranes. Following GTP hydrolysis, membrane-bound GDP-Rab3a is removed from vesicles by the concerted actions of GDI and Hsp90, concomitantly freeing α-syn from membranes. Whether the two proteins dissociate from the membrane separately, or as part of the GDI complex, remains to be resolved. We favor the interpretation that α-syn likely separates from Rab3a quickly after GTP hydrolysis and its membrane retrieval because we did not detect a cytosolic α-syn/Rab3a interaction. Disruption of the GDI complex or its activity by mutant GDI or Hsp90 inhibitors clearly impaired α-syn dissociation. We speculate above on the function of α-syn interactions with membranes or with Rab3a and its recycling machinery, although the relationship between these interactions and neurodegeneration remains unclear. Evidence from animal models and human disease indicates that both reduction and overexpression of α-syn can induce neurodegeneration. Furthermore, alterations in SNARE complexes or deficits in exocytosis could amplify neuronal death pathways brought about by progressive sequestration of α-syn into oligomeric forms.
Acknowledgment
We are grateful to William Balch (Scripps Research Institute) for the generous provision of Rab3a and GDI constructs.
This work was supported in part by Canadian Institutes of Health Research Operating Grants MOP 84501 (to A. T.) and MOP 115056 (to P. E. F.) and a Parkinson Society of Canada operating grant (to A. T.).
This article was selected as a Paper of the Week.
- α-syn
- α-synuclein
- GDI
- GDP dissociation inhibitor
- PD
- Parkinson disease
- ANOVA
- analysis of variance
- CSP
- cysteine string protein
- Tg
- transgenic.
REFERENCES
- 1. Cookson M. R. (2005) The biochemistry of Parkinson's disease. Annu. Rev. Biochem. 74, 29–52 [DOI] [PubMed] [Google Scholar]
- 2. Abeliovich A., Schmitz Y., Fariñas I., Choi-Lundberg D., Ho W. H., Castillo P. E., Shinsky N., Verdugo J. M., Armanini M., Ryan A., Hynes M., Phillips H., Sulzer D., Rosenthal A. (2000) Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252 [DOI] [PubMed] [Google Scholar]
- 3. Cabin D. E., Shimazu K., Murphy D., Cole N. B., Gottschalk W., McIlwain K. L., Orrison B., Chen A., Ellis C. E., Paylor R., Lu B., Nussbaum R. L. (2002) Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J. Neurosci. 22, 8797–8807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nemani V. M., Lu W., Berge V., Nakamura K., Onoa B., Lee M. K., Chaudhry F. A., Nicoll R. A., Edwards R. H. (2010) Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scott D. A., Tabarean I., Tang Y., Cartier A., Masliah E., Roy S. (2010) A pathologic cascade leading to synaptic dysfunction in α-synuclein-induced neurodegeneration. J. Neurosci. 30, 8083–8095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murphy D. D., Rueter S. M., Trojanowski J. Q., Lee V. M. (2000) Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20, 3214–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Irizarry M. C., Kim T. W., McNamara M., Tanzi R. E., George J. M., Clayton D. F., Hyman B. T. (1996) Characterization of the precursor protein of the non-Aβ component of senile plaques (NACP) in the human central nervous system. J. Neuropathol. Exp. Neurol. 55, 889–895 [DOI] [PubMed] [Google Scholar]
- 8. Kahle P. J., Neumann M., Ozmen L., Muller V., Jacobsen H., Schindzielorz A., Okochi M., Leimer U., van Der Putten H., Probst A., Kremmer E., Kretzschmar H. A., Haass C. (2000) Subcellular localization of wild-type and Parkinson's disease-associated mutant α-synuclein in human and transgenic mouse brain. J. Neurosci. 20, 6365–6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wislet-Gendebien S., D'Souza C., Kawarai T., St George-Hyslop P., Westaway D., Fraser P., Tandon A. (2006) Cytosolic proteins regulate α-synuclein dissociation from presynaptic membranes. J. Biol. Chem. 281, 32148–32155 [DOI] [PubMed] [Google Scholar]
- 10. Fortin D. L., Nemani V. M., Voglmaier S. M., Anthony M. D., Ryan T. A., Edwards R. H. (2005) Neural activity controls the synaptic accumulation of α-synuclein. J. Neurosci. 25, 10913–10921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thayanidhi N., Helm J. R., Nycz D. C., Bentley M., Liang Y., Hay J. C. (2010) α-Synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 21, 1850–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weinreb P. H., Zhen W., Poon A. W., Conway K. A., Lansbury P. T., Jr. (1996) NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry 35, 13709–13715 [DOI] [PubMed] [Google Scholar]
- 13. Davidson W. S., Jonas A., Clayton D. F., George J. M. (1998) Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449 [DOI] [PubMed] [Google Scholar]
- 14. Eliezer D., Kutluay E., Bussell R., Jr., Browne G. (2001) Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 307, 1061–1073 [DOI] [PubMed] [Google Scholar]
- 15. Jao C. C., Der-Sarkissian A., Chen J., Langen R. (2004) Structure of membrane-bound α-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. U.S.A. 101, 8331–8336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wislet-Gendebien S., Visanji N. P., Whitehead S. N., Marsilio D., Hou W., Figeys D., Fraser P. E., Bennett S. A., Tandon A. (2008) Differential regulation of wild-type and mutant α-synuclein binding to synaptic membranes by cytosolic factors. BMC Neurosci. 9, 92. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17. Fischer von Mollard G., Südhof T. C., Jahn R. (1991) A small GTP-binding protein dissociates from synaptic vesicles during exocytosis. Nature 349, 79–81 [DOI] [PubMed] [Google Scholar]
- 18. Tandon A., Bannykh S., Kowalchyk J. A., Banerjee A., Martin T. F., Balch W. E. (1998) Differential regulation of exocytosis by calcium and CAPS in semi-intact synaptosomes. Neuron 21, 147–154 [DOI] [PubMed] [Google Scholar]
- 19. Engelender S., Kaminsky Z., Guo X., Sharp A. H., Amaravi R. K., Kleiderlein J. J., Margolis R. L., Troncoso J. C., Lanahan A. A., Worley P. F., Dawson V. L., Dawson T. M., Ross C. A. (1999) Synphilin-1 associates with α-synuclein and promotes the formation of cytosolic inclusions. Nat. Genet. 22, 110–114 [DOI] [PubMed] [Google Scholar]
- 20. Sakisaka T., Meerlo T., Matteson J., Plutner H., Balch W. E. (2002) Rab-αGDI activity is regulated by a Hsp90 chaperone complex. EMBO J. 21, 6125–6135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen C. Y., Sakisaka T., Balch W. E. (2005) Use of Hsp90 inhibitors to disrupt GDI-dependent Rab recycling. Methods Enzymol. 403, 339–347 [DOI] [PubMed] [Google Scholar]
- 22. Chen F., Tandon A., Sanjo N., Gu Y. J., Hasegawa H., Arawaka S., Lee F. J., Ruan X., Mastrangelo P., Erdebil S., Wang L., Westaway D., Mount H. T., Yankner B., Fraser P. E., St George-Hyslop P. (2003) Presenilin 1 and presenilin 2 have differential effects on the stability and maturation of nicastrin in mammalian brain. J. Biol. Chem. 278, 19974–19979 [DOI] [PubMed] [Google Scholar]
- 23. Burstein E. S., Brondyk W. H., Macara I. G. (1992) Amino acid residues in the Ras-like GTPase Rab3A that specify sensitivity to factors that regulate the GTP/GDP cycling of Rab3A. J. Biol. Chem. 267, 22715–22718 [PubMed] [Google Scholar]
- 24. Brondyk W. H., McKiernan C. J., Burstein E. S., Macara I. G. (1993) Mutants of Rab3A analogous to oncogenic Ras mutants. Sensitivity to Rab3A-GTPase-activating protein and Rab3A-guanine nucleotide releasing factor. J. Biol. Chem. 268, 9410–9415 [PubMed] [Google Scholar]
- 25. Schlüter O. M., Khvotchev M., Jahn R., Südhof T. C. (2002) Localization versus function of Rab3 proteins. Evidence for a common regulatory role in controlling fusion. J. Biol. Chem. 277, 40919–40929 [DOI] [PubMed] [Google Scholar]
- 26. Schlüter O. M., Schmitz F., Jahn R., Rosenmund C., Südhof T. C. (2004) A complete genetic analysis of neuronal Rab3 function. J. Neurosci. 24, 6629–6637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lang T., Jahn R. (2008) Core proteins of the secretory machinery. Handb. Exp. Pharmacol. 184, 107–127 [DOI] [PubMed] [Google Scholar]
- 28. Schalk I., Zeng K., Wu S. K., Stura E. A., Matteson J., Huang M., Tandon A., Wilson I. A., Balch W. E. (1996) Structure and mutational analysis of Rab GDP-dissociation inhibitor. Nature 381, 42–48 [DOI] [PubMed] [Google Scholar]
- 29. Luan P., Heine A., Zeng K., Moyer B., Greasely S. E., Kuhn P., Balch W. E., Wilson I. A. (2000) A new functional domain of guanine nucleotide dissociation inhibitor (α-GDI) involved in Rab recycling. Traffic 1, 270–281 [DOI] [PubMed] [Google Scholar]
- 30. Jo E., Darabie A. A., Han K., Tandon A., Fraser P. E., McLaurin J. (2004) α-Synuclein-synaptosomal membrane interactions: implications for fibrillogenesis. Eur. J. Biochem. 271, 3180–3189 [DOI] [PubMed] [Google Scholar]
- 31. Jo E., McLaurin J., Yip C. M., St George-Hyslop P., Fraser P. E. (2000) α-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 275, 34328–34334 [DOI] [PubMed] [Google Scholar]
- 32. Perrin R. J., Woods W. S., Clayton D. F., George J. M. (2000) Interaction of human α-synuclein and Parkinson's disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J. Biol. Chem. 275, 34393–34398 [DOI] [PubMed] [Google Scholar]
- 33. Narayanan V., Scarlata S. (2001) Membrane binding and self-association of α-synucleins. Biochemistry 40, 9927–9934 [DOI] [PubMed] [Google Scholar]
- 34. Zhu M., Li J., Fink A. L. (2003) The association of α-synuclein with membranes affects bilayer structure, stability, and fibril formation. J. Biol. Chem. 278, 40186–40197 [DOI] [PubMed] [Google Scholar]
- 35. Visanji N. P., Wislet-Gendebien S., Oschipok L. W., Zhang G., Aubert I., Fraser P. E., Tandon A. (2011) Effect of Ser-129 phosphorylation on interaction of α-synuclein with synaptic and cellular membranes. J. Biol. Chem. 286, 35863–35873 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36. Jo E., Fuller N., Rand R. P., St George-Hyslop P., Fraser P. E. (2002) Defective membrane interactions of familial Parkinson's disease mutant A30P α-synuclein. J. Mol. Biol. 315, 799–807 [DOI] [PubMed] [Google Scholar]
- 37. Jensen P. H., Nielsen M. S., Jakes R., Dotti C. G., Goedert M. (1998) Binding of α-synuclein to brain vesicles is abolished by familial Parkinson's disease mutation. J. Biol. Chem. 273, 26292–26294 [DOI] [PubMed] [Google Scholar]
- 38. Bussell R., Jr., Eliezer D. (2004) Effects of Parkinson's disease-linked mutations on the structure of lipid-associated α-synuclein. Biochemistry 43, 4810–4818 [DOI] [PubMed] [Google Scholar]
- 39. Kubo S., Nemani V. M., Chalkley R. J., Anthony M. D., Hattori N., Mizuno Y., Edwards R. H., Fortin D. L. (2005) A combinatorial code for the interaction of α-synuclein with membranes. J. Biol. Chem. 280, 31664–31672 [DOI] [PubMed] [Google Scholar]
- 40. Dalfó E., Ferrer I. (2005) α-Synuclein binding to rab3a in multiple system atrophy. Neurosci. Lett. 380, 170–175 [DOI] [PubMed] [Google Scholar]
- 41. Dalfó E., Barrachina M., Rosa J. L., Ambrosio S., Ferrer I. (2004) Abnormal α-synuclein interactions with rab3a and rabphilin in diffuse Lewy body disease. Neurobiol. Dis. 16, 92–97 [DOI] [PubMed] [Google Scholar]
- 42. Dalfó E., Gómez-Isla T., Rosa J. L., Nieto Bodelón M., Cuadrado Tejedor M., Barrachina M., Ambrosio S., Ferrer I. (2004) Abnormal α-synuclein interactions with Rab proteins in α-synuclein A30P transgenic mice. J. Neuropathol. Exp. Neurol. 63, 302–313 [DOI] [PubMed] [Google Scholar]
- 43. Gitler A. D., Bevis B. J., Shorter J., Strathearn K. E., Hamamichi S., Su L. J., Caldwell K. A., Caldwell G. A., Rochet J. C., McCaffery J. M., Barlowe C., Lindquist S. (2008) The Parkinson's disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cooper A. A., Gitler A. D., Cashikar A., Haynes C. M., Hill K. J., Bhullar B., Liu K., Xu K., Strathearn K. E., Liu F., Cao S., Caldwell K. A., Caldwell G. A., Marsischky G., Kolodner R. D., Labaer J., Rochet J. C., Bonini N. M., Lindquist S. (2006) α-Synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313, 324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu J., Zhang J. P., Shi M., Quinn T., Bradner J., Beyer R., Chen S., Zhang J. (2009) Rab11a and HSP90 regulate recycling of extracellular α-synuclein. J. Neurosci. 29, 1480–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fukuda M. (2008) Regulation of secretory vesicle traffic by Rab small GTPases. Cell. Mol. Life Sci. 65, 2801–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goody R. S., Rak A., Alexandrov K. (2005) The structural and mechanistic basis for recycling of Rab proteins between membrane compartments. Cell. Mol. Life Sci. 62, 1657–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khosravi-Far R., Lutz R. J., Cox A. D., Conroy L., Bourne J. R., Sinensky M., Balch W. E., Buss J. E., Der C. J. (1991) Isoprenoid modification of Rab proteins terminating in CC or CXC motifs. Proc. Natl. Acad. Sci. U.S.A. 88, 6264–6268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Farnsworth C. C., Kawata M., Yoshida Y., Takai Y., Gelb M. H., Glomset J. A. (1991) C terminus of the small GTP-binding protein smg p25A contains two geranylgeranylated cysteine residues and a methyl ester. Proc. Natl. Acad. Sci. U.S.A. 88, 6196–6200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Musha T., Kawata M., Takai Y. (1992) The geranylgeranyl moiety but not the methyl moiety of the smg-25A/rab3A protein is essential for the interactions with membrane and its inhibitory GDP/GTP exchange protein. J. Biol. Chem. 267, 9821–9825 [PubMed] [Google Scholar]
- 51. An Y., Shao Y., Alory C., Matteson J., Sakisaka T., Chen W., Gibbs R. A., Wilson I. A., Balch W. E. (2003) Geranylgeranyl switching regulates GDI-Rab GTPase recycling. Structure 11, 347–357 [DOI] [PubMed] [Google Scholar]
- 52. Luan P., Balch W. E., Emr S. D., Burd C. G. (1999) Molecular dissection of guanine nucleotide dissociation inhibitor function in vivo. Rab-independent binding to membranes and role of Rab recycling factors. J. Biol. Chem. 274, 14806–14817 [DOI] [PubMed] [Google Scholar]
- 53. Fernández-Chacón R., Wölfel M., Nishimune H., Tabares L., Schmitz F., Castellano-Muñoz M., Rosenmund C., Montesinos M. L., Sanes J. R., Schneggenburger R., Südhof T. C. (2004) The synaptic vesicle protein CSPα prevents presynaptic degeneration. Neuron 42, 237–251 [DOI] [PubMed] [Google Scholar]
- 54. Chandra S., Gallardo G., Fernández-Chacón R., Schlüter O. M., Südhof T. C. (2005) α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell 123, 383–396 [DOI] [PubMed] [Google Scholar]
- 55. Burré J., Sharma M., Tsetsenis T., Buchman V., Etherton M. R., Südhof T. C. (2010) α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sharma M., Burré J., Bronk P., Zhang Y., Xu W., Südhof T. C. (2012) CSPα knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 31, 829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sharma M., Burré J., Südhof T. C. (2011) CSPα promotes SNARE complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 13, 30–39 [DOI] [PubMed] [Google Scholar]
- 58. Darios F., Ruipérez V., López I., Villanueva J., Gutierrez L. M., Davletov B. (2010) α-Synuclein sequesters arachidonic acid to modulate SNARE-mediated exocytosis. EMBO Rep. 11, 528–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Larsen K. E., Schmitz Y., Troyer M. D., Mosharov E., Dietrich P., Quazi A. Z., Savalle M., Nemani V., Chaudhry F. A., Edwards R. H., Stefanis L., Sulzer D. (2006) α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 26, 11915–11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Park S. M., Jung H. Y., Kim H. O., Rhim H., Paik S. R., Chung K. C., Park J. H., Kim J. (2002) Evidence that α-synuclein functions as a negative regulator of Ca2+-dependent α-granule release from human platelets. Blood 100, 2506–2514 [DOI] [PubMed] [Google Scholar]
- 61. Geng X., Lou H., Wang J., Li L., Swanson A. L., Sun M., Beers-Stolz D., Watkins S., Perez R. G., Drain P. (2011) α-Synuclein binds the KATP channel at insulin-secretory granules and inhibits insulin secretion. Am. J. Physiol. Endocrinol. Metab. 300, E276–E286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim K. S., Park J. Y., Jou I., Park S. M. (2010) Regulation of Weibel-Palade body exocytosis by α-synuclein in endothelial cells. J. Biol. Chem. 285, 21416–21425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Scott D., Roy S. (2012) α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 32, 10129–10135 [DOI] [PMC free article] [PubMed] [Google Scholar]