

Background: Microglial NADPH oxidase 2 (Nox2) expression is critical for nerve injury-induced spinal cord microglia activation and subsequent pain hypersensitivity.
Results: Toll-like receptor 2 (TLR2) is required for Nox2 expression in spinal cord microglia after spinal nerve injury.
Conclusion: TLR2 signaling mediates nerve injury-induced Nox2 expression in spinal cord microglia.
Significance: Targeting TLR2-Nox2 pathway may be a new option for preventing neuropathic pain.
Keywords: Microglia, NADPH Oxidase, p38 MAPK, Reactive Oxygen Species (ROS), Toll-like Receptors (TLR), Neuropathic Pain, Spinal Nerve Injury
Abstract
We have previously reported that NADPH oxidase 2 (Nox2) is up-regulated in spinal cord microglia after spinal nerve injury, demonstrating that it is critical for microglia activation and subsequent pain hypersensitivity. However, the mechanisms and molecules involved in Nox2 induction have not been elucidated. Previous studies have shown that Toll-like receptors (TLRs) are involved in nerve injury-induced spinal cord microglia activation. In this study, we investigated the role of TLR in Nox2 expression in spinal cord microglia after peripheral nerve injury. Studies using TLR knock-out mice have shown that nerve injury-induced microglial Nox2 up-regulation is abrogated in TLR2 but not in TLR3 or -4 knock-out mice. Intrathecal injection of lipoteichoic acid, a TLR2 agonist, induced Nox2 expression in spinal cord microglia both at the mRNA and protein levels. Similarly, lipoteichoic acid stimulation induced Nox2 expression and reactive oxygen species production in primary spinal cord glial cells in vitro. Studies on intracellular signaling pathways indicate that NF-κB and p38 MAP kinase activation is required for TLR2-induced Nox2 expression in glial cells. Conclusively, our data show that TLR2 mediates nerve injury-induced Nox2 gene expression in spinal cord microglia via NF-κB and p38 activation and thereby may contribute to spinal cord microglia activation.
Introduction
A series of studies have demonstrated that spinal cord glial cells play a critical role in the development of neuropathic pain after peripheral nerve injury (1). Activation of spinal cord glia in the absence of peripheral nerve injury enhanced pain sensitivity (2), and inhibition of these cells attenuated pain behavior in a neuropathic pain model (3). It was later reported that proinflammatory cytokines or growth factors expressed by activated spinal cord glia such as TNF-α, IL-1β, IL-6, and BDNF facilitate pain hypersensitivity by inducing central sensitization at the spinal cord level (4, 5). Studies on the mechanisms of spinal cord microglia activation have proposed that various transmembrane receptors, including CX3CR1, a fractalkine receptor (6), and P2X4, an ATP receptor (7), can potentially respond to nerve injury and trigger microglia activation. We have also proposed that Toll-like receptor 2 (TLR2)3 may trigger the proinflammatory activation of spinal cord microglia during peripheral nerve injury (8). Likewise, TLR3 and -4 have been implicated in the activation of spinal cord microglia during the development of nerve injury-induced neuropathic pain (9, 10).
At the level of intracellular signaling, p38 activation seems to be a key event in the activation of microglia upon nerve injury. Microglia activation mostly accompanies p38 activation (11), and the nerve injury-induced proinflammatory cytokine expression in microglia and the subsequent pain hypersensitivity is abrogated by the inhibition of p38 (12). In addition, we have recently discovered that NADPH oxidase 2 (Nox2) is induced in spinal cord microglia after nerve injury and that Nox2-derived reactive oxygen species (ROS) production is required for nerve injury-induced microglia activation and pain hypersensitivity (13). These findings further indicate that Nox2 is an important intracellular mediator of nerve injury-induced spinal cord microglia activation. However, it is currently not clear how Nox2 is up-regulated in spinal cord microglia after nerve injury. Based on the studies implicating TLRs as microglial activation receptors that respond to nerve injury (8–10), we hypothesized that TLRs might be involved in nerve injury-induced Nox2 expression in spinal cord microglia. To investigate this hypothesis, our study design used knock-out mice of different TLR genes that have been implicated in nerve injury-induced spinal cord microglia activation (e.g. TLR2, -3, or -4 knock-out mice). Our data show that TLR2 plays a critical role in nerve injury-induced Nox2 expression in spinal cord microglia.
EXPERIMENTAL PROCEDURES
Animals and Surgery
All surgical and experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee at Seoul National University. The animal treatments were performed in accordance with the guidelines of the International Association for the Study of Pain. All mice were housed in an animal facility with a specific pathogen-free barrier under a 12-h light/dark cycle. Mice were allowed to access food and water ad libitum. Wild-type (WT), TLR2, -3, or -4 knock-out, and MyD88 knock-out mice of C57BL/6 background aged 8–12 weeks were used. Peripheral nerve injury was induced by transecting the L5 spinal nerve (L5 SNT) under anesthesia with pentobarbital sodium (50 mg/kg, intraperitoneal) as described previously (13). In brief, an incision was made in the skin from the spinal processes at L4 to S2 levels. The paraspinal muscles were separated, and the L6 transverse process was partially removed. The L5 spinal nerve was transected carefully upon exposure, and then the surgical site was closed in two layers with surgical staples. The sham operation was performed as described above but in the absence of transecting the L5 spinal nerve. The mice operated on were monitored on a warm pad during recovery.
Real-time RT-PCR
Real-time RT-PCR was performed using SYBR Green PCR Master Mix and an ABI Prism 7300 sequence detection system (Applied Biosystems, Foster City, CA) as described previously (13). The following PCR primer sequences were used: mouse/rat GAPDH (forward), 5′-AGG TCA TCC CAG AGC TGA ACG-3′; mouse/rat GAPDH (reverse), 5′-CAC CCT GTT GCT GTA GCC GTA T-3′; mouse Nox2 (forward), 5′-GAC CCA GAT GCA GGA AAG GAA-3′; mouse Nox2 (reverse), 5′-TCA TGG TGC ACA GCA AAG TGA T-3′; rat Nox2 (forward), 5′-TGC AAG TCA ACA CCC CAA CA-3′; rat Nox2 (reverse), 5′-CGG ACT CAG AGT TGG AGA TGC T-3′. Duplicate reactions were performed, with each containing 10 pmol of primer, 4 μl of cDNA, and 5 μl of 2× SYBR Green PCR Master Mix (Applied Biosystems) in a total volume of 10 μl. The mRNA level of the Nox2 gene was normalized to the corresponding level of GAPDH and represented as the fold induction, which was calculated using the 2−ΔΔCT method as described previously (14). All real-time RT-PCR experiments were performed at least three times, and the mean ± S.E. values are presented unless otherwise noted.
Immunohistochemistry
Mice were deeply anesthetized with pentobarbital sodium (50 mg/kg, intraperitoneal) and transcardially perfused with 4% paraformaldehyde in 0.1 m phosphate buffer (pH 7.4). The L5 spinal cord was removed, postfixed in 4% paraformaldehyde at 4 °C overnight, and transferred to a 30% sucrose phosphate buffer for 48 h. Spinal cord transverse sections (14-μm-thick) were prepared on a gelatin-coated slide glass using a cryocut microtome. The sections were blocked in a solution containing 5% normal goat serum (Jackson ImmunoResearch Laboratories, Bar Harbor, ME), 2% BSA (Sigma), and 0.1% Triton X-100 (Sigma) for 1 h at room temperature. The sections were then incubated overnight at 4 °C with primary antibody for mouse anti-Nox2 (1:100, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-Iba-1 (1:1000, Wako, Osaka, Japan), mouse anti-8-Hydroxyguanosine (8-OHG, 1:1500, Abcam, Cambridge, MA), rabbit anti-glial fibrillary acidic protein (1:5000, DAKO, Glostrup, Denmark), rabbit anti-MAP2 (1:2000, Millipore, Billerica, MA), and rabbit anti-NG2 (1:200, Millipore). After rinsing in 0.1 m PBS, the sections were incubated for 1 h at room temperature with a mixture of FITC-conjugated or Cy3-conjugated secondary antibodies (1:200, Jackson ImmunoResearch Laboratories). The sections were mounted using Vectashield mounting medium (Vector Laboratories, Burlingame, CA), and fluorescent images were obtained with a confocal microscope (LSM700, Carl Zeiss, Germany). Fluorescent signal intensity was quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
Intrathecal Injection
For the administration of lipoteichoic acid (LTA, Sigma) and SB202190 (Sigma), mice were injected under pentobarbital sodium anesthesia (25 mg/kg, intraperitoneal) by direct lumbar puncture between the L5 and L6 vertebrae of the spine, using a 10-μl Hamilton syringe (Hamilton Company, Reno, NV) with a 30-gauge one-half-inch needle as described previously (15). 5 μl of 50 μg/ml LTA in PBS or PBS alone was intrathecally injected in WT and TLR2 knock-out mice. LTA was preincubated with polymyxin B (10 μg/ml) for 30 min at 37 °C before use to prevent LPS contamination. SB202190 was dissolved in dimethyl sulfoxide at 25 mg/ml and diluted in 0.9% saline solution. 10 μl of 1 mg/ml SB202190 in saline solution or vehicle alone was injected in WT mice before L5 SNT. The success of the intrathecal injection was assessed by monitoring the tail-flicking response of the animal when the needle penetrated the subarachnoid space.
Primary Cell Culture
Primary mixed glial and microglial cell cultures were prepared according to previously established procedures (8, 16). In brief, primary spinal cord glial cells were prepared from 7-day-old Sprague-Dawley rats. After the rats were anesthetized, their spines were extirpated, a syringe with PBS (pH 7.4) was inserted, and the spinal cord was pushed out by hydraulic pressure. Upon stripping off the meninges from the spinal cord, the spinal cord was dissociated into single cells by repetitive pipetting and incubation in glial cell culture medium (DMEM supplemented with 10 mm HEPES, 10% FBS, 2 mm l-glutamine, and 1× antibiotic-antimycotic) in a 75-cm2 flask at 37 °C in a 5% CO2 incubator. The medium was changed every 5 days thereafter. After 2 weeks, the glial cells were trypsinized and seeded in six-well plates for subsequent use.
Primary brain glial cell cultures were prepared from postnatal day 1–3 C57BL/6 mice. The meninges were removed from the cerebral hemispheres, dissociated into single cells by trituration, and incubated in glial cell culture medium in a 75-cm2 flask at 37 °C in a 5% CO2 incubator. Primary microglia were harvested from mixed glial cultures on day 14. After shaking at 200 rpm for 3 h on an orbital shaker, the media from the cultures were collected and centrifuged at 800 rpm for 5 min. Microglia were plated in glial culture media. After 15 min, the dishes were washed with medium to remove any unattached astrocytes. The purity of the microglia was routinely monitored and determined to be >98% after immunostaining with Iba-1 antibody (1:1000, Wako).
Western Blot Analysis
Primary rat spinal cord glial cells or mouse brain glial cells stimulated with LTA for various periods of time were harvested with lysis buffer (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1% Triton X-100, 10% glycerol, protease inhibitor mixture (G-Biosciences, St Louis, MO) and phosphatase inhibitor mixture set IV (Millipore)). 20 or 30 μg of the cell lysate from each sample was resolved using electrophoresis on a 10% SDS-PAGE. The proteins separated based on size were transferred to a nitrocellulose membrane, which was then blocked with 5% nonfat dry milk in Tris-buffered saline containing Tween 20 (TBST, 20 mm Tris, pH 7.4, 0.1% Tween 20, and 150 mm NaCl). The membrane were probed overnight with primary antibody for mouse-anti-Nox2 (1:1000, Millipore) or rabbit anti-phospho-eIF4E (Ser-209) (1:1000, Cell Signaling Technology, Danvers, MA) at 4 °C, followed by incubation with horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h prior to ECL (GE Healthcare) treatment. The signal was detected by MicroChemi (DNR Bio-imaging Systems). For the normalization of the antibody signal, the membranes were stripped and reprobed with antibody for β-actin (1:2000, Sigma).
Intracellular ROS Measurement
Intracellular ROS production in spinal cord glial cells was measured using cell-permeable fluorescent dye, CM-H2DCFDA (Life Technologies), after TLR2 stimulation. At 12 h after LTA (2 μg/ml) treatment, CM-H2DCFDA (10 μm) was loaded in glial cells and incubated for 45 min at 37 °C. Upon rinsing the spinal cord glial cells with PBS, the fluorescent intensity was measured at an excitation wavelength of 485 nm and an emission wavelength of 520 nm. The three experiments were independently performed, and each experiment was performed in triplicate.
Statistical Analysis
The statistical significance of differences was assessed using SigmaPlot software (version 11.0, Systat Software, Inc., San Jose, CA). Statistical analysis was performed using the Student's t test. All data are presented as the mean ± S.E., and differences were considered statistically significant when the p value was < 0.05.
RESULTS
Nerve Injury-induced Nox2 Expression Is Abrogated in TLR2 Knock-out Mice
TLR2, -3, or -4 knock-out mice were utilized to evaluate whether TLRs are involved in Nox2 expression in spinal cord microglia after spinal nerve injury. All of these genes have been implicated in spinal cord microglia activation after spinal nerve injury (8–10). L5 spinal nerve axotomy, a well known mouse neuropathic pain model, induced Nox2 mRNA expression in the L5 spinal cord region of WT mice by 2-fold at 12 and 24 h (Fig. 1A). Similar levels of Nox2 mRNA induction were detected in the spinal cord of TLR3 or -4 knock-out mice. However, nerve injury-mediated Nox2 mRNA induction was completely abrogated in the spinal cord of TLR2 knock-out mice. Likewise, Nox2 protein expression, measured by immunohistochemistry, was up-regulated in the dorsal horn of the ipsilateral spinal cord of WT mice at 1 day post injury (dpi) (Fig. 1B). The Nox2-immunoreactivity (IR) mainly co-localized to Iba-1-IR microglia (Fig. 1B, merged panels). In contrast, L5 spinal nerve injury failed to induce Nox2 protein expression in the TLR2 knock-out mice. In the TLR3 and -4 knock-out mice, the levels of Nox2-up-regulation detected in the ipsilateral dorsal horn were similar to that detected in WT mice (Fig. 1B). However, Nox2-IR was not detected in the spinal cord of Nox2 knock-out mice 3 days after L5 SNT, confirming the specificity of the Nox2 antibody (Fig. 1C). The up-regulation of Nox2 accompanied ROS generation in the spinal cord of nerve-injured WT mice but not in TLR2 knock-out mice (Fig. 1D). These data suggest that TLR2, but not TLR3 or -4, is required for nerve injury-induced Nox2 up-regulation in microglia and the subsequent ROS production in the dorsal horn.
FIGURE 1.

L5 SNT-induced Nox2 up-regulation is abrogated in TLR2 knock-out mice. A, Nox2 mRNA expression in the L5 spinal cord segment after L5 SNT was measured by real-time RT-PCR. Total RNA was isolated from the L5 spinal cord tissues of sham-operated control mice and SNT-injured mice (each group, n = 3) at 12 h and 1 dpi (each time point, n = 3). The Nox2 transcript level is presented as the fold induction compared with the levels measured in the control mice of each group. Data are represented as mean ± S.E. (Student's t test; *, p < 0.05; **, p < 0.01, versus sham control in each mice group; #, p < 0.05; ##, p < 0.01, versus TLR2 knock-out mice). B, the L5 spinal cord sections were prepared from WT, TLR2, TLR3, and TLR4 knock-out mice with or without L5 SNT (1 dpi) and then used for immunostaining with Iba-1 and Nox2 antibodies. Representative spinal cord sections are shown (each group, n = 3; scale bar, 100 μm). C, to confirm the specificity of Nox2 immunoreactivity, spinal cord sections from WT and Nox2 knock-out mice at 3 dpi were immunostained with anti-Nox2 antibody (scale bar, 100 μm). D, spinal cord sections were immunostained with antibody against 8-OHG, a nucleotide oxidation marker, at 3 dpi. Compared with TLR2 knock-out mice, 8-OHG-IR in WT mice was dramatically increased following L5 SNT. The fluorescent intensity of 8-OHG-IR signals was measured and presented as the fold induction compared with the corresponding level measured in sham-operated mice. Data are represented as mean ± S.E. (Student's t test, ***, p < 0.001, versus sham control in WT mice, ###, p < 0.001, versus TLR2 knock-out mice, each group, n = 3; scale bar, 100 μm).
TLR2 Stimulation Induces Nox2 Up-regulation in the Spinal Cord in Vivo
To assess whether or not TLR2 activation can induce Nox2 expression in spinal cord microglia in vivo, LTA, a TLR2 agonist, was introduced intrathecally in WT and TLR2 knock-out mice. Twelve h after LTA administration, Nox2 mRNA was up-regulated by 7-fold in the WT spinal cord but was not induced in the TLR2 knock-out spinal cord (Fig. 2A). Similarly, Nox2 protein was up-regulated by the intrathecal injection of LTA in the spinal cord of WT mice but not in that of TLR2 knock-out mice (Fig. 2B). Nox2 was mainly expressed in the Iba-1-positive microglia (Fig. 2B, merged panels). These data indicate that TLR2 activation in spinal cord cells can induce Nox2 expression in microglia.
FIGURE 2.

LTA stimulation induces Nox2 expression in spinal cord microglia in vivo. A, At 12 h after intrathecal injection of 5 μl of LTA (50 μg/ml in PBS, each group, n = 3) or vehicle (Veh, each group, n = 3), total RNA was isolated from L5 spinal cord tissues and used to determine Nox2 mRNA expression by real-time RT-PCR. Data are expressed as mean ± S.E. (Student's t test, **, p < 0.01, versus vehicle control in WT mice group, ##, p < 0.01, versus TLR2 knock-out mice). B, Nox2 protein expression in the ipsilateral L5 spinal cord dorsal horn at 1 day (1 d) after 5 μl of LTA (50 μg/ml in PBS) or vehicle intrathecal injection was evaluated by immunohistochemistry. Spinal cord sections were immunostained with Nox2 and Iba-1 antibodies. Nox2-IR-signals were increased in Iba-1-IR microglia in WT mice after LTA administration, but not in TLR2 knock-out microglia. Vehicle-injected mice served as the control and representative spinal cord sections are shown (each group, n = 3; scale bar, 100 μm). Cont, control.
TLR2 Is Expressed only in Microglia in the Spinal Cord
To identify the cell types that express TLR2 in the spinal cord, immunohistochemistry was performed. When nerve injury was not induced, TLR2 expression was detected at very low levels in the spinal cord, whereas it was up-regulated following L5 SNT (Fig. 3A). Furthermore, the enhanced TLR2-IR mainly co-localized with Iba-1-IR microglia but not with glial fibrillary acidic protein-positive astrocytes, microtubule-associated protein 2 (MAP2)-positive neurons, or NG2-positive oligodendrocyte progenitor cells (Fig. 3B), indicating that TLR2 is expressed exclusively in microglia in the spinal cord. No obvious TLR2-IR signal was detected upon immunostaining spinal cord sections from TLR2 knock-out mice, confirming the specificity of the TLR2 antibody (Fig. 3A). Taken together, these data suggest that LTA introduction in the spinal cord induces microglial Nox2 expression by directly stimulating TLR2 on the spinal cord microglia.
FIGURE 3.

TLR2 is up-regulated in spinal cord microglia by L5 SNT. A, spinal cord sections from sham-operated or L5 SNT-injured WT mice were immunostained with TLR2 antibody. TLR2-IR signals were increased in the ipsilateral spinal cord dorsal horn upon L5 SNT. To confirm the TLR2 antibody specificity, spinal cord sections from TLR2 knock-out mice at 1 dpi were immunostained with anti-TLR2 antibody (scale bar, 100 μm). B, spinal cord sections from L5 SNT-injured mice (1 dpi) were double-immunostained with antibodies against TLR2 and a cell type-specific marker for microglia (Iba-1), astrocytes (glial fibrillary acidic protein; GFAP), neurons (MAP2), and oligodendrocyte precursor cells (NG2) (scale bar, 100 μm). TLR2-IR signals were detected only in Iba-1-IR microglia.
TLR2 Stimulation Induces Nox2 Expression in Primary Spinal Cord Glia
Primary mixed glial cells and microglia were cultured from rat spinal cord and mice cerebra, respectively, and then stimulated with LTA to further investigate TLR2-mediated Nox2 gene expression in spinal cord microglia. After LTA stimulation for 3 h, Nox2 mRNA was induced in primary spinal cord mixed glial cells and primary microglia by 2- and 4-fold, respectively (Fig. 4, A and B). As measured by Western blot assay, LTA stimulation also increased Nox2 protein levels in spinal glial cells (Fig. 4C). In addition, LTA stimulation increased intracellular ROS production in spinal cord glial cells by >2-fold (Fig. 4D). These data demonstrate that TLR2 stimulation induces Nox2 expression and ROS production in microglia.
FIGURE 4.
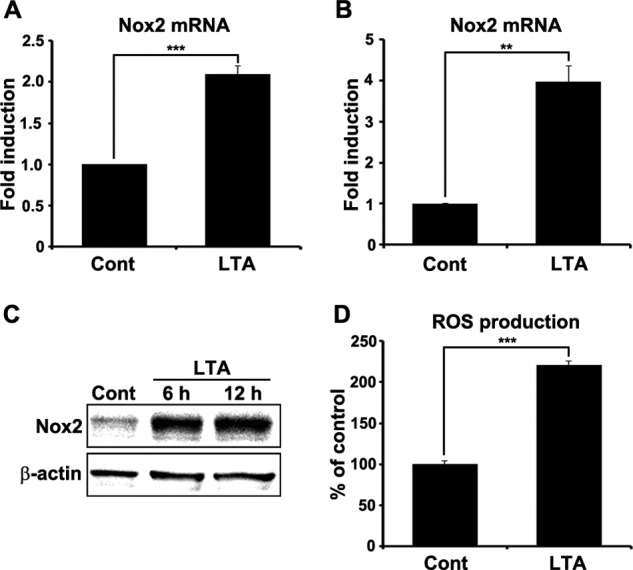
TLR2 stimulation induces Nox2 expression in spinal cord glial cells. Primary spinal cord mixed glial cells (A) and primary microglia (B) were stimulated with LTA (2 μg/ml) for 3 h. Total RNA was prepared from each sample and used for real-time RT-PCR to measure Nox2 mRNA expression. Three independent experiments were performed using primary cells cultured from different donors. The Nox2 transcript level is presented as the fold induction, and data are expressed as mean ± S.E. (Student's t test, **, p < 0.01; ***, p < 0.001). C, Nox2 protein expression was determined by Western blot assay in primary spinal cord glial cells at 6 and 12 h after LTA (2 μg/ml) treatment. β-Actin was used as a loading control. D, intracellular ROS production in spinal cord glial cells was measured using cell permeable fluorescent dye, CM-H2DCFDA (10 μm), after TLR2 stimulation. At 12 h after LTA (2 μg/ml) treatment, intracellular ROS generation was increased in spinal cord glial cells. Data are represented as mean ± S.E. (Student's t test, ***, p < 0.001). Cont, control.
NF-κB and p38 Activation Is Involved in TLR2-activated Nox2 Expression in Glia
We then characterized the intracellular signaling pathways that mediate TLR2-induced Nox2 gene expression. It is well known that TLR2 induces activation of NF-κB and MAP kinases in microglia in vitro (17, 18). Accordingly, we used pharmacological inhibitors to test the involvement of these signaling molecules in TLR2-activated Nox2 mRNA expression. Helenalin, a NF-κB inhibitor, inhibited LTA-induced Nox2 mRNA induction by >80% (Fig. 5A), indicating that NF-κB activation is required for TLR2-induced Nox2 transcription. However, the induction level was not significantly affected by the pretreatment of U0126 (ERK inhibitor), SB202190 (p38 inhibitor), or SP600125 (JNK inhibitor). Similarly, LTA stimulation increased Nox2 protein expression by >5-fold in primary glial cells (Fig. 5B). The protein induction level was partially reduced by the treatment of NF-κB inhibitor by up to 35% but almost completely blocked by p38 inhibitor (Fig. 5C). Neither ERK nor JNK inhibitors significantly affected Nox2 protein expression. These data suggest that p38 activation induces Nox2 expression at the translational level. It is known that p38 regulates translation by activating eukaryotic translation initiation factor 4E (eIF4E) (19). In brain glia, TLR2 stimulation induced activation of eIF4E as early as 30 min, which was measured by phosphorylated eIF4E (Fig. 5D). The TLR2-induced eIF4E phosphorylation was almost completely blocked by p38 inhibitor (Fig. 5, E and F), indicating that p38 MAPK may increase Nox2 expression at the translational level via eIF4E activation.
FIGURE 5.
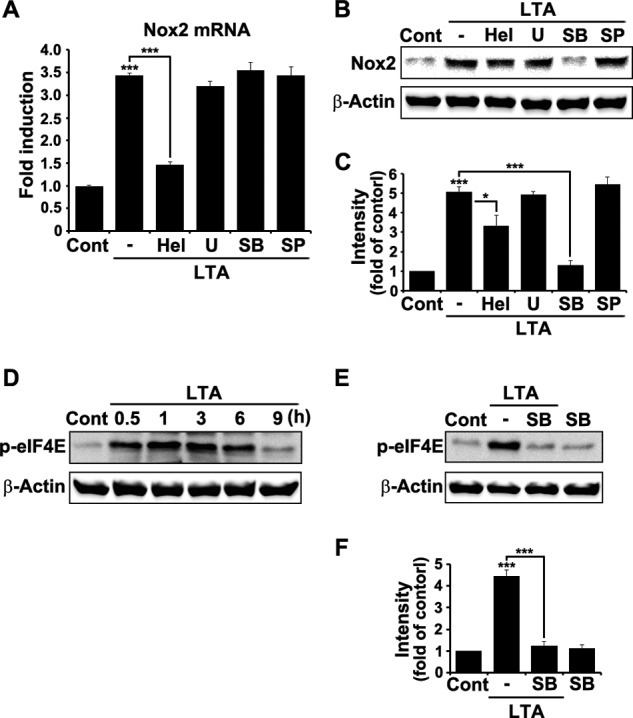
NF-κB and p38 MAP kinase activation is required for Nox2 expression after TLR2 stimulation. A, primary mouse brain glial cells were stimulated with LTA (2 μg/ml) for 3 h in the presence or absence of the following NF-κB or MAPK inhibitors: helenalin (Hel; NF-κB inhibitor, 5 μm), U0126 (U; ERK inhibitor, 5 μm), SB202190 (SB; p38 inhibitor, 5 μm), and SP600125 (SP; JNK inhibitor, 5 μm). The inhibitors were pretreated for 1 h prior to the LTA stimulation. Total RNA was isolated from each sample and used to analyze Nox2 mRNA transcript level by real-time RT-PCR. Data are expressed as mean ± S.E. (Student's t test; ***, p < 0.001). B, brain glial cells were stimulated with LTA (2 μg/ml) for 6 h with or without NF-κB or MAPK inhibitors. Cell lysates were prepared from each sample and used for Western blot analysis to measure Nox2 expression. Three independent experiments were performed, and a representative Western blot image is shown. C, the band intensity of the Nox2 protein was measured and presented as the fold increase compared with the control, which was normalized to the intensity of β-actin (Student's t test; *, p < 0.05; ***, p < 0.001). D, brain glial cells were stimulated with LTA (2 μg/ml) for various time points. Cell lysates were used to measure phosphorylated eIF4E (p-eIF4E) by Western blot analysis. E, glial cells were stimulated with LTA (2 μg/ml) for 1 h in the presence or absence of SB202190. p38 inhibitor was pretreated for 1 h before TLR2 stimulation. Three experiments were independently performed, and a representative Western blot image is shown. F, the band intensity of the p-eIF4E was measured and normalized to the corresponding intensity of β-actin. Data are represented as fold increase compared with the control (Cont; Student's t test, ***, p < 0.001).
To determine whether p38 activation is also required for nerve injury-induced Nox2 expression in spinal cord microglia in vivo, we introduced SB202190 into the spinal cord intrathecally. Upon SB202190 injection, nerve injury-induced Nox2 up-regulation in the spinal cord microglia was nearly completely abrogated (Fig. 6A). These data show that p38 activation is necessary for nerve injury-induced Nox2 expression in spinal cord microglia in vivo. TLR2 transmits intracellular signals via MyD88 (20). In this regard, we tested whether MyD88 is required for nerve injury-induced Nox2 expression using MyD88 knock-out mice. As expected, the spinal cord microglial Nox2 expression was almost completely blocked in the MyD88 knock-out mice (Fig. 6B). Taken together, these data indicate that peripheral nerve injury induces spinal cord microglial Nox2 expression via TLR2-MyD88 signaling pathways.
FIGURE 6.

p38 activation and MyD88 are required for L5 SNT-induced Nox2 expression in spinal cord microglia in vivo. A, spinal cord sections were prepared from sham-operated or L5 SNT-injured WT mice (1 dpi) and double-immunostained with Nox2 and Iba-1 antibodies. SB202190 (10 μg) or vehicle (Veh) was intrathecally injected prior to L5 SNT. Representative images are shown (each group, n = 3; scale bar, 100 μm). B, the L5 spinal cord sections were prepared from WT and MyD88 knock-out mice upon L5 SNT (1 dpi) and then immunostained with Iba-1 and Nox2 antibodies. Representative spinal cord sections are shown (each group, n = 3; scale bar, 100 μm). 1 d, 1 day.
DISCUSSION
The results from this study demonstrate that TLR2 is required for Nox2 expression in spinal cord microglia after L5 spinal nerve injury, playing a critical role in microglia activation and the subsequent induction of neuropathic pain (13). Our data suggest that Nox2 gene expression in spinal cord microglia is due to TLR2-MyD88 activation upon nerve injury. Interestingly, although TLR2, -3, and -4 are all implicated in microglia activation after peripheral nerve injury (8–10), the L5 SNT-induced Nox2 up-regulation was only absent in TLR2 knock-out mice. Meanwhile, Nox2 expression in TLR3 or 4 knock-out mice was comparable to the levels found in WT mice. Even so, nerve injury-induced spinal cord microglia activation and pain induction were still compromised in TLR3 and -4 knock-out mice, (9, 10). These data suggest that the pain-potentiating mechanisms of TLR2, -3, and -4 are not redundant; TLR2 may contribute to pain hypersensitivity via Nox2 induction in spinal cord microglia, whereas TLR3 and -4 may affect central sensitization via other pathways. Unlike TLR2, TLR3, and -4 trigger intracellular signals via TIR-domain-containing adapter-inducing interferon-β (TRIF) in addition to MyD88 (21), which may explain the differences between them. In this regard, it will be worthwhile to investigate if the SNT-induced neuropathic pain is completely blocked in TLR2 and -3 or TLR2 and -4 double knock-out mice.
Although our data from the TLR2 knock-out mice point to the important role of microglial TLR2 in Nox2 expression, at this point, we cannot exclude the possibility that TLR2 on peripheral immune cells may indirectly affect Nox2 expression in spinal cord microglia. However, our study utilized intrathecal LTA administration, which supposedly affects cells in the spinal cord rather than peripheral immune cells, suggesting that TLR2 activation in spinal cord cells leads to Nox2 induction in microglia. In conjunction with these data, our immunohistochemistry data illustrate that TLR2 is solely expressed on microglia in the spinal cord but not in other spinal cord glia or neurons. This is consistent with a previous report that showed that TLR2 expression was only present in microglia of spinal cord tissues from amyotrophic lateral sclerosis patients (22). Given the cellular localization of TLR2 in the spinal cord, it is reasonable to speculate that TLR2 activation on microglia by intrathecal LTA injection directly activates Nox2 expression in microglia. From these data, it can be inferred that TLR2 activation on spinal cord microglia leads to Nox2 expression after L5 SNT. However, it is not clear how microglial TLR2 is activated during peripheral nerve injury. One possibility is that a certain endogenous TLR2 ligand is released from damaged primary afferent neurons after L5 SNT and thereby activates TLR2 on spinal cord microglia. To date, various endogenous TLR2 agonists have been documented, including heat shock proteins, hyaluronic acid, and high mobility group box-1 protein (23). However, there is no evidence implicating any of these TLR2 endogenous agonists in the development of nerve injury-induced neuropathic pain. Instead, other putative microglia activating molecules such as fractalkine, ATP, and glutamate have been reported to be released from damaged sensory neurons after nerve injury (6, 24). However, in our study, fractalkine, ATP, and glutamate were not able to induce Nox2 mRNA expression in primary spinal cord microglia (data not shown). Thus, it is likely that, in addition to the above activators, currently unidentified endogenous TLR2 agonist molecules are involved in spinal cord microglia activation during peripheral nerve injury. Accordingly, future research to determine whether any TLR2 endogenous agonist molecules are released in the spinal cord dorsal horn upon peripheral nerve injury is of importance.
To our knowledge, our in vitro study using primary cultured microglia showed, for the first time, that TLR2 activation on microglia is able to induce Nox2 gene expression. Additionally, our results revealed that NF-κB and p38 MAP kinase differentially regulate TLR2-induced Nox2 gene expression: NF-κB at the transcriptional level versus p38 at the translational level. Transcriptional regulatory mechanisms of Nox2 gene expression have been documented previously. For instance, interferon-γ (IFN-γ) induces Nox2 expression in monocytes by phosphorylating PU.1 in the PKC-dependent pathway (25). In addition, LPS/IFN-γ stimulation can induce Nox2 expression in monocytes via NF-κB activation (26). However, to our knowledge, translational regulation of the Nox2 gene has not been reported. Our in vitro data further contribute to the literature on the regulatory mechanisms of Nox2 gene expression by elucidating novel regulatory mechanisms of Nox2 that is dependent on the TLR2-p38 signaling pathway. Of note, in our in vitro study, NF-κB inhibitor only partially inhibited TLR2-induced Nox2 protein expression although it almost completely blocked mRNA induction. Although there is constitutive Nox2 mRNA expression, it can be assumed that p38 activation alone, in the presence of NF-κB inhibitor, is able to increase Nox2 protein by activating translation from the constitutively expressed Nox2 mRNA. It is well known that p38 MAP kinase is activated in spinal cord microglia after nerve injury (27). p38 activation is a key signaling event that leads to pain-mediating gene expression in spinal cord microglia such as the expression of proinflammatory cytokines, Cox2, and inducible nitric oxide synthase after peripheral nerve injury and the subsequent neuropathic pain induction (28). We have also reported that Nox2-induced ROS production is required for maximal proinflammatory cytokine expression in microglia (13). Based on these reports, a hypothesis that merits further investigation is that the previously established functions of p38 activation on proinflammatory cytokine expression and pain hypersensitivity are at least partly due to its effects on Nox2 expression.
In conclusion, we found that TLR2 is required for Nox2 expression in spinal cord microglia after L5 SNT. Ultimately, this finding suggests that the TLR2-MyD88-p38-Nox2 signaling pathway plays an important role in spinal cord microglial activation during the development of peripheral nerve injury-induced neuropathic pain.
This work was supported by the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology (Grants 2010-0002149, 2010-C00078, 2011-0000934, and 2011-0027667).
- TLR2
- Toll-like receptor 2
- Nox2
- NADPH oxidase 2
- LTA
- lipoteichoic acid
- ROS
- reactive oxygen species
- L5 SNT
- L5 spinal nerve transection
- dpi
- day post injury
- IR
- immunoreactivity
- MAP2
- microtubule-associated protein 2.
REFERENCES
- 1. Watkins L. R., Maier S. F. (2003) Glia: a novel drug discovery target for clinical pain. Nat. Rev. Drug Discov. 2, 973–985 [DOI] [PubMed] [Google Scholar]
- 2. Milligan E. D., Mehmert K. K., Hinde J. L., Harvey L. O., Martin D., Tracey K. J., Maier S. F., Watkins L. R. (2000) Thermal hyperalgesia and mechanical allodynia produced by intrathecal administration of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein, gp120. Brain Res. 861, 105–116 [DOI] [PubMed] [Google Scholar]
- 3. Raghavendra V., Tanga F., DeLeo J. A. (2003) Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J. Pharmacol. Exp. Ther. 306, 624–630 [DOI] [PubMed] [Google Scholar]
- 4. Coull J. A., Beggs S., Boudreau D., Boivin D., Tsuda M., Inoue K., Gravel C., Salter M. W., De Koninck Y. (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021 [DOI] [PubMed] [Google Scholar]
- 5. Kawasaki Y., Zhang L., Cheng J. K., Ji R. R. (2008) Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 28, 5189–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Verge G. M., Milligan E. D., Maier S. F., Watkins L. R., Naeve G. S., Foster A. C. (2004) Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 20, 1150–1160 [DOI] [PubMed] [Google Scholar]
- 7. Tsuda M., Shigemoto-Mogami Y., Koizumi S., Mizokoshi A., Kohsaka S., Salter M. W., Inoue K. (2003) P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424, 778–783 [DOI] [PubMed] [Google Scholar]
- 8. Kim D., Kim M. A., Cho I. H., Kim M. S., Lee S., Jo E. K., Choi S. Y., Park K., Kim J. S., Akira S., Na H. S., Oh S. B., Lee S. J. (2007) A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J. Biol. Chem. 282, 14975–14983 [DOI] [PubMed] [Google Scholar]
- 9. Tanga F. Y., Nutile-McMenemy N., DeLeo J. A. (2005) The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc. Natl. Acad. Sci. U.S.A. 102, 5856–5861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mei X. P., Zhou Y., Wang W., Tang J., Zhang H., Xu L. X., Li Y. Q. (2011) Ketamine depresses toll-like receptor 3 signaling in spinal microglia in a rat model of neuropathic pain. Neurosignals 19, 44–53 [DOI] [PubMed] [Google Scholar]
- 11. Koistinaho M., Koistinaho J. (2002) Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia. 40, 175–183 [DOI] [PubMed] [Google Scholar]
- 12. Ledeboer A., Sloane E. M., Milligan E. D., Frank M. G., Mahony J. H., Maier S. F., Watkins L. R. (2005) Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain 115, 71–83 [DOI] [PubMed] [Google Scholar]
- 13. Kim D., You B., Jo E. K., Han S. K., Simon M. I., Lee S. J. (2010) NADPH oxidase 2-derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury-induced neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 107, 14851–14856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 15. Hylden J. L., Wilcox G. L. (1980) Intrathecal morphine in mice: a new technique. Eur. J. Pharmacol. 67, 313–316 [DOI] [PubMed] [Google Scholar]
- 16. Cho I. H., Hong J., Suh E. C., Kim J. H., Lee H., Lee J. E., Lee S., Kim C. H., Kim D. W., Jo E. K., Lee K. E., Karin M., Lee S. J. (2008) Role of microglial IKKβ in kainic acid-induced hippocampal neuronal cell death. Brain 131, 3019–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin H. Y., Tang C. H., Chen J. H., Chuang J. Y., Huang S. M., Tan T. W., Lai C. H., Lu D. Y. (2011) Peptidoglycan induces interleukin-6 expression through the TLR2 receptor, JNK, c-Jun, and AP-1 pathways in microglia. J. Cell Physiol. 226, 1573–1582 [DOI] [PubMed] [Google Scholar]
- 18. Schachtele S. J., Hu S., Little M. R., Lokensgard J. R. (2010) Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J. Neuroinflammation 7, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang X., Flynn A., Waskiewicz A. J., Webb B. L., Vries R. G., Baines I. A., Cooper J. A., Proud C. G. (1998) The phosphorylation of eukaryotic initiation factor eIF4E in response to phorbol esters, cell stresses, and cytokines is mediated by distinct MAP kinase pathways. J. Biol. Chem. 273, 9373–9377 [DOI] [PubMed] [Google Scholar]
- 20. Takeda K., Akira S. (2004) TLR signaling pathways. Semin. Immunol. 16, 3–9 [DOI] [PubMed] [Google Scholar]
- 21. Kawai T., Akira S. (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 [DOI] [PubMed] [Google Scholar]
- 22. Casula M., Iyer A. M., Spliet W. G., Anink J. J., Steentjes K., Sta M., Troost D., Aronica E. (2011) Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience 179, 233–243 [DOI] [PubMed] [Google Scholar]
- 23. Erridge C. (2010) Endogenous ligands of TLR2 and TLR4: agonists or assistants? J. Leukoc. Biol. 87, 989–999 [DOI] [PubMed] [Google Scholar]
- 24. Milligan E. D., Watkins L. R. (2009) Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 10, 23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mazzi P., Donini M., Margotto D., Wientjes F., Dusi S. (2004) IFN-γ induces gp91phox expression in human monocytes via protein kinase C-dependent phosphorylation of PU.1. J. Immunol. 172, 4941–4947 [DOI] [PubMed] [Google Scholar]
- 26. Anrather J., Racchumi G., Iadecola C. (2006) NF-κB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 281, 5657–5667 [DOI] [PubMed] [Google Scholar]
- 27. Tsuda M., Mizokoshi A., Shigemoto-Mogami Y., Koizumi S., Inoue K. (2004) Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 45, 89–95 [DOI] [PubMed] [Google Scholar]
- 28. Ji R. R., Suter M. R. (2007) p38 MAPK, microglial signaling, and neuropathic pain. Mol. Pain 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]