

Background: Fucoidan was tested as a novel drug for hemophilia, but its effect on platelets has not been studied.
Results: We show that fucoidan activates human and mouse platelets and that activation is blocked in CLEC-2 knock-out murine platelets.
Conclusion: Fucoidan is a novel agonist for the CLEC-2.
Significance: Understanding the effects of fucoidan on platelets can help in designing efficient drugs for hemophilia.
Keywords: Phosphotyrosine Receptor, Phosphotyrosine Signaling, Platelets, Protein Phosphorylation, Receptor Tyrosine Kinase, CLEC-2 Receptor, Fucoidan
Abstract
Fucoidan, a sulfated polysaccharide from Fucus vesiculosus, decreases bleeding time and clotting time in hemophilia, possibly through inhibition of tissue factor pathway inhibitor. However, its effect on platelets and the receptor by which fucoidan induces cellular processes has not been elucidated. In this study, we demonstrate that fucoidan induces platelet activation in a concentration-dependent manner. Fucoidan-induced platelet activation was completely abolished by the pan-Src family kinase (SFK) inhibitor, PP2, or when Syk is inhibited. PP2 abolished phosphorylations of Syk and Phospholipase C-γ2. Fucoidan-induced platelet activation had a lag phase, which is reminiscent of platelet activation by collagen and CLEC-2 receptor agonists. Platelet activation by fucoidan was only slightly inhibited in FcRγ-chain null mice, indicating that fucoidan was not acting primarily through GPVI receptor. On the other hand, fucoidan-induced platelet activation was inhibited in platelet-specific CLEC-2 knock-out murine platelets revealing CLEC-2 as a physiological target of fucoidan. Thus, our data show fucoidan as a novel CLEC-2 receptor agonist that activates platelets through a SFK-dependent signaling pathway. Furthermore, the efficacy of fucoidan in hemophilia raises the possibility that decreased bleeding times could be achieved through activation of platelets.
Introduction
Platelets act as essential mediators in maintaining homeostasis of the circulatory system, and their activation is tightly regulated in vivo under normal physiological conditions. However, when there is vascular damage, exposure of subendothelial collagen facilitates platelet activation, mainly through the glycoprotein VI receptor (GPVI),3 which is essential for hemostasis (1–3). Failure of platelet activation leads to excessive bleeding. Subsequent to platelet activation, coagulation proteins are activated resulting in thrombin generation and fibrin clot formation (5). Deficiencies in coagulation proteins also lead to bleeding diathesis. For example, hemophilia A is a bleeding disorder in which the patient has an increased bleeding tendency due to factor VIII absence or dysfunction (6, 7).
Sulfated polysaccharides of natural origin, which are characterized by their biological activities, have a favorable therapeutic profile in animals and humans because most have minimal side effects. These polysaccharides are composed of a broad range of subclasses similar to heparins, glycoaminoglycans, fucoidan, dermatan, or dextran sulfates (7). Notably, compounds of natural origin exhibit different biological activities depending on the concentration or dose (8, 9). Fucoidan, a nonanticoagulatory sulfated polysaccharide, is derived from the brown seaweed Fucus vesiculosus. When compared with other sulfated polysaccharides, fucoidan demonstrates superior procoagulant properties (7, 10, 11). Pharmacological activity of this compound was tested in several animal models in vitro and in vivo. In humans, in vitro experiments with fucoidan demonstrated several beneficial effects on ulcers and bleeding disorders (11, 12). Non-anticoagulatory sulfated polysaccharides were originally considered as a novel approach to improve coagulation because of their inhibitory effect on physiological anticoagulation, especially fucoidan, which inhibits tissue factor pathway inhibitor and accelerates clotting in hemophilia A and B patients (8). Therefore, fucoidan is in clinical trials as a new candidate drug for hemophilia patients. In prior studies, fucoidan was shown to enhance the survival rate in murine models of hemophilia A and B (8) and a canine model of hemophilia A (9) by decreasing bleeding times. However, the effect of fucoidan on platelet aggregation, which regulates bleeding times, has not been studied.
GPVI signaling in platelets depends on FcRγ chain activation leading to tyrosine kinase pathways resulting in the activation of Syk and PLCγ2 (10). C-type lectin-like receptor 2 (CLEC-2), also linked to activation of Syk and PLCγ2 (11), is highly expressed in platelets and at lower levels in neutrophils and dendritic cells (12, 13). CLEC-2 was identified by using the snake venom protein rhodocytin, purified from Calloselasma rhodostoma (11), as a ligand on affinity chromatography, and later shown to be an endogenous receptor for podoplanin. The crystal structure of rhodocytin shows that CLEC-2 receptors are activated through clustering by this tetrameric ligand (14–16). CLEC-2 and podoplanin are implicated in tumor metastasis (17, 18) and hemostasis and thrombosis (19). Unlike GPVI, CLEC-2 has an immunoreceptor tyrosine-based activation motif (hemITAM) sequence that is phosphorylated by Src and Syk tyrosine kinases, whereas phosphorylation of the ITAM is mediated solely by Src kinases (20). Rhodocytin and podoplanin are the only two known agonists for the CLEC-2 receptor (17, 21, 22).
In the present study, we show that fucoidan causes platelet activation, leading to αIIbβ3-mediated aggregation. We show that fucoidan signals through a tyrosine kinase-dependent signaling pathway downstream of CLEC-2 receptor and propose that this contributes to a decrease in bleeding time in hemophilia animal models. These results support the notion that decreased bleeding times can be achieved in hemophilia patients through activation of platelets.
EXPERIMENTAL PROCEDURES
All animal experiments were performed in compliance with the Institutional Animal Care and Use Committee at Temple University (Philadelphia, PA).
Mice
Platelet specific CLEC-2 knock-out mice were produced as described previously (50). Briefly, the CLEC-2 gene (CLEC1b) was flanked with LoxP sites, and this mouse was cross-bred with a PF4-Cre expressing mouse to drive excision of the gene in only the megakaryocyte/platelet lineage.
Materials
Apyrase (type VII), acetylsalicylic acid, fucoidan (80% pure, with the major contaminant being fucose), and indomethacin were obtained from Sigma. Convulxin was purified as described (23). YM-254890 was a gift from Yamanochi Pharmaceutical Co., Ltd (Ibaraki, Japan). FURA-2 AM was from Molecular Probes (Eugene, OR). PP3 and PP2 were purchased from Biomol Research Laboratories (Plymouth Meeting, PA). OXSI-2 was from Calbiochem (Darmstadt, Germany). Whatman protein nitrocellulose transfer membrane was obtained from Fisher Scientific (Pittsburgh, PA), LI-COR Odyssey blocking buffer was purchased from LI-COR Biosciences (Lincoln, NE). Antibodies to Syk (Tyr525/526), PLCγ2 (Tyr759), Lat (Tyr191), and β-actin were bought from Cell Signaling Technology (Beverly, MA), Total Syk and Lat antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal mouse anti-CLEC-2 antibody was obtained from Santa Cruz Biotechnology. FITC-labeled PAC-1 and mouse IgG isotype control antibodies were obtained from BD Technologies (Franklin Lakes, NJ). P-selectin-PE antibodies were purchased from BD Biosciences.
Preparation of Human Platelets
Blood was collected from informed healthy volunteers in to one-sixth volume of acid/citrate/dextrose (2.5 g of sodium citrate, 2 g of glucose, and 1.5 g of citric acid in 100 ml deionized water). Platelet-rich plasma was obtained by centrifugation at 250 × g for 20 min at ambient temperature and incubated with 1 mm aspirin for 30 min at 37 °C. Platelets were isolated from plasma by centrifugation at 980 × g for 10 min at ambient temperature and resuspended in Tyrode's buffer, pH 6.5 (138 mm NaCl, 2.7 mm KCl, 2 mm MgCl2, 0.42 mm NaH2PO4, 5 mm glucose, 10 mm PIPES (pH 6.5) containing 20 nm PGE1, 10 μm indomethacin, 500 μm EGTA, and 0.2 units/ml apyrase,). Platelets were isolated from Tyrode's buffer, pH 6.5, by centrifugation at 980 × g for 10 min and resuspended in Tyrode's buffer, pH 7.4 (138 mm NaCl, 2.7 mm KCl, 2 mm MgCl2, 0.42 mm NaH2PO4, 5 mm glucose, 10 mm HEPES, and 0.2 units/ml apyrase, pH 7.4). The platelet count was adjusted to 2–2.5 × 108/ml. Approval was obtained from the institutional review board of Temple University for these studies. Informed consent was provided prior to blood donation.
Preparation of Murine Platelets
All mice were maintained in a specific pathogen-free facility, and animal procedures were carried out in accordance with institutional guidelines after the Temple University Animal Care and Use Committee approved the study protocol, or they were performed under UK Home Office Project license (PPL 40/2803). Fccer1g (FcRγ knock-out) or γ-chain-deficient mice were purchased from Taconic (Germantown, NY). Age- and gender-matched wild-type mice were used as controls. Blood was drawn via cardiac puncture into one-tenth volume of 3.8% sodium citrate. Blood was spun at 100 × g for 10 min, and the platelet rich plasma (PRP) was separated. Red blood cells were mixed with 400 μl of 3.8% sodium citrate and spun for a further 10 min at 100 × g. Resulting platelet rich plasmas (PRPs) were combined, and 1 μm PGE1 added and centrifuged for 10 min at 400 × g. Platelet-poor plasma was removed, and the platelet pellet was resuspended in Tyrode's buffer (pH 7.4) containing 0.2 units/ml apyrase. Platelet counts were determined using a Hemavet 950FS blood cell counter (Drew Scientific Inc., Dallas, TX). For aggregation studies, a density of 2 × 108 platelets/ml was used.
Platelet Aggregation
Platelet aggregation was measured using a lumi-aggregometer (Chrono-Log, Havertown, PA) at 37 °C under stirring conditions. A 0.5-ml sample of aspirin-treated washed platelets was stimulated with different agonists, and change in light transmission was measured. Platelets were preincubated with different inhibitors where noted before agonist stimulation. The chart recorder was set for 0.2 mm/s.
Measurement of Intracellular Ca2+ Mobilization
Platelet-rich plasma was incubated with 5 μm Fura-2/AM and 1 mm aspirin for 45 min at 37 °C. Platelets were prepared as described above, and fluorescence was measured in a Perkin-Elmer apparatus with excitation set at 340 nm and emission set at 510 nm. Fluorescence measurements were converted to Ca2+ concentrations using the equation as reported (24).
Flow Cytometry
Washed and aspirin-treated human platelets were used to measure agonist-dependent levels of αIIβb3-activated receptor by PAC-1-FITC antibody and CD62P-PE antibody. Aliquots (0.5 ml) of washed platelet suspension in Tyrode's buffer (pH 7.4) containing 1 mm CaCl2 were preincubated with the indicated concentration of inhibitors for 5 min at 37 °C. Prior to the activation with fucoidan, an aliquot containing 106 platelets was gently mixed with 40 μl of antibody mixture and incubated for 20 min at 37 °C in the dark. Platelets were identified and gated according to the forward and side scatter signals. As control for immunolabeling, platelets were incubated with non-immuno-IgG isotype control antibody. To fix the platelets, 1% parafarmaldehyde dissolved in phosphate-buffered saline was added. A total of 10,000 platelet events were acquired per sample, and the percentage of positive gated cells was analyzed. All determinations were performed on a FACS Caliber flow cytometer (BD Biosciences).
Immunoprecipitation
Washed human platelets (1 × 109/ml) were stimulated with rhodocytin (50 nm) or fucoidan (50 μg/ml). Reactions were terminated by adding equal volume of 2× Nonidet P-40 lysis buffer (20 mm Tris-HCl, pH 7.6, 300 mm NaCl, 2 mm EGTA, 2 mm EDTA, 2% Nonidet P-40, 2 mm PMSF, 5 mm Na3VO4, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 μg/ml pepstatin). Cell debris was removed by centrifugation at 15,000 × g for 10 min. Antibodies against CLEC-2 (5 μg/ml) or control IgG were added to the resultant supernatant and incubated overnight with protein A Sepharose. Precipitated proteins were separated by SDS-PAGE and Western blotted with the 4G10 antibody.
Western Blot Analysis
Platelets were stimulated with agonists in the presence of inhibitors or vehicles for the appropriate time under stirring conditions at 37 °C, and the reaction was stopped by the addition of 0.6 n HClO4. The resulting acid precipitate was collected and kept on ice. The samples were centrifuged at 13,000 × g for 4 min followed by resuspending in 0.5 ml of deionized water. The protein was again pelleted by centrifugation at 13,000 × g for 4 min. The protein pellets were solubilized in sample buffer containing 0.1 m Tris, 2% SDS, 1% (v/v) glycerol, 0.1% bromphenol blue, and 100 mm DTT and then boiled for 10 min. Proteins were resolved by SDS-polyacrylamide gels and transferred to nitrocellulose membrane (Whatman Protran). Membranes were blocked with Odyssey blocking buffer for 1 h at ambient temperature, incubated overnight at 4 °C with the desired primary antibody, and then washed four times with TBS-T. Membranes were then incubated with appropriate secondary infrared dye-labeled antibody for 60 min at room temperature and washed four times with TBS-T. Membranes were examined with a Li-Cor Odyssey infrared imaging system.
Statistical Analysis
Each experiment was repeated at least three times. Results are expressed as means ± S.E. with number of observations (n). Data were analyzed using KaleidaGraph software. Significant differences were determined using Student's t test. Differences were considered significant at p ≤ 0.05.
RESULTS
Fucoidan Causes Platelet Activation
As fucoidan reduced bleeding times in hemophilia animal models (8, 9), we evaluated whether fucoidan can cause platelet activation. Fucoidan caused aggregation of washed human platelets in a concentration-dependent manner (Fig. 1A). Fucoidan caused only shape change at a concentration of 10 μg/ml, whereas maximum aggregation was achieved at 50 μg/ml (Fig. 1A). We also evaluated the effects of fucoidan on other platelet functional responses, including integrin activation (Fig. 1B), dense granule secretion (Fig. 1C), P-selectin expression (Fig. 1D), and thromboxane generation (Fig. 1E) in human platelets. These results show that fucoidan causes platelet activation.
FIGURE 1.

Fucoidan-induced platelet responses. A, washed aspirin-treated human platelets were stimulated with increasing concentrations of fucoidan at 37 °C under stirring conditions. Platelet aggregation was measured using a Lumi aggregometer. The traces are representative of data from at least three independent experiments. B, fucoidan-induced αIIbβ3 activation was measured by activating washed, aspirin-treated human platelets with fucoidan (50 μg/ml) and analyzed with FITC-PAC1 antibody by flow cytometry. C, dense granule secretion was assessed by measuring fucoidan-induced (50 μg/ml) ATP release from human platelets. D, α granule expression was measured by CD62P (P-selectin) expression in response to fucoidan (50 μg/ml) and was analyzed using flow cytometry. Graphs represent mean ± S.E. of % positive cells from at least three different experiments (***, p ≤ 0.05). E, washed human platelets without aspirin treatment were stimulated in a platelet aggregometer, and levels of TXB2 were determined according to established protocol (4).
Role of Src Family Kinase (SFK)-dependent Pathways in Fucoidan-induced Platelet Activation
Many platelet surface receptors, which mediate positive functional responses, are coupled to either Gq-dependent or tyrosine kinase-dependent pathways (25). To determine the signaling mechanism involved in fucoidan-induced platelet activation, we used the selective Gq inhibitor YM-254890 (26, 27) or the pan SFK inhibitor PP2 (28). Platelets pretreated with YM-254890 had only a minimum reduction in aggregation in comparison with the full inhibition of aggregation to AYPGKF (Fig. 2A), suggesting that the Gq pathway plays a minimal role in the activation of platelets by fucoidan, presumably through the feedback action of TxA2. As shown in Fig. 2B, fucoidan-induced platelet activation was completely abolished by PP2 but not by its inactive analog, PP3. These data demonstrate that SFK-dependent pathways are required for fucoidan-induced platelet activation.
FIGURE 2.
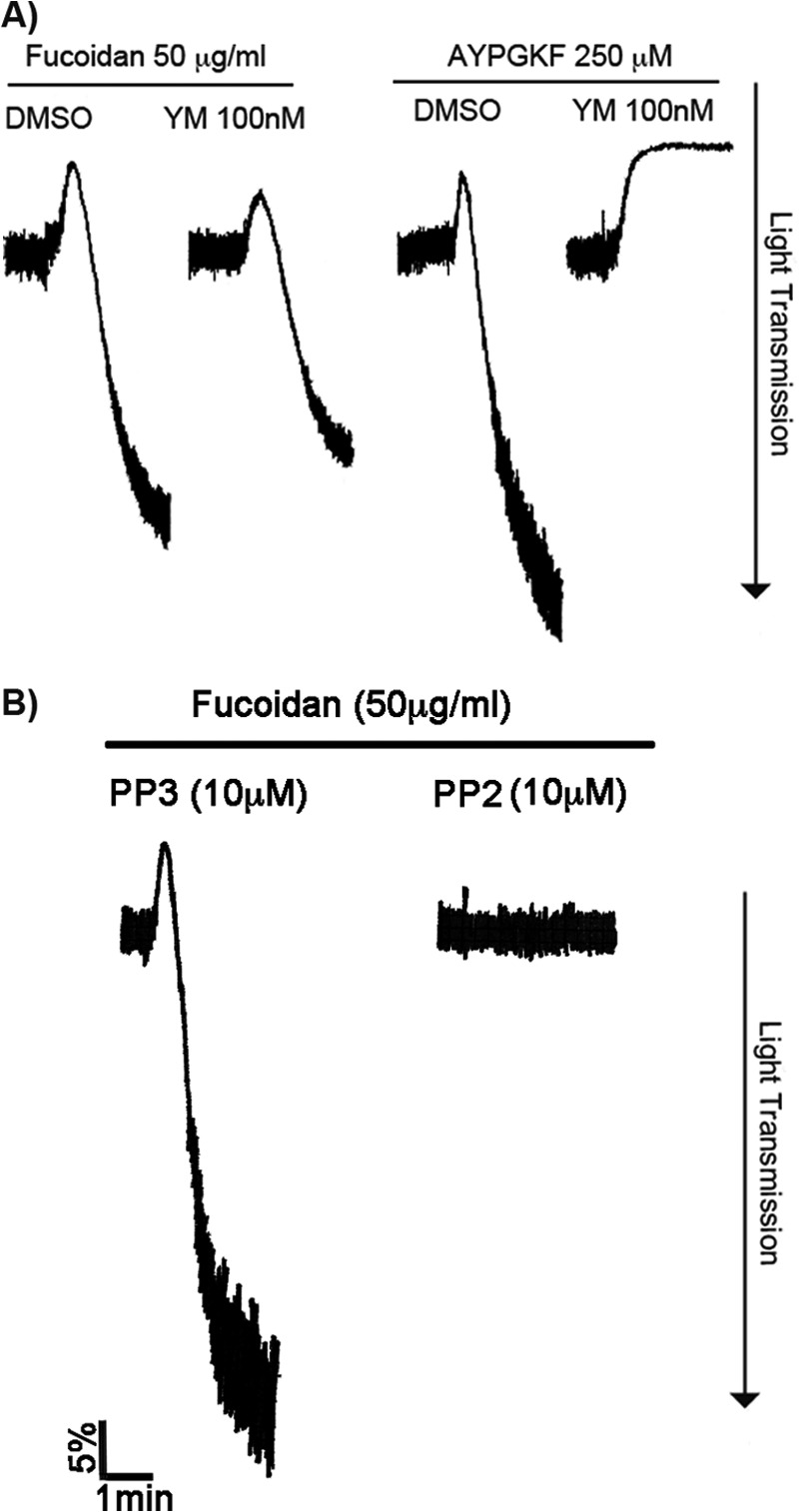
Role of Src family tyrosine kinase (SFK)-dependent pathways in fucoidan-induced platelet activation. A, washed, aspirin-treated human platelets were incubated with YM-254890 (50 nm), a selective Gq inhibitor, for 5 min and stimulated with fucoidan (50 μg/ml) for 3 min at 37 °C under stirred conditions. Platelet aggregation was measured using lumi-aggregometer. The traces are representative of data from at least three independent experiments. B, washed, aspirin-treated human platelets were pretreated with SFK inhibitor PP2 (10 μm) or PP3 (control) at 37 °C for 5 min followed by stimulation with fucoidan (50 μg/ml) for 3 min under stirred conditions. Platelet aggregation was measured by aggregometry. The tracings are representative of data from at least three independent experiments. DMSO, dimethyl sulfoxide.
It is known that SFK-mediated signaling pathways involve activation and phosphorylation of Syk and Lat in platelets (29, 30). Therefore, we evaluated the phosphorylation status of Syk and Lat following stimulation of platelets with fucoidan. As shown in Fig. 3A, Syk and Lat phosphorylations occurred as early as 15 s and dephosphorylated in a time-dependent manner. Fucoidan-induced Syk and Lat phosphorylations were abolished by pretreatment of platelets with PP2 (Fig. 3B). These data support our observation that fucoidan-mediated signaling pathways involve Syk and Lat activation downstream of SFKs. Intracellular Ca2+ mobilization was also measured in response to fucoidan in the presence of the pan SFK inhibitor PP2. PP2 completely blocked calcium mobilization compared with control (PP3) platelets (Fig. 3C). These data suggest that fucoidan activates a SFK-dependent pathway leading to intracellular Ca2+ mobilization.
FIGURE 3.

Effect of SFK inhibition on fucoidan-induced platelet activation. A, fucoidan-induced phosphorylation of Syk and Lat. Washed, aspirin-treated human platelets were stimulated with fucoidan (50 μg/ml) for different time periods. Samples were subjected to SDS-PAGE and analyzed for phosphorylation of Syk and Lat. The results are representative of data from platelets at least three independent experiments. B, washed, aspirin-treated human platelets were stimulated with fucoidan (50 mg/ml) in the presence of the SFK inhibitor PP2 for 30 s, and the effect on Syk (Y525/26) and LAT (Y191) phosphorylation were analyzed. C, washed, aspirin-treated human platelets loaded with 5 μm FURA-2 AM were pretreated with PP3 or PP2 (10 μm) and activated with fucoidan (50 μg/ml) to measure intracellular Ca2+ mobilization.
Syk Mediates Platelet Activation by Fucoidan
The functional role of Syk in signaling by fucoidan was investigated by pretreating platelets with the Syk inhibitors OXSI-2 or Go6976 (31, 32). Fucoidan-induced platelet activation was completely inhibited when treated with the Syk inhibitors, which was measured by percentage PAC1 binding using flow cytometry (Fig. 4A). To determine whether or not OXSI-2 was affecting the activity of SFKs, phosphorylation of Tyr416 (marker of SFK activation) was examined under similar experimental conditions. As demonstrated in Fig. 4B, SFK phosphorylation was not effected when a Syk inhibitor, OXSI-2 or Go6976, was used. The critical role of Syk in signaling by fucoidan was illustrated by the lack of PLCγ2 phosphorylation, which is an important downstream signaling molecule (Fig. 4, C and D). Furthermore, fucoidan-induced intracellular Ca2+ mobilization was completely abolished in the presence of either Syk inhibitor OXSI-2 or Go6976 (Fig. 4E). Inhibition of fucoidan-induced platelet activation by OXSI-2 or Go6976 suggests that Syk is an important downstream signaling molecule in fucoidan-induced platelet activation.
FIGURE 4.

The role of Syk in fucoidan-induced aggregation, calcium mobilization, and αIIbβ3 activation. A, washed, aspirin-treated human platelets were pretreated with vehicle (dimethyl sulfoxide), OXSI-2 (2 μm), or Go 6976 (1 μm) for 5 min at 37 °C prior to activation with convulxin (CVX; 100 ng/ml) or fucoidan (50 μg/ml) (US-unstimulated). αIIbβ3 expression was analyzed with PAC-1-FITC antibody. Graphs are represented mean ± S.E. of % positive cells from three different experiments (***, p < 0.01). B, the effect of Syk inhibitors (OXSI-2 or Go 6976) on SFK phosphorylation induced by fucoidan (50 μg/ml) in human platelets was analyzed. C, time course analysis of PLCγ2 tyrosine phosphorylation from aspirin-treated human platelets stimulated with fucoidan (50 μg/ml). D, platelets were stimulated with 50 μg/ml of fucoidan for 1 min. PLCγ2 tyrosine phosphorylation was measured in the presence of OXSI2, Go6976, or dimethyl sulfoxide. E, washed, aspirin-treated human platelets loaded with 5 μm FURA-2 AM were pretreated with OXSI-2 (2 μm) or Go 6976 (1 μm) and activated with fucoidan (50 μg/ml) to measure the effect of Syk inhibition in Ca2+ mobilization.
Fucoidan Mediates Platelet Functional Responses through CLEC-2
The major receptors that trigger tyrosine kinase pathways in platelets are GPVI and CLEC-2 (33–35). Upon ligation of collagen to GPVI, the FcRγ chain ITAM becomes phosphorylated by SFK members to initiate downstream signaling (36). Data presented in Figs. 2 and 3 suggest that the profile of activation by fucoidan is similar to that of GPVI. Previously, it was shown that FcRγ chain-null platelets are not only unresponsive to collagen but also show no surface expression of GPVI (37). To evaluate whether fucoidan signals through GPVI, we made use of FcRγ chain-null murine platelets. Wild-type murine platelets aggregate in response to collagen-related peptide (10 μg/ml) (Fig. 5A), whereas addition of collagen-related peptide to FcRγ chain-null murine platelets failed to induce platelet aggregation (Fig. 5A). Interestingly, fucoidan-induced aggregation observed in FcRγ chain-null murine platelets was similar when compared with wild-type murine platelets (Fig. 5A). FcγRIIA is not present in murine platelets but does exist in human platelets. We therefore tested the role of this receptor by using the IV.3 monoclonal antibody that binds and blocks FcγRIIA. Blocking of FcγRIIA by IV.3 antibody did not affect fucoidan-induced platelet activation (Fig. 5B). These data suggest that fucoidan does not mediate its effects primarily through the GPVI or FcγRIIA receptor.
FIGURE 5.
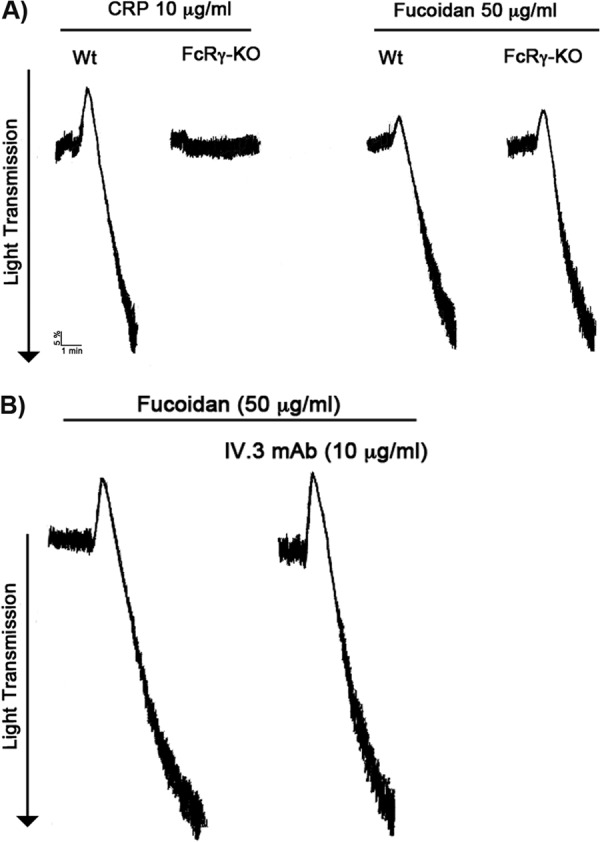
Fucoidan does not activate GPVI or FcγRIIA. A, fucoidan-induced platelet aggregation on FcRγ chain-null murine platelets. Wild-type or FcRγ chain-null murine platelets were stimulated with fucoidan (50 μg/ml) and CRP (Collagen Related Peptide) allowed to aggregate for 3 min at 37 °C under stirred conditions in a Lumi-aggregometer. The traces are representative of data from at least three independent experiments. B, human platelets were stimulated with fucoidan (50 μg/ml) in the absence and presence of IV.3 monoclonal antibody (10 μg/ml) for 3 min. The traces are representative of data from at least three independent experiments.
CLEC-2, a C-type lectin receptor, has been shown to signal independently of the FcRγ chain in platelets (16, 34). The presence of Syk phosphorylation (38) and detectable levels of Lat phosphorylation in platelets activated with fucoidan (Fig. 3) suggested that CLEC-2 (39) may be a possible receptor candidate for fucoidan on platelets. To determine whether CLEC-2 is the receptor that mediates fucoidan-induced platelet activation, platelet-specific (PF4-Cre) CLEC-2 receptor knock-out mice platelets were used. Wild-type murine platelets aggregated in response to fucoidan, but fucoidan failed to induce platelet activation in CLEC-2 receptor null mice (Fig. 6A). Furthermore, tyrosine phosphorylation of the CLEC2 receptor was observed in immunoprecipitation studies when washed human platelets where activated with either rhodocytin or fucoidan (Fig. 6B), suggesting that fucoidan mediates platelet functional responses through the CLEC-2 receptor. We thus propose a model for the signaling events induced by fucoidan through the CLEC-2 receptor (Fig. 7).
FIGURE 6.
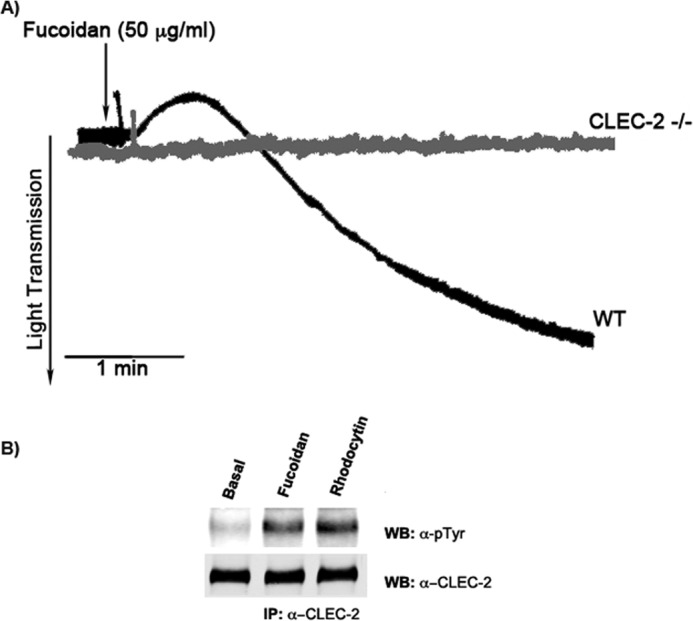
Fucoidan-induced platelet responses are mediated by CLEC-2 receptor. A, wild-type or CLEC-2 knock-out platelets (2 × 108/ml) were stimulated with fucoidan (50 μg/ml) and allowed to aggregate at 37 °C under stirred conditions in a Lumi-aggregometer (n = 10). B, washed human platelets (1 × 109/ml) were stimulated with 100 nm rhodocytin or 50 μg/ml fucoidan. Reaction was terminated by addition of an equal volume of 2× lysis buffer. Platelet lysates were precleared, and detergent-insoluble debris were removed using centrifuge. Antibodies against CLEC-2 were added to the resultant supernatant and incubated overnight with protein A Sepharose. Precipitated proteins were separated by SDS-PAGE and Western blotted (WB) with the phosphotyrosine antibody. The data are a representation of three experiments.
FIGURE 7.

Model depicting tyrosine kinase-dependent signaling pathway in platelets activated with fucoidan. Fucoidan activates platelets through an SFK-dependent pathway through the CLEC-2 receptor. YP represents tyrosine phosphorylation sites on the cytoplasmic domain of CLEC-2 receptor.
DISCUSSION
Fucoidan, a sulfated polysaccharide from a brown seaweed, decreases bleeding time and clotting time in hemophilia (8, 9). Decreased bleeding times in the hemophilia animal models by in vivo administration of fucoidan highlights the beneficial effect of fucoidan as a novel treatment (9). Furthermore, in vitro studies by using platelet poor plasma from hemophilia animal models and human patients have showed that fucoidan inhibits tissue factor pathway inhibitor, thereby contributing to an increase in the extrinsic coagulation pathway activity (8). The effect of fucoidan on platelets, however, has not been studied. In the current work, we investigated whether or not fucoidan induces platelet activation and if so, what is the receptor that is mediating these signaling events. Our results suggest that fucoidan induces platelet activation (Fig. 1) through a tyrosine kinase-dependent pathway (outlined in Fig. 7). Using pharmacological inhibitors, we have shown that SFKs and Syk are crucial for fucoidan-induced platelet activation (Figs. 2 and 3). Our data from platelet-specific CLEC-2 receptor knock-out murine platelets suggest that CLEC-2 is the physiological receptor for fucoidan on platelets (Fig. 6A), thereby providing a novel CLEC-2 receptor agonist other than rhodocytin and podoplanin.
Previous studies suggest that fucoidan-induced platelet activation was regulated by two important factors, molecular weight of the compound and the difference in sulfate content (40). Our results suggest that fucoidan, derived from the brown seaweed Fucus vesiculosus, with a high molecular weight of 150–200 kDa and low sulfate content of 8–11%, induces platelet activation through a tyrosine kinase-dependent pathway that is mediated through the CLEC-2 receptor (Figs. 1 and 6). Previously, fucoidan has been show to have antithrombotic effects (41–43). The reason for such opposite effects of fucoidan on platelets may be due to different molecular weights of fucoidan. When platelets are stimulated with fucoidan, we see a lag phase, which is reminiscent of GPVI- or CLEC-2-mediated activation. Recent studies have shown that the antibody to CLEC-2 induces dimerization of the receptor, generating a weak intracellular signal. The further clustering of CLEC-2 with a secondary antibody induces rapid and powerful activation. Similarly, tetrameric rhodocytin and polymeric podoplanin induce a much greater degree of receptor clustering (14, 15, 21). Low molecular weight fucoidan may bind to CLEC-2 but may not be able to induce the clustering of the receptors that is necessary for platelet activation (37), whereas high molecular weight fucoidan induces rapid receptor clustering and platelet activation. This clustering effect is also dependent on the concentration of high molecular weight fucoidan added to platelets as shown in Fig. 1.
In our effort to elucidate signaling pathways by which fucoidan activates platelets, we have characterized a tyrosine kinase-dependent pathway, which leads to αIIbβ3 activation (Fig. 1B). We also show that key signaling molecules, SFKs and Syk, are crucial for fucoidan-induced platelet activation (Figs. 2–4). Downstream of the GPVI or CLEC-2 receptor, Syk kinase has been shown to play an important role in activating other effectors, which ultimately leads to the activation of αIIbβ3 (38, 44). Inhibition of Syk following GPVI stimulation shuts down all functional responses. Consistent with the previous studies, we observed significant inhibition of PAC-1 binding in fucoidan-stimulated platelets in the presence of the Syk inhibitor OXSI-2 (32) or G06976 (Fig. 4A) (31). Platelet activation with fucoidan in the presence of the tyrosine kinase inhibitor PP2 also showed inhibition of platelet activation (Fig. 2). These results suggest for the first time that fucoidan-induced platelet activation depends on SFK- and Syk-dependent pathways. Fucoidan is known to activate cells such as macrophages and nerve cells (45–47), but the receptor that mediates these signaling events has not been elucidated. Fucoidan is considered as one of the agonists for scavenger receptor A in macrophages (35) and other cell lines, but to our knowledge, scavenger receptor A has not been identified in platelets. Low molecular weight fucoidan was also been reported to bind to P-selectin (Molecular Imaging and Contrast Agent Database). In resting platelets, however, P-selectin is located on the inner wall of α granules (48, 49) and is only exposed on the surface of platelets after activation. Therefore, fucoidan must first activate platelets through a receptor to expose P-selectin. Considering all our results, it is evident that fucoidan activates platelets through a tyrosine kinase-dependent receptor. The major receptors that can trigger tyrosine kinase pathways in platelets are GPVI, FcγRIIA, and CLEC-2 (16, 34). Of these, the GPVI receptor signaling depends on the FcRγ chain (37). However, our data show that platelets lacking the FcRγ chain are unresponsive to collagen-related peptide but still retain their ability to aggregate in response to fucoidan (Fig. 5A). Also, fucoidan is able to induce human platelet activation in the presence of the monoclonal antibody IV.3 (Fig. 5B), which blocks FcγRIIA, and fucoidan also induces activation of murine platelets, which lack FcγRIIA. These results suggest that fucoidan does not mediate signaling through GPVI or FcγRIIA. CLEC-2 is a C-type lectin receptor that has been shown to signal independently of the FcRγ chain in platelets (16, 34). We observed inhibition of platelet functional responses in platelet-specific (PF4) CLEC-2 receptor knock-out platelets when stimulated with fucoidan (Fig. 6A). To further support our conclusion that fucoidan induces platelet activation through the CLEC-2 receptor, we observed fucoidan-induced tyrosine phosphorylation of the CLEC-2 receptor (Fig. 6B). These results show for the first time that fucoidan mediates platelet activation through the CLEC-2 receptor. These results not only support a role for fucoidan as a novel drug for hemophilia treatment but also provide a new efficient agonist for the CLEC-2 receptor on platelets other than podoplanin and rhodocytin. In conclusion, fucoidan is a novel CLEC-2 receptor agonist that activates platelets through an SFK-dependent signaling pathway (as outlined in Fig. 7). The stimulatory action of fucoidan on platelets strongly indicates that it may play an important role in platelet activation under diseased conditions such as hemophilia.
This work was supported in part by Grants HL60683 and HL93231 from the National Institutes of Health (to S. P. K.).
- GPVI
- glycoprotein VI receptor
- SFK
- Src family kinase
- CLEC-2
- C-type lectin like receptor 2
- Gq
- heterotrimaric GTP-binding protein that stimulates phospholipase C
- PP2
- 4-amino-5-(4-chloro-phenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- PP3
- 4-amino-7-phenylpyrazolo[3,4-d]pyramidine
- PLC
- Phospholipase-C
- FcR
- Fc receptor
- PRP
- platelet rich plasma
- ITAM
- immunoreceptor tyrosine-based activation motif.
REFERENCES
- 1. Packham M. A. (1994) Role of platelets in thrombosis and hemostasis. Can J. Physiol. Pharmacol. 72, 278–284 [DOI] [PubMed] [Google Scholar]
- 2. Broos K., De Meyer S. F., Feys H. B., Vanhoorelbeke K., Deckmyn H. (2012) Blood platelet biochemistry. Thromb. Res. 129, 245–249 [DOI] [PubMed] [Google Scholar]
- 3. Clemetson K. J. (2012) Platelets and primary haemostasis. Thromb. Res. 129, 220–224 [DOI] [PubMed] [Google Scholar]
- 4. Shankar H., Garcia A., Prabhakar J., Kim S., Kunapuli S. P. (2006) P2Y12 receptor-mediated potentiation of thrombin-induced thromboxane A2 generation in platelets occurs through regulation of Erk1/2 activation. J. Thromb. Haemost. 4, 638–647 [DOI] [PubMed] [Google Scholar]
- 5. Shuman M. A., Levine S. P. (1978) Thrombin generation and secretion of platelet Factor 4 during blood clotting. J. Clin. Invest. 61, 1102–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zimmerman T. S., Fulcher C. A. (1985) Factor VIII procoagulant protein. Clin. Haematol. 14, 343–358 [PubMed] [Google Scholar]
- 7. Zimmerman T. S., Ratnoff O. D., Powell A. E. (1971) Immunologic differentiation of classic hemophilia (factor 8 deficiency) and von Willebrand's dissase, with observations on combined deficiencies of antihemophilic factor and proaccelerin (factor V) and on an acquired circulating anticoagulant against antihemophilic factor. J. Clin. Invest. 50, 244–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu T., Scallan C. D., Broze G. J., Jr., Patarroyo-White S., Pierce G. F., Johnson K. W. (2006) Improved coagulation in bleeding disorders by Non-Anticoagulant Sulfated Polysaccharides (NASP). Thromb. Haemost. 95, 68–76 [PubMed] [Google Scholar]
- 9. Prasad S., Lillicrap D., Labelle A., Knappe S., Keller T., Burnett E., Powell S., Johnson K. W. (2008) Efficacy and safety of a new-class hemostatic drug candidate, AV513, in dogs with hemophilia A. Blood 111, 672–679 [DOI] [PubMed] [Google Scholar]
- 10. Tsuji M., Ezumi Y., Arai M., Takayama H. (1997) A novel association of Fc receptor γ-chain with glycoprotein VI and their co-expression as a collagen receptor in human platelets. J. Biol. Chem. 272, 23528–23531 [DOI] [PubMed] [Google Scholar]
- 11. Suzuki-Inoue K., Fuller G. L., García A., Eble J. A., Pöhlmann S., Inoue O., Gartner T. K., Hughan S. C., Pearce A. C., Laing G. D., Theakston R. D., Schweighoffer E., Zitzmann N., Morita T., Tybulewicz V. L., Ozaki Y., Watson S. P. (2006) A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood 107, 542–549 [DOI] [PubMed] [Google Scholar]
- 12. Sobanov Y., Bernreiter A., Derdak S., Mechtcheriakova D., Schweighofer B., Düchler M., Kalthoff F., Hofer E. (2001) A novel cluster of lectin-like receptor genes expressed in monocytic, dendritic and endothelial cells maps close to the NK receptor genes in the human NK gene complex. Eur. J. Immunol. 31, 3493–3503 [DOI] [PubMed] [Google Scholar]
- 13. Colonna M., Samaridis J., Angman L. (2000) Molecular characterization of two novel C-type lectin-like receptors, one of which is selectively expressed in human dendritic cells. Eur. J. Immunol. 30, 697–704 [DOI] [PubMed] [Google Scholar]
- 14. Watson A. A., Eble J. A., O'Callaghan C. A. (2008) Crystal structure of rhodocytin, a ligand for the platelet-activating receptor CLEC-2. Protein Sci. 17, 1611–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hooley E., Papagrigoriou E., Navdaev A., Pandey A. V., Clemetson J. M., Clemetson K. J., Emsley J. (2008) The crystal structure of the platelet activator aggretin reveals a novel (αβ)2 dimeric structure. Biochemistry 47, 7831–7837 [DOI] [PubMed] [Google Scholar]
- 16. Watson A. A., Christou C. M., James J. R., Fenton-May A. E., Moncayo G. E., Mistry A. R., Davis S. J., Gilbert R. J., Chakera A., O'Callaghan C. A. (2009) The platelet receptor CLEC-2 is active as a dimer. Biochemistry 48, 10988–10996 [DOI] [PubMed] [Google Scholar]
- 17. Christou C. M., Pearce A. C., Watson A. A., Mistry A. R., Pollitt A. Y., Fenton-May A. E., Johnson L. A., Jackson D. G., Watson S. P., O'Callaghan C. A. (2008) Renal cells activate the platelet receptor CLEC-2 through podoplanin. Biochem. J. 411, 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki-Inoue K. (2011) Essential in vivo roles of the platelet activation receptor CLEC-2 in tumour metastasis, lymphangiogenesis and thrombus formation. J. Biochem. 150, 127–132 [DOI] [PubMed] [Google Scholar]
- 19. May F., Hagedorn I., Pleines I., Bender M., Vögtle T., Eble J., Elvers M., Nieswandt B. (2009) CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood 114, 3464–3472 [DOI] [PubMed] [Google Scholar]
- 20. Spalton J. C., Mori J., Pollitt A. Y., Hughes C. E., Eble J. A., Watson S. P. (2009) The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC-2 signaling in platelets. J. Thromb. Haemost. 7, 1192–1199 [DOI] [PubMed] [Google Scholar]
- 21. Watson A. A., O'Callaghan C. A. (2011) Molecular Analysis of the Interaction of the Snake Venom Rhodocytin with the Platelet Receptor CLEC-2. Toxins 3, 991–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suzuki-Inoue K., Kato Y., Inoue O., Kaneko M. K., Mishima K., Yatomi Y., Yamazaki Y., Narimatsu H., Ozaki Y. (2007) Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J. Biol. Chem. 282, 25993–26001 [DOI] [PubMed] [Google Scholar]
- 23. Francischetti I. M., Saliou B., Leduc M., Carlini C. R., Hatmi M., Randon J., Faili A., Bon C. (1997) Convulxin, a potent platelet-aggregating protein from Crotalus durissus terrificus venom, specifically binds to platelets. Toxicon 35, 1217–1228 [DOI] [PubMed] [Google Scholar]
- 24. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 25. Brass L. (2010) Understanding and evaluating platelet function. Hematology Am. Soc. Hematol. Educ. Program 387–396 [DOI] [PubMed] [Google Scholar]
- 26. Uemura T., Kawasaki T., Taniguchi M., Moritani Y., Hayashi K., Saito T., Takasaki J., Uchida W., Miyata K. (2006) Biological properties of a specific Gα q/11 inhibitor, YM-254890, on platelet functions and thrombus formation under high-shear stress. Br. J. Pharmacol. 148, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kawasaki T., Uemura T., Taniguchi M., Takasaki J. (2006) [Pharmacological properties of a specific Gq/11 inhibitor, YM-254890]. Nihon Yakurigaku Zasshi 128, 23–31 [DOI] [PubMed] [Google Scholar]
- 28. Li Z., Zhang G., Liu J., Stojanovic A., Ruan C., Lowell C. A., Du X. (2010) An important role of the SRC family kinase Lyn in stimulating platelet granule secretion. J. Biol. Chem. 285, 12559–12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Speich H. E., Grgurevich S., Kueter T. J., Earhart A. D., Slack S. M., Jennings L. K. (2008) Platelets undergo phosphorylation of Syk at Y525/526 and Y352 in response to pathophysiological shear stress. Am. J. Physiol. Cell Physiol. 295, C1045–1054 [DOI] [PubMed] [Google Scholar]
- 30. Pasquet J. M., Gross B., Quek L., Asazuma N., Zhang W., Sommers C. L., Schweighoffer E., Tybulewicz V., Judd B., Lee J. R., Koretzky G., Love P. E., Samelson L. E., Watson S. P. (1999) LAT is required for tyrosine phosphorylation of phospholipase cγ2 and platelet activation by the collagen receptor GPVI. Mol. Cell Biol. 19, 8326–8334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Getz T. M., Mayanglambam A., Daniel J. L., Kunapuli S. P. (2011) Go6976 abrogates GPVI-mediated platelet functional responses in human platelets through inhibition of Syk. J. Thromb. Haemost. 9, 608–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bhavaraju K., Kim S., Daniel J. L., Kunapuli S. P. (2008) Evaluation of [3-(1-methyl-1H-indol-3-yl-methylene)-2-oxo-2,3-dihydro-1H-indole-5-sulfonamide] (OXSI-2), as a Syk-selective inhibitor in platelets. Eur. J. Pharmacol. 580, 285–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watson S. P., Auger J. M., McCarty O. J., Pearce A. C. (2005) GPVI and integrin αIIb β3 signaling in platelets. J. Thromb. Haemost. 3, 1752–1762 [DOI] [PubMed] [Google Scholar]
- 34. Watson S. P., Herbert J. M., Pollitt A. Y. (2010) GPVI and CLEC-2 in hemostasis and vascular integrity. J. Thromb. Haemost. 8, 1456–1467 [DOI] [PubMed] [Google Scholar]
- 35. Hsu H. Y., Chiu S. L., Wen M. H., Chen K. Y., Hua K. F. (2001) Ligands of macrophage scavenger receptor induce cytokine expression via differential modulation of protein kinase signaling pathways. J. Biol. Chem. 276, 28719–28730 [DOI] [PubMed] [Google Scholar]
- 36. Ezumi Y., Shindoh K., Tsuji M., Takayama H. (1998) Physical and functional association of the Src family kinases Fyn and Lyn with the collagen receptor glycoprotein VI-Fc receptor γ chain complex on human platelets. J. Exp. Med. 188, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jarvis G. E., Atkinson B. T., Snell D. C., Watson S. P. (2002) Distinct roles of GPVI and integrin α(2)β(1) in platelet shape change and aggregation induced by different collagens. Br. J. Pharmacol. 137, 107–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hughes C. E., Pollitt A. Y., Mori J., Eble J. A., Tomlinson M. G., Hartwig J. H., O'Callaghan C. A., Fütterer K., Watson S. P. (2010) CLEC-2 activates Syk through dimerization. Blood 115, 2947–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hughes C. E., Auger J. M., McGlade J., Eble J. A., Pearce A. C., Watson S. P. (2008) Differential roles for the adapters Gads and LAT in platelet activation by GPVI and CLEC-2. J. Thromb. Haemost. 6, 2152–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dürig J., Bruhn T., Zurborn K. H., Gutensohn K., Bruhn H. D., Béress L. (1997) Anticoagulant fucoidan fractions from Fucus vesiculosus induce platelet activation in vitro. Thromb. Res. 85, 479–491 [DOI] [PubMed] [Google Scholar]
- 41. Millet J., Jouault S. C., Mauray S., Theveniaux J., Sternberg C., Boisson Vidal C., Fischer A. M. (1999) Antithrombotic and anticoagulant activities of a low molecular weight fucoidan by the subcutaneous route. Thromb. Haemost. 81, 391–395 [PubMed] [Google Scholar]
- 42. Trento F., Cattaneo F., Pescador R., Porta R., Ferro L. (2001) Antithrombin activity of an algal polysaccharide. Thromb. Res. 102, 457–465 [DOI] [PubMed] [Google Scholar]
- 43. Durand E., Helley D., Al Haj Zen A., Dujols C., Bruneval P., Colliec-Jouault S., Fischer A. M., Lafont A. (2008) Effect of low molecular weight fucoidan and low molecular weight heparin in a rabbit model of arterial thrombosis. J. Vasc. Res. 45, 529–537 [DOI] [PubMed] [Google Scholar]
- 44. Suzuki-Inoue K., Wilde J. I., Andrews R. K., Auger J. M., Siraganian R. P., Sekiya F., Rhee S. G., Watson S. P. (2004) Glycoproteins VI and Ib-IX-V stimulate tyrosine phosphorylation of tyrosine kinase Syk and phospholipase Cγ2 at distinct sites. Biochem. J. 378, 1023–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brandenburg L. O., Konrad M., Wruck C. J., Koch T., Lucius R., Pufe T. (2010) Functional and physical interactions between formyl-peptide-receptors and scavenger receptor MARCO and their involvement in amyloid β 1–42-induced signal transduction in glial cells. J. Neurochem. 113, 749–760 [DOI] [PubMed] [Google Scholar]
- 46. Gao Y., Li C., Yin J., Shen J., Wang H., Wu Y., Jin H. (2012) Fucoidan, a sulfated polysaccharide from brown algae, improves cognitive impairment induced by infusion of Aβ peptide in rats. Environ. Toxicol. Pharmacol. 33, 304–311 [DOI] [PubMed] [Google Scholar]
- 47. Jhamandas J. H., Wie M. B., Harris K., MacTavish D., Kar S. (2005) Fucoidan inhibits cellular and neurotoxic effects of β-amyloid (A β) in rat cholinergic basal forebrain neurons. Eur. J. Neurosci. 21, 2649–2659 [DOI] [PubMed] [Google Scholar]
- 48. Koedam J. A., Cramer E. M., Briend E., Furie B., Furie B. C., Wagner D. D. (1992) P-selectin, a granule membrane protein of platelets and endothelial cells, follows the regulated secretory pathway in AtT-20 cells. J. Cell Biol. 116, 617–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mazurov A. V., Vinogradov D. V., Khaspekova S. G., Krushinsky A. V., Gerdeva L. V., Vasiliev S. A. (1996) Deficiency of P-selectin in a patient with grey platelet syndrome. Eur. J. Haematol. 57, 38–41 [DOI] [PubMed] [Google Scholar]
- 50. Finney B. A., Schweighoffer E., Navarro-Núñez L., Bénézech C., Barone F., Hughes C. E., Langan S. A., Lowe K. L., Pollitt A. Y., Mourao-Sa D., Sheardown S., Nash G. B., Smithers N., Reis e Sousa C., Tybulewicz V. L., Watson S. P. (2012) CLEC-2 and Syk in the megakaryocytic/platelet lineage are essential for development. Blood 119, 747–756 [DOI] [PMC free article] [PubMed] [Google Scholar]