

Background: The FACT complex reorganizes nucleosomes and functions in DNA replication, transcription, and DNA repair.
Results: Myogenin interacts with FACT, which is required for histone depletion at promoters and initiation of gene expression.
Conclusion: Myogenin recruits the FACT complex to muscle-specific genes where FACT depletes nucleosomes at promoters, thus facilitating gene expression.
Significance: The recruitment of FACT by myogenin is crucial to activate myogenin-dependent genes.
Keywords: Chromatin Regulation, Chromatin Remodeling, Myogenesis, RNA Polymerase II, Skeletal Muscle, FACT, MyoD, SPT16, SSRP1, Myogenin
Abstract
Facilitates chromatin transcription (FACT) functions to reorganize nucleosomes by acting as a histone chaperone that destabilizes and restores nucleosomal structure. The FACT complex is composed of two subunits: SSRP1 and SPT16. We have discovered that myogenin interacts with the FACT complex. Transfection of FACT subunits with myogenin is highly stimulatory for endogenous muscle gene expression in 10T1/2 cells. We have also found that FACT subunits do not associate with differentiation-specific genes while C2C12 cells are proliferating but are recruited to muscle-specific genes as differentiation initiates and then dissociate as differentiation proceeds. The recruitment is dependent on myogenin, as knockdowns of myogenin show no recruitment of the FACT complex. These data suggest that FACT is involved in the early steps of gene activation through its histone chaperone activities that serve to open the chromatin structure and facilitate transcription. Consistent with this hypothesis, we find that nucleosomes are depleted at muscle-specific promoters upon differentiation and that this activity is dependent on the presence of FACT. Our results show that the FACT complex promotes myogenin-dependent transcription and suggest that FACT plays an important role in the establishment of the appropriate transcription profile in a differentiated muscle cell.
Introduction
Gene regulation in skeletal muscle is controlled by a family of highly related basic helix-loop-helix transcription factors, the myogenic regulatory factors (MRFs).3 The MRF family includes Myf5, MyoD, myogenin, and Myf6 (also known as MRF4). The MRFs dimerize with E proteins and bind E box sequences (CANNTG) in the regulatory regions of muscle genes (1). The MRFs work in conjunction with the MEF2 family of MADS box transcription factors (2). MEF2 proteins alone do not have myogenic activity but synergize with the MRFs to enhance gene expression during myogenesis (3, 4).
The MRFs play overlapping but non-redundant roles in myogenesis. As revealed by mouse knockouts, Myf5 and MyoD function early in myogenesis to confer a myogenic fate on mesodermal progenitor cells (5). Myf6 has roles in both early and late events in myogenesis (6). Myogenin functions later in myogenesis to stimulate specified myoblasts to differentiate into functional myofibers. Null mutations in myogenin cause lethality, making it unique among the MRFs (7, 8). In myogenin-null mice, myoblasts are specified, but muscle fibers form poorly (9).
Chromatin modifications and chromatin-modifying enzymes have been shown to play essential roles with the MRFs in establishing required transcriptional control during myogenesis. MyoD can remodel chromatin and activate transcription from silent loci much more efficiently than myogenin (10). Acetyltransferases interact with MyoD and acetylate promoter histones as well as MyoD itself (reviewed in Ref. 1). The activity of the MEF2 proteins is controlled by interactions with the class II histone deacetylases, which function to repress differentiation (reviewed in Ref. 11). The ATP-dependent chromatin-remodeling enzyme SWI/SNF interacts with MyoD, myogenin, and MEF2 proteins and is required for muscle differentiation via its ability to alter chromatin structure at endogenous, muscle-specific loci (12–16).
The facilitates chromatin transcription (FACT) complex was first identified as a complex required for transcript elongation on nucleosomal templates. It is a histone chaperone that is crucial for nucleosomal reorganization during transcription (17, 18), DNA replication (19, 20), and DNA repair (21, 22). FACT acts by inducing either a structural change or stabilizing an existing alternative nucleosomal structure (23, 24), an activity termed “nucleosomal reorganization” (25). FACT has a high affinity for H2A-H2B dimers and can induce displacement of these dimers from nucleosomes in vitro (26, 27).
Mammalian FACT is made up of two subunits, SSRP1 (structure-specific recognition protein 1) and SPT16 (suppressor of Ty 16), which have both shown to be essential for nucleosomal reorganization (18, 24). FACT promotes transcription by RNAPII by several mechanisms. In the yeast Saccharomyces cerevisiae, FACT has been shown to be required for the eviction of histones from the GAL1–10 and PHO5 promoters (27, 29), and it promotes binding of the TATA-binding protein (TBP) to chromatin (30). FACT has also been shown to travel with elongating RNAPII and promote efficient elongation (31). Less is understood about the function of FACT in mammalian cells, but several studies have implicated FACT in promoting muscle-specific gene expression. In smooth muscle, FACT has been seen to interact with MKL1, where it synergistically activates its transcriptional activity (32). MKL1 is expressed in a variety of different cell types preceding cellular differentiation (33) and functions as an activator of serum response factor, a MADS box transcription factor that is responsible for controlling genes involved in cell proliferation and differentiation. It has also been shown that SSRP1 interacts with serum response factor, thus leading to an increase in DNA binding activity of serum response factor and an increased activation of its promoters, implying that SSRP1 is a coregulator of serum response factor-dependent transcription (33).
We have discovered that the FACT complex interacts with myogenin and promotes differentiation-specific muscle gene expression. Although the physical interaction is restricted to myogenin, FACT is highly stimulatory for myogenin-dependent gene expression in the presence of both myogenin and MyoD. FACT is recruited to muscle-specific genes upon differentiation through the interaction with myogenin and travels with RNA polymerase II during elongation of these genes. Nucleosomes are depleted at these promoters coincident with the arrival of FACT, and we show that SSRP1 is required for this depletion and for expression of differentiation-specific genes. This work aids in our understanding of the factors that control the differentiation program of skeletal muscle and advances our knowledge of the role and recruitment of FACT in skeletal muscle.
EXPERIMENTAL PROCEDURES
Cell Culture
The RD1 (ATCC), HEK293 (ATCC), 10T1/2 (ATCC), and proliferating C2C12 (ATCC) cell lines were grown in DMEM supplemented with 10% fetal bovine serum (Hyclone) in a humidified CO2 incubator at 37 °C according to standard protocols (34). To induce differentiation in C2C12 cells and 10T1/2 cells, cells were grown to 70% confluence, and the medium was switched to DMEM supplemented with 2% horse serum (Hyclone). C2C12 cells were grown in differentiation medium for the number of days indicated in each experiment. Primary myoblasts were isolated and propagated as in Ref. 35.
Tandem Affinity Purification (TAP)
Stable cell lines expressing N-TAP myogenin were grown in 150-cm plates and harvested by washing in PBS. Four grams of wet cell pellet was subjected to cryogenic lysis, and TAP-containing protein complexes were purified on IgG-coated Dynabeads (as detailed in Ref. 36). Affinity purification buffer consisted of 20 mm Hepes (pH 7.4), 300 mm NaCl, 2 mm MgCl2, 0.1% Tween, and protease inhibitors. Isolated proteins were resolved by SDS-PAGE, visualized by Coomassie staining, subjected to in-gel trypsin digestion, analyzed by LC-MS/MS with a Thermo LTQ mass spectrometer, and identified by database searching with an in-house version of Mascot (as detailed in Ref. 37, 38).
Cloning
To clone murine genes, RNA from C2C12 cells was extracted with TRIzol (Invitrogen) and used to generate cDNA through the use of the Superscript III first strand synthesis system for an RT-PCR kit with oligo(dT) primers (Invitrogen). N-TAP myogenin was cloned by amplifying myogenin from cDNA generated from C2C12 cells after 2 days of differentiation (D2). The primers used were Myog EcoRI F 5′ TCGAATTCATGGAGCTGTATGAGACATCC 3′ and Myog EcoRI R 5′ ACGAATTCTCAGTTGGGCATGGTTT 3′. The resulting product was digested with EcoRI and cloned into pZome1N (EUROSCARF).
Murine SSRP1 was amplified from cDNA generated from undifferentiated C2C12 cells using SSRP1 FL F 5′ATGGCAGAGACATTGGAGTTCAACGA 3′ and SSRP1 FL R 5′ TTCATCAGATCCCGAGGC. The resulting PCR product was TOPO-cloned using the pcDNA 3.1/V5-His TOPO TA expression kit (Invitrogen).
Full-length myogenin was amplified from cDNA generated from differentiated C2C12 cells using Myog F BamHI 5′ AGGATCCATGGAGCTGTATGAGACATCC 3′ and Myog R EcoR1 5′ ACGAATTCTCAGTTGGGCATGGTTT 3′. The resulting PCR product was cloned into pcDNA3.1 V5-FLAG (a gift of S. Dent, M.D. Anderson Cancer Center). The myogenin truncations were amplified and cloned as above using the following primers: the N-terminal region with the βHLH domain (amino acids 1–136) used Myog F BamHI as above, and Myog βHLH R EcoRI 5′ GGAATTCTCAGAGCAAGGCCTGTAG 3′ and the C-terminal region with the βHLH domain (amino acids 59–224) used Myog βHLH F BamH1 5′ CGGGATCCGAGCATTGTCCAGGCCAGT 3′ and Myog R EcoR1 as above. All resulting clones were confirmed by sequencing.
Coimmunoprecipitations
HEK293 cells were transiently transfected with plasmids expressing myogenin, MyoD, SSRP1, and SPT16. EMSV-myogenin (provided by Diane Edmondson, The University of Texas Medical School at Houston) and pEMCIIS (provided by Andrew Lassar, Harvard Medical School) were used for expressing myogenin and MyoD, respectively. Plasmids mSSRP1-V5 (this laboratory) and pCMV-4-SPT16 (a gift of Takanori Kihara and Masayuki Murata, University of Tokyo) were used to express SSRP1 and SPT16. Following the transfection, whole cell extracts were made in radioimmunoprecipitation assay buffer. 300 μg of extract was used for each immunoprecipitation with 1 μg of antibody. The antibodies used included anti-V5 (Invitrogen), anti-MyoG (F5D, Developmental Studies Hybridoma Bank), anti-MyoD (5.8A, Santa Cruz Biotechnology), anti-SSRP1 (10D1, Biolegend), and anti-SPT16 (8D2, Biolegend). Following an overnight incubation, antibody-antigen complexes were collected with protein A-agarose beads (Invitrogen). The beads were washed with radioimmune precipitation assay buffer and resuspended in protein loading dye. Immunoprecipitated samples with appropriate controls were loaded onto SDS-PAGE gels and transferred to PVDF membranes for Western blot analysis. For each immunoprecipitation, the blot was probed with both the reciprocal factor to test for the coimmunoprecipitation and with the antibody used for the immunoprecipitation to confirm that the immunoprecipitation was successful. All immunoprecipitations were performed at least twice to confirm the results.
Western Blot Analysis
Cell extracts were made by lysing PBS-washed cell pellets in radioimmune precipitation assay buffer supplemented with protease inhibitors (Complete protease inhibitor, Roche). Following incubation on ice, clear lysates were obtained by centrifugation. Protein concentrations were determined by Bradford's assay (Bio-Rad). For each sample, 30 μg of protein was loaded on each gel. Proteins were transferred onto a PVDF membrane using a tank blotter (Bio-Rad). The membranes were then blocked using 5% milk and 1× Tris Buffered Saline with Tween 20 and incubated with primary antibody overnight at 4 °C. Membranes were then washed with 1× TBST and incubated with the corresponding secondary antibody. Membranes were again washed with 1× TBST, incubated with chemiluminescent substrate according to the protocol of the manufacturer (SuperSignal, Pierce), and visualized by autoradiography. The antibodies used include anti-SSRP1 (10D1, Biolegend), anti-SPT16 (8D2, Biolegend), anti-V5 (Invitrogen), anti-FLAG (Sigma), anti-MyoD (5.8A, Santa Cruz Biotechnology), anti-MyoG (F5D, Developmental Studies Hybridoma Bank), and anti-GAPDH, (Chemicon).
Cell Transfections and Luciferase Assays
HEK293 and 10T1/2 cells were transiently transfected with calcium phosphate according to standard protocols (34). For the luciferase assay, the vectors pGL3 basic (Promega) and pGL3(+) (Promega) were used as the negative and positive controls, respectively. The Lmod2 reporter has been described previously (39), and the TRE reporter was a generous gift of the Olson laboratory (University of Texas Southwestern Medical Center). The plasmids EMSV-myogenin (a gift of D. Edmondson, University of Texas Medical School at Houston) and pEMCIIs (a gift of Andrew Lassar, Harvard Medical School) were used for expressing myogenin and MyoD, respectively. Plasmids mSSRP1-V5 (this laboratory) and pCMV-4-SPT16 (a gift of Takanori Kihara and Masayuki Murata, University of Tokyo) were used to express SSRP1 and SPT16. The plasmids pcDNA-MEF2C (a gift of Eric N. Olson, University of Texas Southwestern Medical Center) and E12-pCLBABE (provided by S. Tapscott, Fred Hutchinson Cancer Center, Addgene catalog no. 20918) were used for expressing MEF2C and E12, respectively. Luciferase activity was determined using the dual-luciferase reporter assay system (Promega). 10T1/2 cells were seeded at a density of 5 × 103 cells/well in 96-well plates and transfected with 0.2 μg of DNA. Transfections were normalized to Renilla luciferase. Transfections were performed in triplicates, and all data sets were repeated at least twice.
For detection of endogenous gene expression, cells were seeded at a density of 5 × 104 cells/well in 6-well plates and transfected with 2 μg of plasmid DNA. Cells were maintained in growth medium for 1 day post-transfection. When the cells reached confluency, low-serum medium (differentiation medium) was placed on the cells for 24 h prior to harvesting RNA. Transfections were performed in triplicates, and all data sets were repeated at least twice. Primers used were as in Ref. 40.
Quantitative Reverse Transcriptase Polymerase Chain Reaction
RNA was isolated from cells by TRIzol extractions (Invitrogen). 2 μg of total RNA were reverse-transcribed with MultiScribeTM MuLV reverse transcriptase (Applied Biosystems). cDNA equivalent to 40 ng was used for quantitative PCR amplification with SYBR Green PCR Master Mix (Applied Biosystems). Samples in which no reverse transcriptase was added (no RT) were included for each RNA sample. The relative levels of expression of genes were normalized according to those of Hprt. Quantitative PCR data were calculated using the comparative Ct method (Applied Biosystems). Standard deviations from the mean of the [Δ]Ct values were calculated from three independent RNA samples. Primers to the coding region of Acta1, Lmod2, Tnni2, and Hprt were described in Ref. 35. Tnnt1 was amplified with primers Tnnt1 F 5′ GGAGAAGATGCGGAAGGAG 3′ and Tnnt1 R 5′ CAGTCTGTCGCTTCCCAC 3′. All quantitative PCRs were performed in triplicates, and three independent RNA samples were assayed for each time point.
Chromatin Immunoprecipitation
ChIP assays were performed and quantified as described previously (40). Antibodies against the following proteins were used: myogenin (F5D, Developmental Studies Hybridoma Bank), SSRP1 (10D1, Biolegend), SPT16 (8D2, Biolegend), RNAPII (H-224, SCBT), phosphoserine 2 form of RNAPII (H5, Convance), H2A (2578, Cell Signaling Technology), H2B (2934, Cell Signaling Technology), and H3 (1791, Abcam). Rabbit IgG (Santa Cruz Biotechnology) was used as a nonspecific control. Primers spanning the promoters of Tnni2, Lmod2, and IgH were described in Ref. 40 and used to detect promoter enrichment. Values of [Δ][Δ]Ct were calculated using the following formula on the basis of the comparative Ct method: [Δ]Ct, template (antibody)- [Δ]Ct, template (IgG) = [Δ][Δ]Ct. Fold enrichments were determined using the formula 2 −[Δ][Δ]Ct. (experimental)/2 −[Δ][Δ]Ct (reference, IgH). S.E. from the mean was calculated from replicate [Δ][Δ]Ct values. The non-coding region of the IgH locus was used detect nonspecific binding and normalize the fold enrichments for the individual promoters. All ChIP assays shown are representative of at least three individual experiments.
shRNA Knockdown
Myogenin and SSRP1 knockdown lines were constructed with shRNA constructs designed by the RNAi Consortium in the pLOK.1 plasmid (Open Biosystems). Five constructs targeting murine myogenin, five constructs targeting murine SSRP1, and one scrambled control were linearized, transfected into C2C12 cells, and selected with puromycin (2 μg/ml). Pooled clones were selected and propagated.
Immunohistochemistry
Cells were immunostained with antibodies against myosin heavy chain (MF-20, Developmental Studies Hybridoma Bank) and DAPI as described in (35). The fusion index was calculated as the ratio of nuclei in myosin heavy chain-positive myotubes to the total number of nuclei in the field for ten random fields.
RESULTS
Myogenin Interacts with the FACT Complex
To identify interacting partners of myogenin, a rapid single-step purification using the PrA moiety of the TAP was performed with TAP-tagged myogenin (36). The activity of the fusion protein was verified by testing the ability of TAP-myogenin to activate muscle-specific luciferase reporter constructs. It was found that TAP-myogenin activated these reporters comparably to myogenin (Fig. 1A). As we found that constitutive overexpression of myogenin induced terminal differentiation in proliferating C2C12 cells (data not shown), we reasoned that a rhabdomyosarcoma cell line could be used for this analysis. Rhabdomyosarcoma is a pediatric cancer thought to arise from skeletal muscle precursors. Rhabdomyosarcoma cells often express high levels of MyoD and myogenin yet do not differentiate. Although muscle differentiation is blocked in these cells, it is likely that many of the components of a normal muscle cell would still be present. Novel blocks to the differentiation program could also be identified in such an approach, as was observed with TAP-MyoD in RD cells, a rhabdomyosarcoma cell line (41). Thus, we used a similar approach with the RD cell line to analyze the interaction profile of myogenin. Stable cell lines expressing N-TAP myogenin were selected, amplified, harvested for the PrA-based purification, and then coenriching proteins were identified by mass spectrometry. At a 0.1% false discovery rate, 66 proteins were found to coenrich with N-TAP myogenin, which included putative interacting proteins SSRP1 and SPT16 of the FACT complex.
FIGURE 1.
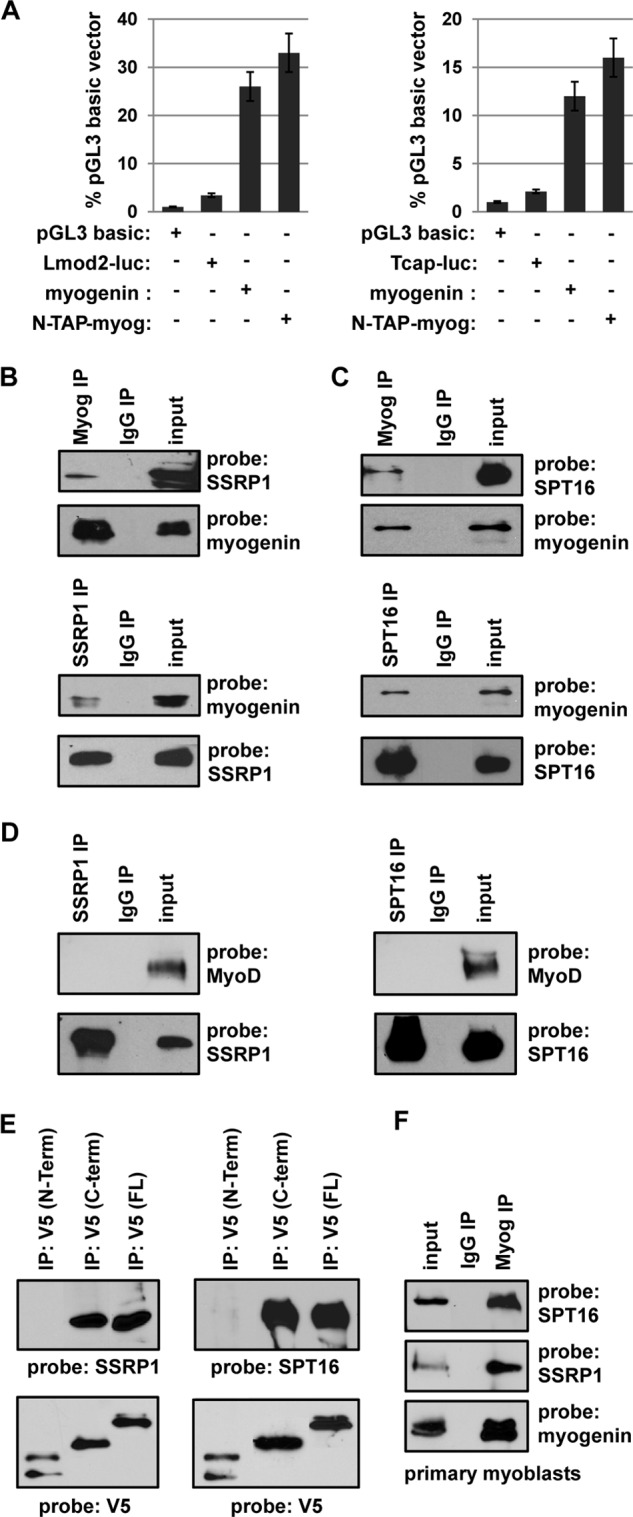
Myogenin interacts with SSRP1 and SPT16. A, N-TAP myogenin is transcriptionally active. 10T1/2 cells were transfected with the indicated constructs and assayed for luciferase activity. The luciferase reporter constructs were Lmod2-luc, which contains the Lmod2 promoter, and Tcap-luc, which contains the Tcap promoter. All data were normalized to the pGL3 basic value, which was set to 1. Experiments were performed in triplicate. B and C, HEK cells were transfected with expression constructs for myogenin (EMSV-myog), SSRP1, or SPT16. B, extracts were immunoprecipitated (IP) with antibodies against myogenin (F5D) and SSRP1 and probed with antibodies against SSRP1 and myogenin. C, extracts were immunoprecipitated with antibodies against myogenin or SPT16 and probed with antibodies against SPT16 and myogenin. D, MyoD does not interact with SSRP1 or SPT16. HEK cells were transfected with expression constructs for MyoD, SSRP1, or SPT16 and immunoprecipitated for SSRP1 or SPT16 and probed with antibodies against MyoD and the reciprocal factor. E, SSRP1 and SPT16 interact with the C terminus of myogenin. HEK cells were transfected with expression constructs for the N terminus of myogenin-V5 (1–136), C terminus of myogenin-V5 (amino acids 59–224), full-length (FL) myogenin-V5 (amino acids 1–224) and either SSRP1 or SPT16. Extracts were immunoprecipitated with antibodies against V5 and probed with antibodies against SPT16 or SSRP1. Blots were also probed with V5 to confirm the immunoprecipitation. F, myogenin interact with SSRP1 and SPT16 in vivo. Extracts from primary myoblasts differentiated for 2 days were immunoprecipitated for myogenin and probed with antibodies against SPT16, SSRP1, and myogenin.
To confirm the interaction of FACT with myogenin, HEK293 cells were transfected with expression constructs for myogenin, SSRP1, and/or SPT16. Extracts were immunoprecipitated with antibodies against myogenin, SSRP1, or SPT16, and a Western blot analysis was performed with antibodies against the reciprocal factor to detect the coimmunoprecipitation. Blots were also probed with the initial antibody used to confirm the immunoprecipitation. The results showed that reciprocal interactions could be detected between myogenin and both SSRP1 (Fig. 1B) and SPT16 (C). As the MRF family is highly related, we were also interested in determining whether MyoD could bind SSRP1 or SPT16. HEK293 cells were transfected with expression constructs for MyoD, SSRP1, or SPT16. Extracts were immunoprecipitated with antibodies against SSRP1 and SPT16, and a Western blot analysis was performed with antibodies against MyoD to detect the coimmunoprecipitation and with antibodies against the reciprocal factor to confirm the immunoprecipitation. An interaction with MyoD could not be detected in these experiments (Fig. 1D). Thus, the interaction of SSRP1 and SPT16 appears to be specific to myogenin.
As the interaction was specific to myogenin, we next sought to determine the interaction domain of myogenin. The MRF family shares a high homology in the βHLH region but have divergent N- and C-terminal domains. An N-terminal construct that included the basic helix-loop-helix region (amino acids 1–136), a C-terminal construct that also included the basic helix-loop-helix domain (amino acids 59–224) and full-length myogenin (amino acids 1–224) were used to map the interaction domain. The constructs were cotransfected with either SSRP1 or SPT16 in HEK293 cells, immunoprecipitated with antibodies against the V5 epitope fused to the myogenin constructs, and probed with antibodies against SSRP1, SPT16, or V5. We found that SSRP1 and SPT16 interacted with the C-terminal domain of myogenin (Fig. 1E). The results suggest that the interaction is not with the βHLH domain, as both truncation constructs contained this domain, but rather with the unique C-terminal domain of myogenin, which has been shown previously to have distinct activities from the C-terminal domain of MyoD (42).
To confirm that the interaction between myogenin and the FACT complex occurs in vivo, we performed the coimmunoprecipitations in primary myoblasts differentiated for 2 days. We found that the interaction could also be detected with endogenous myogenin, SSRP1, and SPT16 (Fig. 1F).
SSRP1 and SPT16 Do Not Stimulate Plasmid-based Muscle-specific Reporters
To determine how the interaction with FACT affects myogenin-dependent transcription, we first used a plasmid-based reporter assay to investigate if SSRP1 and SPT16 could stimulate muscle-specific luciferase reporter constructs. The two reporter vectors used for this assay were the Leiomodin 2 (Lmod2) muscle-specific reporter and the TRE reporter vector, which contains a multimerized series of four E boxes from the Ckm promoter. Reporter constructs were transiently transfected with SSRP1, SPT16, and myogenin into 10T1/2 cells, a fibroblast line. As shown, neither the Lmod2 reporter (Fig. 2A) nor the TRE reporter (B) showed stimulation of muscle-specific reporters in the presence of SSRP1 or SPT16. Activation was observed with the transfection of myogenin, but the addition of SSRP1, SPT16, or both SSRP1 and SPT16 together resulted in no additional activation (Fig. 2, A and B). To confirm that a stimulation of myogenin activity could be observed in this assay, MEF2C was transfected with myogenin for the Lmod2 reporter, which contains a MEF2 binding site. We found that MEF2C robustly enhanced the activation by myogenin for the Lmod2 reporter (Fig. 2A). For the TRE reporter, which lacks a MEF2 binding site, an E protein, E12, was transfected with myogenin. Again, a coactivation with myogenin was observed (Fig. 2B). Each of these experiments were repeated with MyoD, and we again observed no stimulation of MyoD activity with exogenous SSRP1 or SPT16 (data not shown).
FIGURE 2.
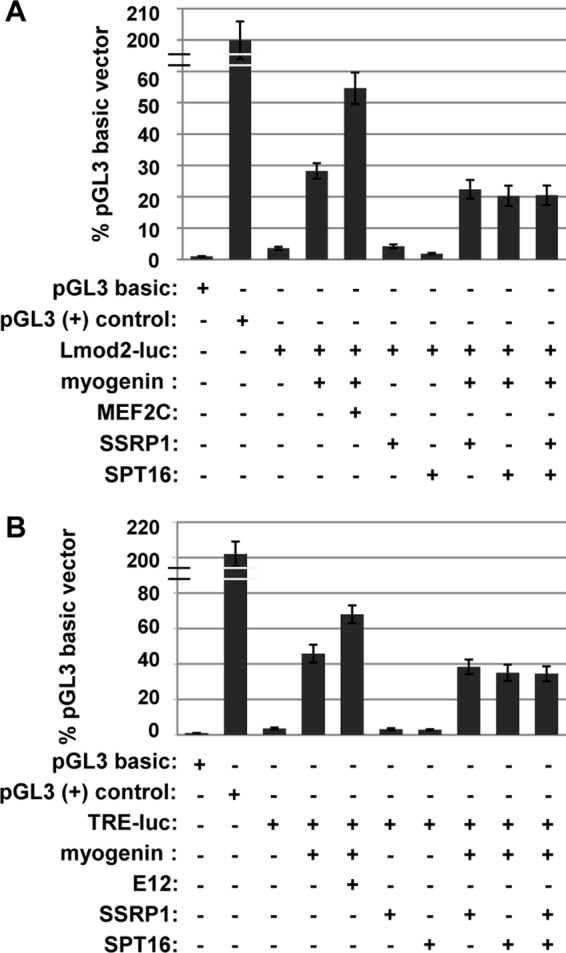
SSRP1 and SPT16 do not activate plasmid-borne muscle-specific luciferase reporters in the presence of myogenin. 10T1/2 cells were transfected with the indicated constructs and assayed for luciferase activity. The luciferase reporter constructs were Lmod2-luc, which contains the Lmod2 promoter (A), and TRE-luc, a multimerized E box from the MCK promoter (B). All data were normalized to the pGL3 basic value, which was set to 1. Experiments were performed in triplicate.
SSRP1 and SPT16 Stimulate Endogenous Muscle-specific Genes in the Presence of Myogenin
It was surprising that no activation was observed in the plasmid-based reporter assay, but we reasoned that the FACT complex might require a nucleosomal context to show activity. We again used the 10T1/2 cell line, which activates endogenous muscle-specific gene expression when transfected with exogenous MRFs such as MyoD or myogenin. To determine how SSRP1 and SPT16 would affect myogenin-dependent activation of endogenous muscle genes, 10T1/2 cells were transfected with myogenin, SSRP1, or SPT16 alone or in combination. To confirm the transfections, gene expression of each of the transfected constructs was assayed by quantitative real-time PCR. We found that myogenin could activate muscle-specific genes such as Acta1, but this activity was stimulated by the addition of SSRP1 or SPT16 (Fig. 3A). Addition of SSRP1 and SPT16 without myogenin did not lead to activation of muscle-specific genes (Fig. 3A). The highest activation was observed when both SSRP1 and SPT16 were added (Fig. 3B). The experiments were repeated with MyoD, and we found that SSRP1 and SPT16 also promoted myogenin-dependent muscle gene expression in the presence of MyoD (Fig. 3C). In fact, the overall levels of gene expression observed were higher than that seen for myogenin in the presence of SSRP1 and SPT16. This was a surprising result, given that we had found that MyoD does not interact with SSRP1 or SPT16, but it is known that expression of MyoD correlates with the expression of myogenin, as myogenin is one of the targets of MyoD. We confirmed that myogenin is expressed in 10T1/2 cells transfected with MyoD by quantitative real time PCR (Fig. 3D). To understand the contribution of myogenin, we also examined a gene, troponin I, skeletal, slow 1 (Tnnt1), known to be activated by MyoD and not myogenin (43). In the 10T1/2 assay, we found that transfection of MyoD, but not myogenin, activated Tnnt1 and the addition of SSRP1 and SPT16 had no effect on the activation by MyoD (Fig. 3E). We also examined Id3 and NP1, which are known to be MyoD dependent, and again saw no additional activation by SSRP1 and SPT16 (data not shown). The data suggest that MyoD and myogenin, together with the FACT complex, lead to the highest level of gene activation on muscle-specific genes regulated by both MRFs.
FIGURE 3.

SSRP1 and SPT16 cooperatively promote endogenous myogenin dependent gene activation. 10T1/2 cells were transfected with expression constructs for myogenin (EMSV-myog), MyoD (pEMCIIS), SSRP1 (pcDNA Ssrp1 V5), and SPT16 (pCMV-4-SPT16) and assayed for muscle-specific gene expression by quantitative PCR. All data were normalized to Hprt. A, SSRP1 and SPT16 independently stimulate gene expression in the presence of myogenin. B, SSRP1 and SPT16 cooperatively stimulate gene expression in the presence of myogenin. C, SSRP1 and SPT16 also cooperatively stimulate myogenin-dependent gene expression in the presence of MyoD. D, MyoD stimulates myogenin expression in 10T1/2 cells. Myogenin expression was assayed in the experiment shown in C. E. SSRP1 and SPT16 do not activate a MyoD target gene that is not stimulated by myogenin.
Detection of Endogenous Expression of FACT in Skeletal Muscle Cells
To relate our results to events that occur in a muscle cell, we wanted to first confirm the expression pattern for SSRP1 and SPT16 in the murine myoblast cell line C2C12. It has been reported that SSRP1 and SPT16 are expressed in proliferating cells but are down-regulated upon differentiation (44). Included in this previous analysis was expression profiling showing that SPT16 was down-regulated immediately upon differentiation, whereas SSRP1 levels gradually decreased during differentiation. However, SSRP1 has also been reported to be associated with muscle-specific genes in both myoblasts and myotubes (45). We compared the protein expression levels of SSRP1 and SPT16 during a time course of differentiation in comparison to the expression of myogenin (Fig. 4A). Consistent with prior studies, we found that SPT16 was down-regulated upon differentiation but observed that the down-regulation occurred after 3 days of differentiation. SSRP1 was also modestly down-regulated after 3 days of differentiation. Myogenin was up-regulated immediately upon differentiation. Thus, it appears that myogenin and SPT16 are co-expressed in muscle cells until 3 days after differentiation initiates. We also examined the expression pattern of SSRP1 and SPT16 in primary myoblasts and found that the expression pattern was similar for SSRP1 (Fig. 4B). Surprisingly, we found that SPT16 levels were reduced but still detectable after 5 days of differentiation in primary myoblasts, indicating that SPT16 is not down-regulated as rapidly in primary myoblasts as compared with C2C12 cells (Fig. 4B). We have shown previously that differentiation-specific genes regulated by myogenin initiate expression at 2 days of differentiation but do not reach full expression levels until 6 days of differentiation (40). This suggests that FACT contributes to the expression of myogenin-dependent genes, as the promoters initiate gene expression.
FIGURE 4.
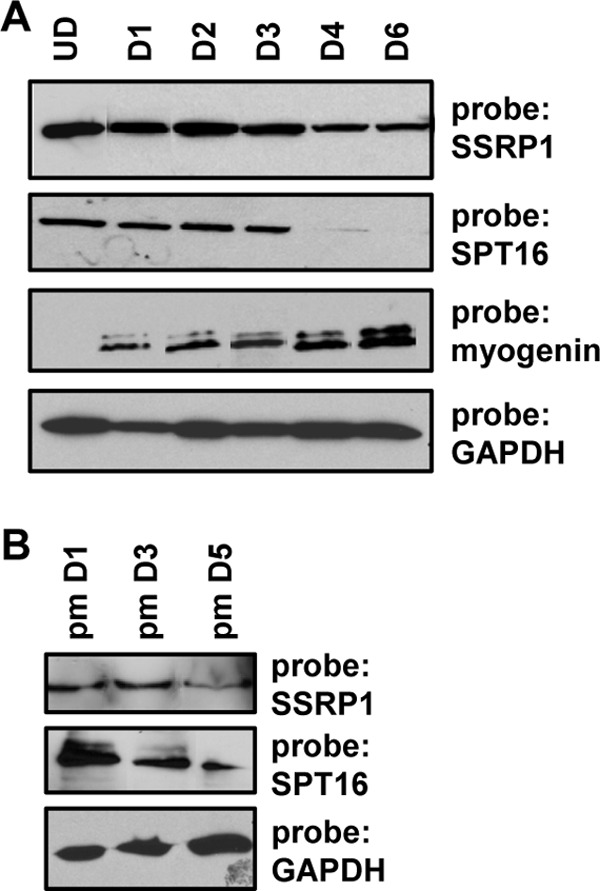
SSRP1 and SPT16 are expressed in proliferating cells but down-regulated during differentiation. A, C2C12 cells were differentiated for the indicated number of days and harvested for protein. Extracts were probed with antibodies against SSRP1, SPT16, MYOG, and GAPDH. UD, undifferentiated or proliferating cells; D1-D6, cells differentiated for the indicated number of days. B, primary myoblasts were differentiated as described in A. Extracts were probed with antibodies against SSRP1, SPT16, and GAPDH.
Myogenin-dependent Genes Show an Association of SSRP1 and SPT16 during the Initial Stages of Differentiation
To determine whether FACT was recruited to muscle-specific genes, we assayed for the presence of SPT16 and SSRP1 at two genes that we have shown are dependent on myogenin in vivo, Tnni2 and Lmod2. A ChIP assay was carried out in C2C12 cells at different stages of differentiation (undifferentiated, day 2, and day 6) for both SSRP1 and SPT16. We have shown previously that myogenin is detectable at these promoters after 2 days of differentiation (40). We found that SSRP1 and SPT16 were not associated with the promoters in undifferentiated cells (Fig. 5A), consistent with the inactivity of the genes tested at this stage (40). We observed that SSRP1 and SPT16 associate with these genes at 2 days of differentiation (Fig. 5A). We have shown previously that myogenin arrives at these promoters at this time point (40), consistent with the arrival of FACT. When the experiment was repeated after 6 days of differentiation, no association of SSRP1 or SPT16 was observed (Fig. 5A). At this time point, we have shown previously that myogenin is still associated with these promoters (40). The lack of association of the FACT complex was consistent with the down-regulation of SPT16 at this time point. Although SSRP1 was still present, we observed that it was no longer associated with these genes after 6 days of differentiation. As FACT is known to promote efficient elongation by RNAPII and travel with the elongating form of the enzyme, we also assayed for the association of FACT in the coding region of Tnni2 after 2 days of differentiation. Chromatin immunoprecipitation assays for RNAPII, SSRP1, and SPT16 were assayed with primers corresponding to the promoter region, intron 1 and intron 5 of Tnni2. We found that RNAPII was recruited with SSRP1 and SPT16 to the promoter after 2 days of differentiation (Fig. 5B). RNAPII, SSRP1, and SPT16 could be detected throughout the coding region (Fig. 5B). To confirm that the RNAPII detected was the elongating form of RNAPII, we repeated the ChIP experiments with antibodies against the C-terminal domain of the largest subunit of RNAPII, RBP1, phosphorylated at serine 2. The C-terminal domain of RBP1 goes through a cycle of phosphorylation events, culminating in the phosphorylation of serine 2 in the elongating form of RNAPII (46). We found that the serine 2 phosphorylated form of RNAPII was associated with the coding region of the gene with enhanced enrichment toward the 3′ end of the gene, similar to the enrichments of SSRP1 and SPT16 (Fig. 5C). Thus, it appears that the FACT complex travels with elongating RNAPII at muscle-specific genes, as has been seen in other systems.
FIGURE 5.

FACT binds muscle-specific genes. A, the FACT complex associates with muscle-specific genes in the early stages of differentiation. ChIP assays were performed on C2C12 cells while proliferating (undifferentiated, UD), after differentiation for 2 days (D2), and after differentiation for 6 days (D6) with antibodies against SSRP1 and SPT16 and primers against the Tnni2 and Lmod2 promoters. All fold enrichment values are normalized to IgH. B, FACT associates with the promoter and coding region of Tnni2 at 2 days of differentiation (D2), coincident with the localization of RNAPII. ChIP assays were performed on C2C12 cells at D2 with antibodies against SSRP1, SPT16, and RNAPII and primers spanning the promoter and coding region of Tnni2. Data are presented as in A. C, FACT localization is coincident with the elongating form of RNAPII in the coding region of Tnni2. ChIP assays were performed as in B with antibodies against the serine 2 phosphorylated C-terminal domain of RBP1, SSRP1, and SPT16 and analyzed as in B.
Myogenin Is Required for Recruitment of FACT to Muscle-specific Genes
To confirm that the interaction with myogenin promotes the recruitment of FACT to muscle-specific genes, we asked whether myogenin was required for the recruitment of FACT. Stable cell lines expressing shRNA constructs for myogenin were assayed for the presence of myogenin at the RNA (Fig. 6A) and protein level (B) to confirm the knockdown. We next assayed for FACT association with muscle-specific genes by chromatin immunoprecipitation at 2 days of differentiation. We found that in cells lacking myogenin, SSRP1 was not recruited to target promoters (Fig. 6C).
FIGURE 6.
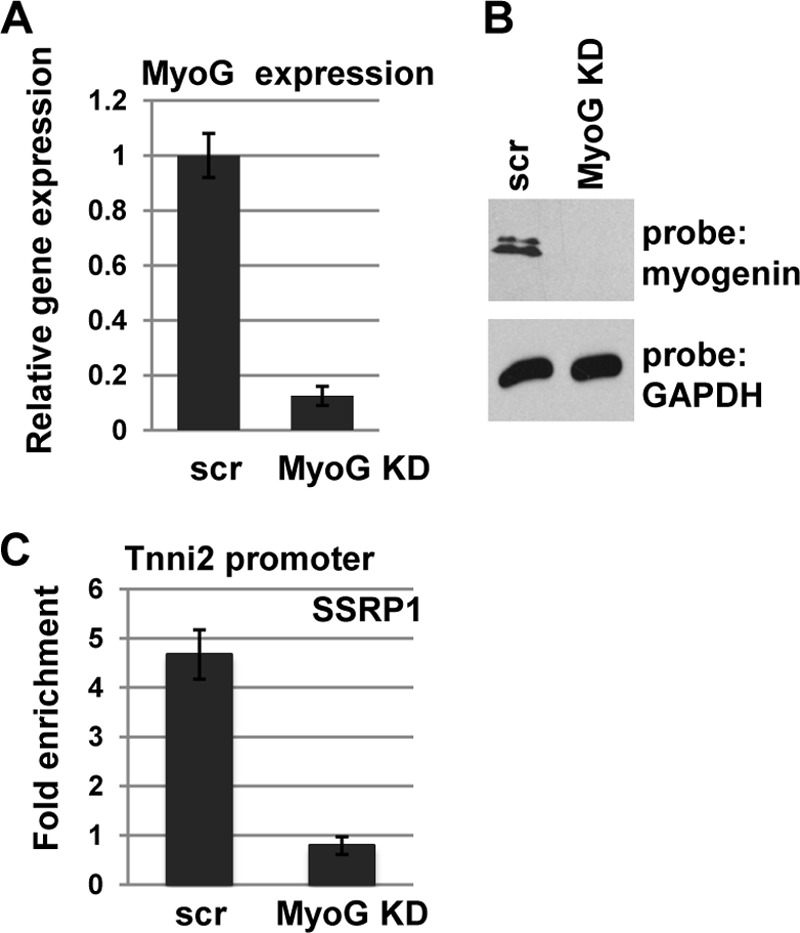
Myogenin is required for recruitment of SSRP1 to muscle-specific genes. A, stable cell lines expressing shRNA constructs were used to deplete myogenin. Depletion was confirmed by analysis of the level of RNA by quantitative real-time PCR (A) and protein by Western blotting (B). C, chromatin immunoprecipitation assays were performed on differentiated C2C12 cells (D2) transfected with scrambled shRNA constructs (scr) or shRNA constructs against myogenin with antibodies against SSRP1 and primers corresponding to the Tnni2 promoter.
Histone Occupancy Is Depleted at Muscle-specific Promoters upon Differentiation
To determine the function of FACT on muscle-specific genes, we assayed for the presence of histones on muscle-specific gene promoters. ChIP assays were performed for H2A, H2B, and H3 and analyzed for Tnni2 and Lmod2 promoter regions. The level of histone association in proliferating cells was compared with the levels observed at day 2 and day 6. We found that promoter occupancy of histone H2A (Fig. 7A), histone H2B (B), and histone H3 (C) were sharply reduced at 2 days of differentiation. Surprisingly, we found that although a modest additional decrease in histone levels could be observed at day 6, the vast majority of histone depletion occurred at 2 days of differentiation, consistent with the arrival of SSRP1 and SPT16.
FIGURE 7.
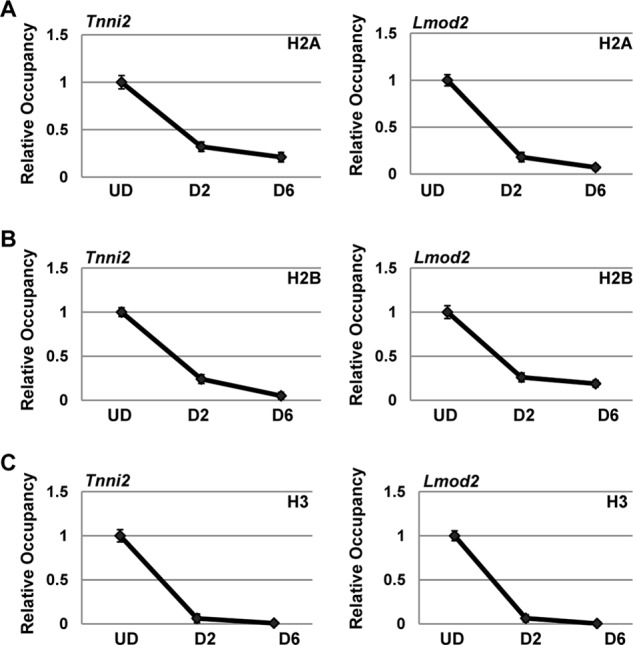
Histone occupancy at muscle-specific gene promoters is highly reduced during gene activation. ChIP assays were performed on C2C12 cells while proliferating (undifferentiated, UD), after differentiation for 2 days (D2), and after differentiation for 6 days (D6) with antibodies against H2A (A), H2B (B), and H3 (C) and primers against the Tnni2 and Lmod2 promoters. Data are shown as relative occupancy with the fold enrichment (normalized to IgH) set at 1 for the UD sample.
SSRP1 Is Required for the Histone Depletion Observed at Muscle-specific Promoters
To confirm that FACT was required for the histone depletion observed at muscle-specific promoters, we asked whether SSRP1 was required. Stable cell lines expressing shRNA constructs against SSRP1 were assayed for expression of SSRP1 by RNA and protein expression. We found that one of the shRNA constructs showed a high level of knockdown (86%), but this cell line could not be propagated (data not shown). This result was not entirely surprising, as FACT is known to be essential (47). However, we were also able to identify two shRNA constructs that resulted in significant knockdown (∼70%) while allowing cell viability. Data from the two shRNA constructs gave equivalent results, so data from only one shRNA construct are shown. We confirmed the down-regulation of SSRP1 by RNA (Fig. 8A) and protein (B) analysis. Cell lines expressing these shRNA constructs were assayed for histone occupancy. A ChIP analysis for histones H2A and H2B in cell lines differentiated for 2 days that expressed a scrambled shRNA control or an shRNA construct against SSRP1 showed that these histones, which are normally depleted upon gene activation, were not depleted at promoter regions when SSRP1 levels were reduced (Fig. 8C). Thus, these data indicate that the presence of SSRP1is required for the depletion of histones at muscle-specific promoters.
FIGURE 8.
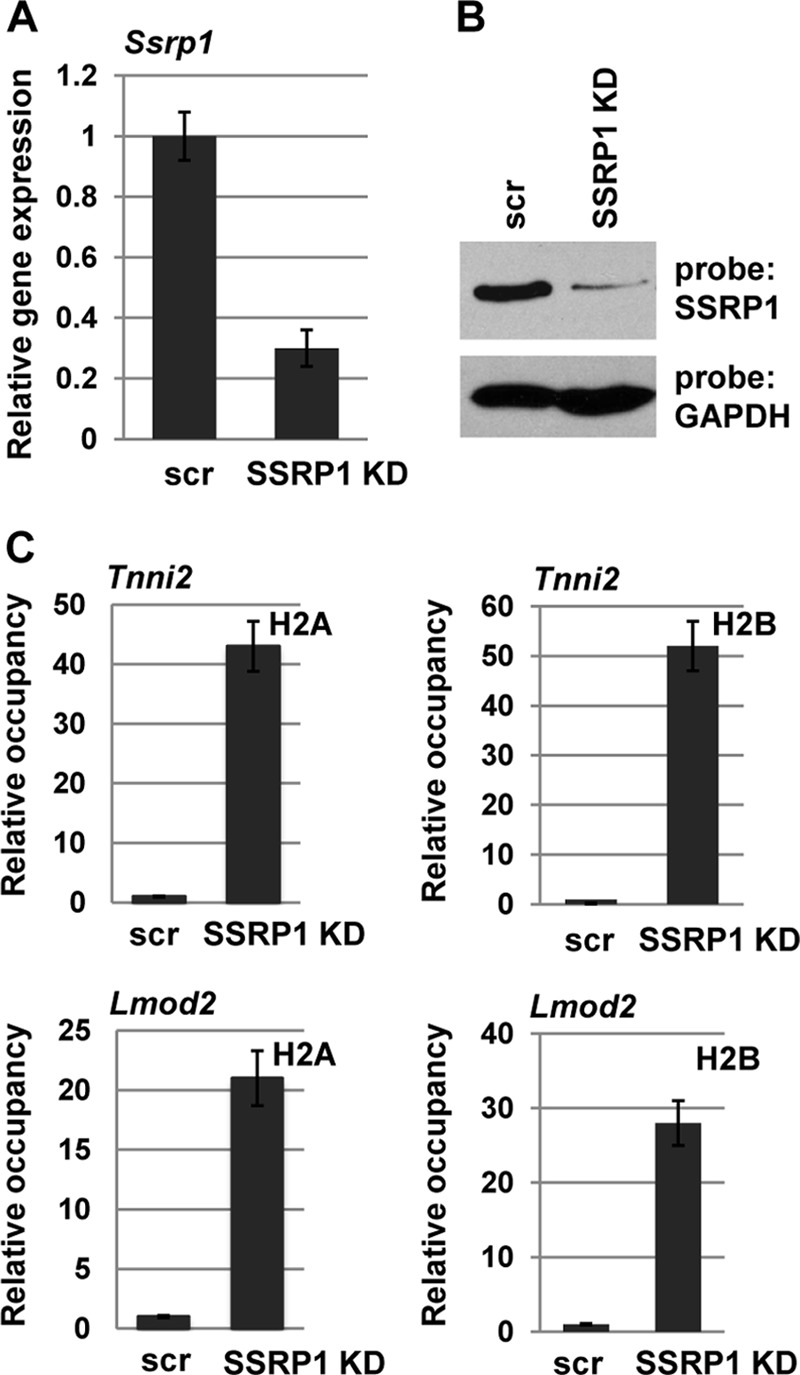
SSRP1 is required for histone depletion at muscle-specific promoters. Stable cell lines expressing shRNA constructs were used to deplete SSRP1. Depletion was confirmed by analysis of the level of RNA by quantitative real-time PCR (A) and protein by Western blotting (B). C, histone levels were assayed by ChIP assays on C2C12 cells transfected with scrambled shRNA constructs (scr) or shRNA constructs against SSRP1 that were differentiated for 2 days. Antibodies against H2A and H2B and primers against the promoter regions of Tnni2 and Lmod2 were used. Data are shown as relative occupancy with the fold enrichment (normalized to IgH) set at 1 for the scr sample.
SSRP1 Is Required for Activation of Muscle-specific Genes
Finally, we asked whether FACT promotes differentiation. We first examined the effect of SSRP1 on myogenin-dependent activation of muscle-specific genes. Using the cell lines expressing shRNA constructs against SSRP1, we assayed for gene expression of muscle-specific genes. Gene expression was assayed in cells expressing scrambled controls (scr shRNA) and shRNA constructs against SSRP1 after 4 days of differentiation. The two cell lines that showed 70% reductions in SSRP1 expression, as described above, were assayed individually, and both produced equivalent results. Data for the same clone as shown in Fig. 8 are shown here. We found that expression of Lmod2 and Tnni2 was greatly reduced in cells depleted for SSRP1 (Fig. 9A). We also assayed for expression of Acta1, which had shown enhanced activation with FACT in the 10T1/2 system. Here, we saw that Acta1 was also down-regulated in C2C12 cells when SSRP1 was depleted (Fig. 9A). We also examined myotube formation in cells depleted for SSRP1. Myotube formation was greatly reduced in cells depleted for SSRP1, as detected by immunostaining for myosin heavy chain-positive cells (Fig. 9B). The fusion index is shown in Fig. 9C. Thus, we show that FACT is required for both the histone depletion at promoters and the appropriate expression of muscle-specific genes to promote differentiation.
FIGURE 9.

SSRP1 promotes differentiation. A, SSRP1 is required for activation of myogenin-dependent muscle-specific genes. Gene expression for muscle specific genes was assayed by quantitative PCR in differentiated (D4) C2C12 cells transfected with scrambled shRNA constructs (scr) or shRNA constructs against SSRP1. Shown are data for Tnni2, Lmod2, and Acta1. All data were normalized to Hprt1. B, SSRP1 is required for myotube formation and MyHC expression. Stable cell lines expressing shRNA constructs targeting SSRP1 or a scrambled control (scr) were differentiated for 4 days and stained with antibodies against myosin heavy chain (MyHC) and DAPI. Fluorescent images are ×100 magnification. Scale bars = 50 μm. C, the fusion index for day 4 myotubes was calculated from the ratio of the number of nuclei in one field for ten random fields.
DISCUSSION
We have found that myogenin interacts with the FACT complex. The interaction with myogenin recruits FACT to muscle-specific genes bound by myogenin, where FACT promotes nucleosome disassembly at promoters, thus promoting gene expression. Our data suggest that FACT plays a role in removing nucleosomes from muscle-specific promoter regions to allow the general transcription machinery to access the DNA sequence. A role for FACT in promoting chromatin disassembly from promoters has been shown for the PHO5 promoter in yeast, but our work is the first to show this in mammalian cells for muscle-specific genes. The activity of histone disassembly at promoters is distinct from the well established role of FACT in chromatin reassembly behind an elongating RNAPII. A promoter-specific role for FACT in activating gene expression has been suggested by studies that showed that FACT is required for TBP recruitment to the GAL1 and HO promoters (31, 48). These dual processes link the process of initiation and elongation (31).
We show here the novel finding that FACT is required for efficient promoter activation of mammalian muscle differentiation-specific genes. Given this role, it was initially surprising that FACT was down-regulated as these genes were activated. However, we show that the role of FACT appears to be in the initial stages of promoter disassembly and activation. Once nucleosomes are removed from the promoter and the genes are active, we hypothesize that FACT is no longer needed to maintain gene expression at these promoters. The observed down-regulation of SPT16 and, to a lesser extent, SSRP1, is consistent with prior data showing that FACT is expressed in proliferating cells and down-regulated in differentiated cells (44). However, we have also observed that SPT16 is not down-regulated as rapidly in primary myoblasts as in C2C12 cells. This result again strengthens the importance of the role we describe here for FACT in establishing the differentiation program in skeletal muscle.
The specific interaction of FACT with myogenin and not MyoD is intriguing, as it suggests that the recruitment of FACT may be one mechanism that myogenin uses to initiate gene expression. However, it is clear from our data that the presence of both MyoD and myogenin leads to the highest activation in the presence of FACT. MyoD is a more robust transactivator than myogenin and has been shown to be bound to many of the promoters occupied by myogenin (39, 40, 49, 50). Thus, it has been difficult to understand the unique role of myogenin at these genes. Clearly, many genes are dependent on the presence of myogenin in vivo, as the null mutation of myogenin results in lethality (7, 8).
It has been shown that MyoD can activate genes in a repressed chromatin environment, whereas myogenin cannot (10). This suggests that the interaction with FACT is not sufficient to enable myogenin to activate genes in a highly repressed chromatin environment. However, our data clearly show that the interaction with FACT is required for myogenin to activate target genes from an inactive state. This suggests that MyoD and myogenin cooperate to activate specific genes by independently recruiting distinct chromatin modification and remodeling complexes.
Our data show that the interaction with myogenin recruits FACT to muscle-specific promoters, but we also show that FACT travels with the elongating RNAPII at these genes, consistent with the well established role of FACT in promoting elongation by reassembling nucleosomes in the wake of RNAPII. It has been shown recently that recruitment of the SSRP1 subunit of FACT coincides with expression in both myoblasts and myotubes (45). In this study, robust association of SSRP1 was observed at Acta1, Trim63, Myog, and Myh3, genes up-regulated in myotubes that have all been shown to be regulated by myogenin. SSRP1 was also associated with genes expressed in myotubes that are not targets of myogenin, and the basis of this recruitment is not currently understood.
In summary, we have shown here that FACT contributes to the initiation of transcription at muscle-specific genes. The works reveals a unique function of myogenin in recruiting the FACT complex to promote nucleosome disassembly and gene expression. Recent work has shown that BAF60c, a subunit of the SWI/SNF chromatin remodeling complex, facilitates MyoD binding to target genes (28). BAF60c phosphorylation, mediated by p38α/β, then recruits SWI/SNF to remodel chromatin and promote the binding of the MRFs to target genes (13). Our work suggests that the binding of myogenin then recruits the FACT complex, which acts to disrupt nucleosomes at the promoters to, presumably, promote recruitment of the general transcription machinery. The stepwise contribution of each of these factors remains to be determined, but it is clear that multiple chromatin modifying complexes are required to establish appropriate transcriptional control at muscle-specific genes. Future studies will be required to understand the precise order of events required to initiate gene expression at target genes and to establish the precise role of FACT in initiating transcription at myogenin-dependent genes.
Acknowledgments
We thank Takanori Kihara, Osaka University, and Masayuki Murata, University of Tokyo for the pCMV-4-SPT16 expression construct. We also thank Rhonda Bassel-Duby and Eric Olson (University of Texas Southwestern) for providing the TRE reporter and MEF2C expression plasmid, Shuang Zhang for cloning the N-TAP myogenin construct, Shuang Zhang and Meiling Zhang for construction and characterization of the stable N-TAP myogenin cell lines used for the TAP purification, Bo Zhu for assistance with the immunostaining protocol, Brian Heine for cloning of the myogenin truncation constructs, and the University of Arkansas for Medical Science Proteomics Facility for mass spectrometric support.
This work was supported, in whole or in part, by National Institute of Health Grants R01DA025755, F32GM093614, UL1RR029884, P30GM103450, and P20GM103429.
- MRF
- myogenic regulatory factor
- RNAPII
- RNA polymerase II
- TAP
- tandem affinity purification.
REFERENCES
- 1. Berkes C. A., Tapscott S. J. (2005) MyoD and the transcriptional control of myogenesis. Semin. Cell Dev. Biol. 16, 585–595 [DOI] [PubMed] [Google Scholar]
- 2. Blais A., Tsikitis M., Acosta-Alvear D., Sharan R., Kluger Y., Dynlacht B. D. (2005) An initial blueprint for myogenic differentiation. Genes Dev. 19, 553–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Molkentin J. D., Black B. L., Martin J. F., Olson E. N. (1995) Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell 83, 1125–1136 [DOI] [PubMed] [Google Scholar]
- 4. Wang D. Z., Valdez M. R., McAnally J., Richardson J., Olson E. N. (2001) The Mef2c gene is a direct transcriptional target of myogenic bHLH and MEF2 proteins during skeletal muscle development. Development 128, 4623–4633 [DOI] [PubMed] [Google Scholar]
- 5. Rudnicki M. A., Schnegelsberg P. N., Stead R. H., Braun T., Arnold H. H., Jaenisch R. (1993) MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 75, 1351–1359 [DOI] [PubMed] [Google Scholar]
- 6. Kassar-Duchossoy L., Gayraud-Morel B., Gomès D., Rocancourt D., Buckingham M., Shinin V., Tajbakhsh S. (2004) Mrf4 determines skeletal muscle identity in Myf5:Myod double-mutant mice. Nature 431, 466–471 [DOI] [PubMed] [Google Scholar]
- 7. Hasty P., Bradley A., Morris J. H., Edmondson D. G., Venuti J. M., Olson E. N., Klein W. H. (1993) Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364, 501–506 [DOI] [PubMed] [Google Scholar]
- 8. Nabeshima Y., Hanaoka K., Hayasaka M., Esumi E., Li S., Nonaka I., Nabeshima Y. (1993) Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 364, 532–535 [DOI] [PubMed] [Google Scholar]
- 9. Venuti J. M., Morris J. H., Vivian J. L., Olson E. N., Klein W. H. (1995) Myogenin is required for late but not early aspects of myogenesis during mouse development. J. Cell Biol. 128, 563–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gerber A. N., Klesert T. R., Bergstrom D. A., Tapscott S. J. (1997) Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin. A mechanism for lineage determination in myogenesis. Genes Dev. 11, 436–450 [DOI] [PubMed] [Google Scholar]
- 11. Potthoff M. J., Olson E. N. (2007) MEF2. A central regulator of diverse developmental programs. Development 134, 4131–4140 [DOI] [PubMed] [Google Scholar]
- 12. de la Serna I. L., Carlson K. A., Imbalzano A. N. (2001) Mammalian SWI/SNF complexes promote MyoD-mediated muscle differentiation. Nat. Gen. 27, 187–190 [DOI] [PubMed] [Google Scholar]
- 13. de la Serna I. L., Ohkawa Y., Berkes C. A., Bergstrom D. A., Dacwag C. S., Tapscott S. J., Imbalzano A. N. (2005) MyoD targets chromatin remodeling complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol. Cell. Biol. 25, 3997–4009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simone C., Forcales S. V., Hill D. A., Imbalzano A. N., Latella L., Puri P. L. (2004) p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat. Gen. 36, 738–743 [DOI] [PubMed] [Google Scholar]
- 15. Ohkawa Y., Marfella C. G., Imbalzano A. N. (2006) Skeletal muscle specification by myogenin and Mef2D via the SWI/SNF ATPase Brg1. EMBO J. 25, 490–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ohkawa Y., Yoshimura S., Higashi C., Marfella C. G., Dacwag C. S., Tachibana T., Imbalzano A. N. (2007) Myogenin and the SWI/SNF ATPase Brg1 maintain myogenic gene expression at different stages of skeletal myogenesis. J. Biol. Chem. 282, 6564–6570 [DOI] [PubMed] [Google Scholar]
- 17. Orphanides G., LeRoy G., Chang C. H., Luse D. S., Reinberg D. (1998) FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92, 105–116 [DOI] [PubMed] [Google Scholar]
- 18. Orphanides G., Wu W. H., Lane W. S., Hampsey M., Reinberg D. (1999) The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400, 284–288 [DOI] [PubMed] [Google Scholar]
- 19. Wittmeyer J., Joss L., Formosa T. (1999) Spt16 and Pob3 of Saccharomyces cerevisiae form an essential, abundant heterodimer that is nuclear, chromatin-associated, and copurifies with DNA polymerase alpha. Biochemistry 38, 8961–8971 [DOI] [PubMed] [Google Scholar]
- 20. Wittmeyer J., Formosa T. (1997) The Saccharomyces cerevisiae DNA polymerase α catalytic subunit interacts with Cdc68/Spt16 and with Pob3, a protein similar to an HMG1-like protein. Mol. Cell. Biol. 17, 4178–4190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bruhn S. L., Pil P. M., Essigmann J. M., Housman D. E., Lippard S. J. (1992) Isolation and characterization of human cDNA clones encoding a high mobility group box protein that recognizes structural distortions to DNA caused by binding of the anticancer agent cisplatin. Proc. Natl. Acad. Sci. U.S.A. 89, 2307–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yarnell A. T., Oh S., Reinberg D., Lippard S. J. (2001) Interaction of FACT, SSRP1, and the high mobility group (HMG) domain of SSRP1 with DNA damaged by the anticancer drug cisplatin. J. Biol. Chem. 276, 25736–25741 [DOI] [PubMed] [Google Scholar]
- 23. Winkler D. D., Muthurajan U. M., Hieb A. R., Luger K. (2011) Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J. Biol. Chem. 286, 41883–41892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winkler D. D., Luger K. (2011) The histone chaperone FACT. Structural insights and mechanisms for nucleosome reorganization. J. Biol. Chem. 286, 18369–18374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Formosa T. (2008) FACT and the reorganized nucleosome. Mol. Biosyst. 4, 1085–1093 [DOI] [PubMed] [Google Scholar]
- 26. Belotserkovskaya R., Oh S., Bondarenko V. A., Orphanides G., Studitsky V. M., Reinberg D. (2003) FACT facilitates transcription-dependent nucleosome alteration. Science 301, 1090–1093 [DOI] [PubMed] [Google Scholar]
- 27. Xin H., Takahata S., Blanksma M., McCullough L., Stillman D. J., Formosa T. (2009) yFACT induces global accessibility of nucleosomal DNA without H2A-H2B displacement. Mol. Cell 35, 365–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Forcales S. V., Albini S., Giordani L., Malecova B., Cignolo L., Chernov A., Coutinho P., Saccone V., Consalvi S., Williams R., Wang K., Wu Z., Baranovskaya S., Miller A., Dilworth F. J., Puri P. L. (2012) Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J. 31, 301–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ransom M., Williams S. K., Dechassa M. L., Das C., Linger J., Adkins M., Liu C., Bartholomew B., Tyler J. K. (2009) FACT and the proteasome promote promoter chromatin disassembly and transcriptional initiation. J. Biol. Chem. 284, 23461–23471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Biswas D., Yu Y., Prall M., Formosa T., Stillman D. J. (2005) The yeast FACT complex has a role in transcriptional initiation. Mol. Cell. Biol. 25, 5812–5822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mason P. B., Struhl K. (2003) The FACT complex travels with elongating RNA polymerase II and is important for the fidelity of transcriptional initiation in vivo. Mol. Cell. Biol. 23, 8323–8333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kihara T., Kano F., Murata M. (2008) Modulation of SRF-dependent gene expression by association of SPT16 with MKL1. Exp. Cell Res. 314, 629–637 [DOI] [PubMed] [Google Scholar]
- 33. Spencer J. A., Baron M. H., Olson E. N. (1999) Cooperative transcriptional activation by serum response factor and the high mobility group protein SSRP1. J. Biol. Chem. 274, 15686–15693 [DOI] [PubMed] [Google Scholar]
- 34. Pollard J. W., Walker J. M. (1997) Basic Cell Culture Protocols, 2nd Ed., Humana Press, Totawa, NJ [Google Scholar]
- 35. Londhe P., Davie J. K. (2011) γ Interferon modulates myogenesis through the major histocompatibility complex class II transactivator, CIITA. Mol. Cell. Biol. 31, 2854–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Byrum S., Smart S. K., Larson S., Tackett A. J. (2012) Analysis of stable and transient protein-protein interactions. Methods Mol. Biol. 833, 143–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Byrum S. D., Raman A., Taverna S. D., Tackett A. J. (2012) ChAP-MS. A method for identification of proteins and histone posttranslational modifications at a single genomic locus. Cell Reports 2, 198–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Byrum S., Avaritt N. L., Mackintosh S. G., Munkberg J. M., Badgwell B. D., Cheung W. L., Tackett A. J. (2011) A quantitative proteomic analysis of FFPE melanoma. J. Cutan. Pathol. 38, 933–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Davie J. K., Cho J. H., Meadows E., Flynn J. M., Knapp J. R., Klein W. H. (2007) Target gene selectivity of the myogenic basic helix-loop-helix transcription factor myogenin in embryonic muscle. Dev. Biol. 311, 650–664 [DOI] [PubMed] [Google Scholar]
- 40. Londhe P., Davie J. K. (2011) Sequential association of myogenic regulatory factors and E proteins at muscle-specific genes. Skelet. Muscle 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang Z., MacQuarrie K. L., Analau E., Tyler A. E., Dilworth F. J., Cao Y., Diede S. J., Tapscott S. J. (2009) MyoD and E-protein heterodimers switch rhabdomyosarcoma cells from an arrested myoblast phase to a differentiated state. Genes Dev. 23, 694–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bergstrom D. A., Tapscott S. J. (2001) Molecular distinction between specification and differentiation in the myogenic basic helix-loop-helix transcription factor family. Mol. Cell. Biol. 21, 2404–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ishibashi J., Perry R. L., Asakura A., Rudnicki M. A. (2005) MyoD induces myogenic differentiation through cooperation of its NH2- and COOH-terminal regions. J. Cell Biol. 171, 471–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garcia H., Fleyshman D., Kolesnikova K., Safina A., Commane M., Paszkiewicz G., Omelian A., Morrison C., Gurova K. (2011) Expression of FACT in mammalian tissues suggests its role in maintaining of undifferentiated state of cells. Oncotarget 2, 783–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vethantham V., Yang Y., Bowman C., Asp P., Lee J. H., Skalnik D. G., Dynlacht B. D. (2012) Dynamic loss of H2B ubiquitylation without corresponding changes in H3K4 trimethylation during myogenic differentiation. Mol. Cell. Biol. 32, 1044–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Buratowski S. (2009) Progression through the RNA polymerase II CTD cycle. Mol. Cell 36, 541–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cao S., Bendall H., Hicks G. G., Nashabi A., Sakano H., Shinkai Y., Gariglio M., Oltz E. M., Ruley H. E. (2003) The high-mobility-group box protein SSRP1/T160 is essential for cell viability in day 3.5 mouse embryos. Mol. Cell. Biol. 23, 5301–5307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Biswas D., Dutta-Biswas R., Mitra D., Shibata Y., Strahl B. D., Formosa T., Stillman D. J. (2006) Opposing roles for Set2 and yFACT in regulating TBP binding at promoters. EMBO J. 25, 4479–4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cao Y., Yao Z., Sarkar D., Lawrence M., Sanchez G. J., Parker M. H., MacQuarrie K. L., Davison J., Morgan M. T., Ruzzo W. L., Gentleman R. C., Tapscott S. J. (2010) Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev. Cell 18, 662–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tai P. W., Fisher-Aylor K. I., Himeda C. L., Smith C. L., Mackenzie A. P., Helterline D. L., Angello J. C., Welikson R. E., Wold B. J., Hauschka S. D. (2011) Differentiation and fiber type-specific activity of a muscle creatine kinase intronic enhancer. Skelet. Muscle 1, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]