

Background: The functional importance of the C2 insert-containing isoform of nonmuscle myosin II-C is not known.
Results: During the neuritogenesis of Neuro-2a cells, NM II-C1C2 becomes the predominant isoform of NM II-C. Decreasing NM II-C1C2 expression leads to shortening of neurites.
Conclusion: NM II-C1C2 plays a role in the later stage of neuritogenesis.
Significance: This work contributes to an understanding of the functional importance of NM II-C1C2.
Keywords: Alternative Splicing, Cell Biology, Molecular Cell Biology, Myosin, Neurodifferentiation
Abstract
The functional role of the C2 insert of nonmuscle myosin II-C (NM II-C) is poorly understood. Here, we report for the first time that the expression of the C2 insert-containing isoform, NM II-C1C2, is inducible in Neuro-2a cells during differentiation both at mRNA and protein levels. Immunoblot and RT-PCR analysis reveal that expression of NM II-C1C2 peaks between days 3 and 6 of differentiation. Localization of NM II-C1C2 in Neuro-2a cells suggests that the C2 insert-containing isoform is localized in the cytosol and along the neurites, specifically at the adherence point to substratum. Inhibition of endogenous NM II-C1C2 using siRNA decreases the neurite length by 43% compared with control cells treated with nonspecific siRNA. Time lapse image analysis reveals that neurites of C2-siRNA-treated cells have a net negative change in neurite length per minute, leading to a reduction of overall neurite length. During neuritogenesis, NM II-C1C2 can interact and colocalize with β1-integrin in neurites. Altogether, these studies indicate that NM II-C1C2 may be involved in stabilizing neurites by maintaining their structure at adhesion sites.
Introduction
Neuritogenesis is powered mainly by two types of molecular motors: microtubule-associated motors and actin-associated motors (1). Microtubule-based motors are essential for vesicular transport of building materials during neurite assembly, whereas actin-based motors are responsible for cell motility, neurite outgrowth, and directionality of the growth cone (2–4). Neurite sprouting, extension, and branching in neuritogenesis are essential processes during development of the mammalian nervous system. Neurite sprouting results in the breakdown of neuroblast symmetry, and neurite extension is the resultant of two vectorial processes, mainly forward and backward velocity (1, 5). Neuroblast symmetry is broken as a localized bud forms, and neurites sprout from buds on the cell surface. As cells differentiate, these neurites extend and form axons and dendrites. Actin microfilaments and actin-based motors are responsible for generation of the force that is required for neurite sprouting and extension (6).
Several studies have been reported that implicate nonmuscle myosin IIs (NM IIs)2 in neuritogenesis (7–11). The number of members of the NM II family has grown due to the discovery of a new gene and alternative splicing of NM IIs. The functional importance of individual isoforms of NM II is being investigated both in vivo and in vitro in several laboratories (12–17). NM IIs belong to the conventional Class II myosins and are hexameric proteins composed of two heavy chains of 230 kDa and two pairs of light chains, referred to as the 20-kDa regulatory myosin light chain (RLC20) and the 17-kDa essential myosin light chain. These myosins form bipolar filaments that slide actin filaments to produce force or maintain tension that is needed to drive important cellular functions, such as cell polarity, cell migration, and cytokinesis (18–20). Studies from several laboratories revealed that three different genes (Myh9, Myh10, and Myh14) code for three isoforms of nonmuscle myosin heavy chain (NMHC) (II-A, II-B, and II-C, respectively) in mice (21). Enzymatic activity of these NM IIs is regulated by phosphorylation and dephosphorylation of RLC20, which are catalyzed by enzymes such as myosin light chain kinase and Rho kinase, and myosin phosphatase (22–28).
Similar to NM II-B, the pre-mRNA of the NMHC II-C gene undergoes alternative splicing to generate four isoforms: NMHC II-C0, -C1, -C2, and -C1C2. These isoforms have been expressed as proteins, and their biochemical properties have been studied in vitro. It has been reported that baculovirus-expressed heavy meromyosin II-C0 or -C1 has actin-activated MgATPase activity and can propel actin filaments following RLC20 phosphorylation, whereas heavy meromyosin II-C2 and -C1C2 are constitutively activated, independent of RLC20 phosphorylation. The C1 insert, encoding 8 amino acids, is widely expressed in a variety of tissues, such as lung, liver, kidney, and brain, whereas expression of the C2 exon, encoding 41 amino acids, is restricted to neuronal tissues in the mouse (29–31). The functional importance of NM II-C protein both in vivo and in vitro has been reported (32, 33), but that of individual isoforms of NM II-C is still incomplete. In an in vitro study, the C1 insert-containing isoform was shown to be involved in cytokinesis in tumor cells (30), whereas no functional study of the C2 insert-containing isoform has been reported thus far.
Here we report the first examination of neuritogenesis in the absence of the C2 insert-containing isoform, NM II-C1C2. We show that inhibition of NM II-C1C2 causes several defects in neuritogenesis: shortening of neurite length, lack of neurite branching, and reduction in the number of neurites per cell. We demonstrate that these defects result from the failure of stable adherence of neurites to the substratum. We offer evidence that NM II-C1C2, which is the major isoform of NM II-C in differentiated neurons, interacts with β1-integrin during neuritogenesis. This interaction may delineate the relationship between stable adherence and neuritogenesis.
EXPERIMENTAL PROCEDURES
Identification and Quantification of the C2 Insert in Mouse Neuro-2a Cells
Total RNA from Neuro-2a cells was isolated using the RNeasy minikit (Qiagen, Valencia, CA). 1 μg of isolated total RNA was reverse transcribed using random hexamers and the Gene-Amp RNA PCR core kit (Applied Biosystems, Branchburg, NJ). The resulting cDNA was amplified by PCR using the primer sets flanking the C2 inserted region: forward primer (P1), 5′- CAGCGCCCCAGGAACCTGCG-3′; reverse primer (P2), 5′-GCTCCAGGGCCTGGATCATCTT-3′. The PCR profile included 35 cycles; the first four cycles are denaturation at 94 °C for 1 min, annealing at 65 °C for 1 min, and extension at 72 °C for 1 min, and the remaining 31 cycles follow denaturation at 94 °C for 30 s, annealing at 60 °C for 30 s, and extension at 72 °C for 30 s. To check genomic DNA contamination in RNA samples, we performed cDNA synthesis in the absence of reverse transcriptase, which was used as a negative control for the RT-PCR experiment. Products generated by RT-PCR were analyzed on a 1.8% agarose gel. The slower migrating bands, 694 bp, were extracted from the gel and digested with PstI, which confirmed the insertion of the C2 insert. Sequences of primers flanking the C1 insert (P3 and P4), Within the C1 and C2 insert sequence (P5 and P6), at the C2 insert junction (P7), at the C1 insert junction (P8), Within NMHC II-A (P9 and P10), X NMHC II-B (P11 and P12), and X GAPDH (P13 and P14) were as follows: P3, 5′-GCCCATGTGGCATCATCTCCA-3′; P4, 5′-CTCCCACGATGTAGCCAGCA-3′; P5, 5′-GCCTCCGTCAGCACCATGTCTTAT-3′; P6, 5′-CGTGGGTGCACAGAGAGACC-3′; P7, 5′-CGATGCCCTCCACATCCTTCCAG-3′; P8, 5′-GGTGTCCCTGGGGAGCTAGAGC-3′; P9, 5′-GCACATGTGGCCTCCTCACAC-3′; P10, 5′-ATGTGGAAGGTCCGCTCCTCT-3′; P11, 5′-GGGACTTGAGTGAGGAGCTG-3′; P12, 5′- GCTTTGAACCTTTTCGCTTG-3′; P13, 5′-GACAACTTTGGCATTGTGGAA-3′; P14, 5′-ACACATTGGGGGTAGGAACA-3′. We used the same PCR program to amplify the amplicons for the above primers. We used primers P1 and P6 for real-time PCR to quantify the amount of NMHC II-C1C2 mRNA using the SYBER Green PCR Master Mix kit (Applied Biosystems). The program includes an initial 10 min at 95 °C and then 40 cycles of 15 s at 95 °C for denaturation and 1 min at 60 °C for annealing and extension. Similarly, the pairs of P9 and P10 and of P11 and P12 were used to quantify NMHC II-A and II-B mRNA, respectively. After each cycle, a melting curve analysis was performed to check that no primer dimers or nonspecific products were formed. -Fold induction of a gene was calculated according to a published method (34).
Plasmid Constructs and siRNAs
Plasmid constructs containing GFP-tagged full-length cDNA of mouse NMHC II-C0, II-C1, II-C2, and II-C1C2 were kind gifts from Dr. Robert S. Adelstein (National Institutes of Health, Bethesda, MD). Expression of all four full-length cDNAs encoding NMHC IIs in the Neuro-2a cell line was confirmed by immunoblot analysis and fluorescence microscopy. siRNA (C2-siRNA) specific for the mouse C2 insert sequence was custom synthesized by Sigma. Sequence for duplex C2-siRNAs was as follows: sense strand, 5′-r(CAGUUCACUUUACUUGGCU)d(TT)-3′; antisense strand, 5′-r(AGCCAAGUAAAGUGAACUG)d(TT)-3′. To inhibit all NMHC II-C mRNA, siRNA sequence was designed on the 3′-UTR region, and sequence was as follows: sense stand, 5′-UUGAACGUGAUGUCAAGAd(TT)-3′; antisense strand, 5′-UCUUGACAUCCACGUUCAAd(TT)-3′. Nonspecific siRNA duplex was obtained from Sigma (catalogue numbers SIC001 and SIC002).
Cell Culture, Transfection, and Immunofluorescence Microscopy
The mouse neuroblastoma cell line, Neuro-2a, was obtained from ATCC (Manassas, VA) and grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (growth medium). To induce differentiation, cells were first cultured in growth medium for 24 h after seeding, and then growth medium was replaced with medium consisting of DMEM with 1.5% DMSO and 0.5% fetal bovine serum (differentiation medium (DM)) according to published protocols (35). For DNA/siRNA transfection, 1 μg of plasmid DNA or 33 pmol of siRNA was transfected to 2 × 105 undifferentiated Neuro-2a cells using LipofectamineTM 2000 (Invitrogen). Then cells were cultured in DM. The transfection efficiency for DNA and siRNA was estimated using a fluorescence microscope (Olympus IX-51) by visualizing the signal coming from a GFP plasmid and Alexa 488-tagged siRNA, respectively. DNA transfection was almost 60%. siRNA could knock down mRNA by >90% as detected by RT-PCR analysis within 24 h post-transfection. For the rescue experiment, 24 h post-3′-UTR-siRNA-treated cells were transfected with GFP-tagged NMHC II-C constructs, and the cells were cultured for an additional 72 h in DM. For immunostaining studies, Neuro-2a cells were grown on chamber slides (BD Biosciences) and were fixed with 4% paraformaldehyde at room temperature for 30 min, permeabilized with 0.5% Triton X-100 for 10 min, and treated with 5% normal goat serum in PBS (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) for 60 min at room temperature followed by incubation with NMHC II-C2 (gift from Dr. Robert S. Adelstein) and β1-integrin (Santa Cruz Biotechnology, Inc.), or fascin (at 1:500 dilution; Santa Cruz Biotechnology, Inc.) antibodies overnight at 4 °C. To stain actin filament, Oregon Green-labeled phalloidin (Invitrogen) was added at room temperature for 1 h. The secondary antibody, Alexa 594 goat anti-rabbit IgG and/or Alexa 488 goat anti-mouse IgG, was incubated with cells at room temperature for 1 h. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) from Invitrogen. After washing, chamber slides were mounted using a Prolong Gold antifade kit (Invitrogen). The images were collected using a Nikon C1 confocal microscope (Nikon, Tokyo, Japan).
Electrophoresis, Immunoblotting, and Immunoprecipitation
Extracts of Neuro-2a cells were prepared for SDS-PAGE as described previously (31). Briefly, cells on tissue culture plates were washed twice with cold phosphate-buffered saline and directly lysed with Laemmli sample buffer. Proteins were separated by SDS-PAGE on 8% polyacrylamide Tris-glycine gels, transferred to a polyvinylidene difluoride membrane (Millipore Corp., Billerica, MA), and blocked in 5% nonfat milk and 0.05% Tween 20 in phosphate-buffered saline. The upper part of the blot was incubated with antibodies to the C2 amino acid sequence (0.27 μg/ml) of NMHC II-C, the C-terminal sequence of NMHC II-A (Cell Signaling Technology (Danvers, MA) or a gift from Dr. Robert S. Adelstein), or C-terminal sequence of NMHC II-B (Cell Signaling), and the lower part of the blot was incubated with antibody to β-actin (Sigma) at 4 °C overnight, washed, and then incubated with horseradish peroxidase-conjugated secondary antibodies against mouse or rabbit IgG (Fisher) at room temperature for 1 h. Blots were treated with SuperSignal® West Femto luminol enhancer solution (Fisher). Luminescence signal was captured on Biomax MR film (Eastman Kodak Co.). For immunoprecipitation, an extract of Neuro-2a cells was prepared using a buffer composed of 50 mm Tris-HCl (pH 8.0), 60 mm KCl, 10 mm MgCl2, 5 mm ATP, 4 mm EDTA, 1 mm dithiothreitol, 1% Nonidet P-40, 0.5 mm phenylmethylsulfonyl fluoride, and protease inhibitors (chymostatin, 1-chloro-3-tosylamido-7-amino-2-heptanone, l-(tosylamido-2-phenyl)-ethyl chloromethyl ketone, leupeptin, phenylmethylsulfonyl fluoride, aprotinin, benzamidine, and pepstatin A) from Sigma, at 4 °C. The lysates were sedimented at 10,000 × g for 10 min, and the supernatant was incubated with antibody specific to the C2 insert or β1-integrin as reported previously (36). The immunoprecipitates were fractionated by SDS-PAGE on 8% polyacrylamide Tris-glycine gels, and subsequently, the blot was probed with antibody specific to either beta 1-integrin or to the C2 insert, NMHC II-A, and NMHC II-B. Protein concentrations were determined using a protein assay kit (Bio-Rad).
Time Lapse Imaging
Time lapse imaging of neuritogenesis was performed using a Nikon Ti-E microscope supported by the NIS-D software (Nikon, Tokyo). Images were recorded every 5 min for 48 h for both nonspecific and C2-siRNA-treated Neuro-2a cells using a CCD camera (Digital Sight DS-Qi1MC, Nikon) supported by the NIS-D program. All time lapse imaging was performed under 5% CO2 and at 37 °C in a stage incubator. To record neuritogenesis, movies were made taking five frames/s for up to 28 h, whereas for cytokinesis, a movie was made taking two frames/s. Change of neurite length (CNL)/min was calculated using the equation, CNL/min = (Ltn + 1 − Ltn)/(tn + 1 − tn), where (tn + 1 − tn) = 10, Ltn is length of neurite at the nth time point (tn), and Ltn + 1 is the length of the next time point (tn + 1).
Statistical Analysis
Data were expressed as the mean ± S.E. Statistical significance was tested with one-way analysis of variance followed by the Bonferroni test. The differences were considered to be significant if p was <0.05.
RESULTS
Expression of the C2-inserted Isoforms of NMHC II-C in Differentiated Neuro-2a Cells
Jana et al. (31) reported that exon C2, encoding 41 amino acids in the mouse, was inserted into the loop 2 region of NMHC II-C due to alternative splicing of NMHC II-C pre-mRNA. This C2 insert was shown to be expressed only in mouse and human brain tissue. To further investigate the expression profile of the C2 insert and its biological significance, we used the mouse neuroblastoma cell line, Neuro-2a, which is a model for neuronal differentiation. The differentiation method (1.5% DMSO and 0.5% serum) used for Neuro-2a cells induced neuritogenesis: sprouting, neurite elongation, and branching. Neurite length during neuritogenesis was quantified and is shown in supplemental Fig. S1, A and B. Using RT-PCR analysis with primers specific to the C2 insert, we found that the expression of C2 insert-containing mRNA is inducible in Neuro-2a cells during differentiation, as detected by generation of a 297 bp band (Fig. 1A). We differentiated Neuro-2a cells until day 12, and during this period, expression of the C2-inserted NMHC mRNA showed differential expression. Quantification of C2-inserted NMHC II-C mRNA by real-time PCR revealed that 3–6-day postdifferentiated Neuro-2a cells had an almost 18 ± 2-fold increase in expression compared with undifferentiated Neuro-2a cells (Fig. 1B). The expression of both NMHC II-A and NMHC II-B mRNA remained unchanged during differentiation of Neuro-2a cells (Fig. 1, A and B). As shown in supplemental Fig. S2, we found that no splicing into loop 2 of NMHC II-B occurred during the Neuro-2a differentiation. Although trace or small amounts of splicing into loop 1 of NMHC II-B occurred in Neuro-2a cells, expression of the B1 insert remained unchanged during this time period of differentiation, and its expression was less than 10% of total NM II-B. We did not observe alternative splicing of NMHC II-A in either loop.3
FIGURE 1.

Expression of NMHC II-C2 is induced during differentiation. Total RNA from undifferentiated and differentiated Neuro-2a cells at the indicated time was subjected to RT-PCR analysis for NMHC II isoforms using primers specific for each isoform. A, expression of the NMHC IIs, as indicated. GAPDH was used as a control for cDNA in the PCR. B, quantification of NMHC II-A, NMHC II-B, and C2 insert-containing NMHC II-C mRNA, which was done by quantitative real-time PCR, and -fold induction during differentiation compared with undifferentiated cells (Und) was calculated using the equation, -fold induction = 2−ΔΔCt, where Ct represents the threshold cycle number for a gene. C, cell lysates from differentiated Neuro-2a cells at the indicated times were subjected to immunoblot using anti-C2 insert, NMHC II-A, NMHC II-B, and actin antibodies. Note that the expression of the C2 insert-containing isoform increased with the differentiation, whereas that of II-A and II-B remained unchanged. Actin was used as a loading control. Results show representative blots from a total of three experiments. Quantification of C2 insert-containing NMHC II-Cs and NMHC II-A/II-B expression is shown in D, considering band intensity of the immunoblot in undifferentiated cells as 1. Results are expressed as mean ± S.E. (error bars) from three independent experiments. *, p < 0.05 for undifferentiated versus 3/6-day differentiated cells. d, days.
We then examined the expression of NMHC II isoforms at the protein level and also quantified their relative difference in expression in the undifferentiated and differentiated Neuro-2a cells using ImageJ software. Fig. 1C shows immunoblots probed with antibodies specific to NMHC II-A, II-B, C2 insert, and actin. Quantification of band intensity of II-C2 immunoblot (Fig. 1D) by ImageJ software revealed that 6 days after the addition of DM, Neuro-2a cells expressed 50 ± 5-fold more II-C2 compared with undifferentiated cells. In contrast, the immunoblots detecting NMHC II-A and II-B showed almost no change in the expression at the protein level (Fig. 1D), correlating with their mRNA level, which showed no significant change between differentiated and undifferentiated Neuro-2a cells (Fig. 1B). These results suggest that expression of NMHC II-A and II-B both at mRNA and protein levels remains essentially unchanged, whereas the C2 insert-containing NMHC II-C isoform(s) increases during differentiation of Neuro-2a cells.
NMHC II-C1 and NMHC II-C1C2 Are the Major Isoforms of NMHC II-C in Neuro-2a Cells
We investigated the expression of another insert, C1, in NMHC II-C mRNA, which was shown to be expressed in neuronal tissue (31). We found that the C1 insert was expressed in both undifferentiated and differentiated Neuro-2a cells (panels 1 and 2 of Fig. 2A, lane 2, versus panels 3–7). Because both inserts, C1 and C2, were detected during Neuro-2a differentiation, four possible NMHC II-C mRNA isoforms (NMHC II-C0 (with no insert), NMHC II-C1 (with only C1 insert), NMHC II-C2 (with only C2 insert), and NMHC II-C1C2 (with both C1 and C2 inserts)) could be expressed in Neuro-2a cells. Using different pairs of primers for NMHC II-C (flanking C1 and C2 exons, located within C1 and C2 exons, or spanning the C2 exon (see primer location in Table 1)), we analyzed the abundance of each NMHC II-C isoform mRNA during the differentiation of Neuro-2a cells (Fig. 2A). Using primers P3 and P4, flanking the C1 exon, RT-PCR generated a band of 214 bp, but not 190 bp, in both undifferentiated and differentiated Neuro-2a cells, suggesting that C1-containing NMHC II-C mRNAs (i.e. either NMHC II-C1 or NMHC II-C1C2 or both) may be expressed in Neuro-2a cells (Fig. 2A, panel 1).
FIGURE 2.
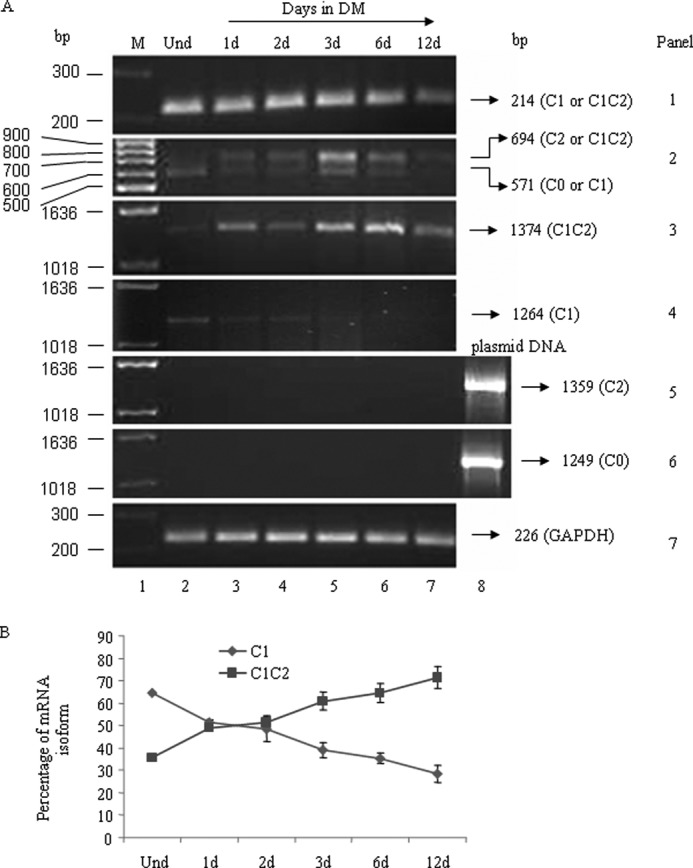
Quantification of NMHC II-C mRNA isoforms. Total RNA from undifferentiated (lane 2) and differentiated Neuro-2a cells at the indicated time (after the addition of DM, lanes 3–7) was subjected to RT-PCR analysis for NMHC II-C isoforms using primers specific for each NMHC II-C isoform. The panels in A show the expression of each isoform of NMHC II-C, as indicated in a representative gel. GAPDH was used as a control for cDNA in the PCR. Plasmid DNA encoding NMHC II-C0 or NMHC II-C2 (lane 8) was used as positive control for PCR in panels 5 and 6. Note that C1 and C1C2 mRNA are the major isoforms expressed in 3–6-day-differentiated Neuro-2a cells. B, quantification of C1 and C1C2 mRNA was expressed as a percentage of total NMHC II-C mRNA (see panel 2 in A). Percentage of an isoform = (intensity of band corresponding to an isoform/total intensity of two bands generated using C2 flanking primers) × 100. Band intensity was measured using ImageJ software. Note that the NMHC II-C1C2 population was increased with differentiation, whereas that of NMHC II-C1 decreased. Results are expressed as mean ± S.E. (error bars) from three independent experiments. d, days.
TABLE 1.
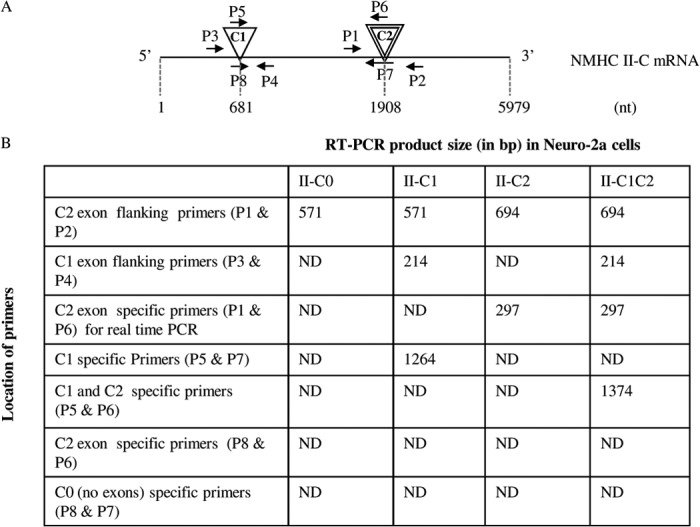
Inserts and primers
A, schematic diagram of NMHC mRNA. Locations of C1 and C2 inserts and primers are denoted with triangles and arrows, respectively. Dashed lines represent nucleotide number. Note that C1 exon (24 nucleotides in size) and C2 exon (123 nucleotides in size) are included after nucleotides 681 and 1908 of NMHC II-C mRNA, respectively. B, summary of RT-PCR analysis from Figs. 1 and 2 for each NMHC II-C isoform in Neuro-2a cells using isoform-specfic primers. ND, not detectable.
Using primers P1 and P2, flanking the C2 exon, RT-PCR generated one band of 571 bp in undifferentiated Neuro-2a cells and two bands of 694 and 571 bp in differentiated cells, suggesting that undifferentiated cells do not express the C2-containing isoform, whereas differentiated cells do (Fig. 2A, panel 2). Hence, using these primer sets, we may conclude that Neuro-2a cells show the expression of both NMHC II-C1 and II-C1C2.
To confirm the presence of NMHC II-C1 and II-C1C2, we designed primers specific for each isoform. For NMHC II-C1C2, primers P5 and P6 (located in the C1 and C2 inserts) were used for RT-PCR analysis. A band of 1374 bp was seen in differentiated Neuro-2a cells (Fig. 2A, panel 3). For NMHC II-C1, the reverse primer, P7, was located at the junction of the C2 insert region (spanning exons 15 and 16 of NMHC II-C; see Table 1), and the forward primer, P5, matched the sequence of the C1 insert. Using P5 and P7, RT-PCR generated a band of 1264 bp representing NMHC II-C1 (Fig. 2A, panel 4). Digestion with Pst1 enzyme confirmed the identity of the PCR bands (data not shown). To quantify the abundance of the two isoforms, we measured the PCR band intensity (Fig. 2A, panel 2) using ImageJ software. Quantification revealed that 64.6 ± 4.1% of total NMHC II-C was NMHC II-C1C2, and 35.4 ± 4.1% was NMHC II-C1 at day 6 during differentiation (Fig. 2B). We used primers P8 (spanning the exons that flank the C1 exon) and P7 and used primers P8 and P6 to check if NMHC II-C0 and NMHC II-C2 were expressed, respectively. Panels 5 and 6 rule out the expression of II-C0 and II-C2 because no band was seen in Neuro-2a cells, respectively. These data suggest that Neuro-2a cells express the NMHC II-C1C2 as the predominant isoform of NMHC II-C during differentiation.
Designing Small Interfering RNA and Measuring Its Specificity against NMHC II-C1 and II-C1C2
We were then interested to study the functional importance of two isoforms, NM II-C1 and II-C1C2, in Neuro-2a cells. We used small interfering RNA (siRNA) to knock down the expression of each isoform.
We designed siRNA in the sequence of the C2 insert to target NMHC II-C1C2 mRNA alone in differentiated Neuro-2a cells. To test whether NMHC II-C1C2 has a critical role in Neuro-2a differentiation, siRNA was transfected into Neuro-2a cells prior to the onset of differentiation. Fig. 3, A and C, shows that both endogenous NMHC II-C1C2 and ectopically expressed NMHC II-C1C2-GFP were down-regulated at 24 h post-transfection with siRNA in C2-siRNA-treated cells compared with nonspecific siRNA-treated cells. We quantified the percentage of inhibition in the expression of NM II-C1C2 protein by measuring the immunoblot band intensity and fluorescence intensity (Fig. 3, B and D). C2-siRNA treatment led to a reduction of NM II-C1C2 by 90% of the endogenous level and by 80% after ectopic expression. Specificity of C2-siRNA was further confirmed by its inability to down-regulate the expression of exogenous GFP-tagged NMHC II-C1, II-C0,3 or endogenous NMHC II-A and II-B (Fig. 3A). We checked the NMHC II-C1C2 mRNA level at 24–72 h after C2-siRNA transfection in Neuro-2a cells (supplemental Fig. S3). C1C2 mRNA was reduced by almost 90% at 24 h.
FIGURE 3.

Inhibition of NM II-C1C2 isoform expression interferes with neuritogenesis. A, immunoblots of two different amounts of Neuro-2a cell lysates with the indicated antibodies. Note that C2-siRNA specifically inhibited NMHC II-C1C2 expression but not NMHC II-A or NMHC II-B expression. Cell lysates were prepared at 72 h after siRNA transfection. B, -fold inhibition by siRNA treatment was calculated by measuring band intensity using ImageJ software and considering the band intensity of NMHC II-C1C2 in nonspecific siRNA-treated cells as 1. C, microscopy images of Neuro-2a cells transfected with GFP-tagged NMHC II-C1C2 and nonspecific siRNA or C2-siRNA. Top left panel, green fluorescence signal coming from NMHC II-C1C2-GFP in nonspecific siRNA-treated cells; bottom left panel, inhibition of fluorescence intensity in C2-siRNA-treated cells. Their corresponding bright field images are shown in the right panels. -Fold inhibition in fluorescence intensity was calculated using NIS-D software and is shown in D. E, bright field images of nonspecific siRNA-treated (left panels) and C2-siRNA-treated (right panels) Neuro-2a cells. Top and bottom panels, 3 and 6 days postdifferentiation, respectively. Neurite length was measured using ImageJ software. Quantification is shown in F. G, pie chart shows that more than 65% cells are round after C2-siRNA treatment. H, schematic representation of time schedule for the addition of siRNA and DM and time lapse microscopy. Scale bar, 100 μm. Results are expressed as mean ± S.E. (error bars) from three independent experiments. *, p < 0.05, nonspecific versus C2-siRNA. d, days.
We designed siRNA (C1-siRNA), covering the region between nucleotides 1904 and 1922 of NMHC II-C mRNA, to deplete mRNA that does not contain the C2 exon either in the presence of growth medium or DM. RT-PCR and fluorescence microscopy were used to monitor endogenous NMHC II-C1 mRNA and exogenous protein levels of NM II-C1, respectively, in siRNA-treated Neuro-2a cells. Supplemental Fig. S4A shows that C1-siRNA specifically inhibited the expression of NMHC II-C1. Expression of GFP-tagged NMHC II-C1C2 in siRNA-treated Neuro-2a cells was not inhibited, suggesting that C1-siRNA specifically inhibited the expression of NMHC II-C1 (supplemental Fig. S4B).
Lower Expression of NMHC II-C1C2 Leads to Reduced Neurite Length
We focused on exploring the effect of suppressing each of the NMHC II-C isoforms in differentiated Neuro-2a cells on neurite length and number of neurites per cell during neuritogenesis. We observed that C2-siRNA against C2 exon had a substantial effect on neurite length, number of neurites/cell, and percentage of cells having no neurites compared with the effect of nonspecific siRNA (Fig. 3E). We quantified neurite length at 3 and 6 days after siRNA treatment. At 3 and 6 days, the average length of neurites was 48.6 and 43.11 μm, respectively, in C2-siRNA-treated cells, whereas neurite length was 81.11 and 101.7 μm, respectively, in nonspecific siRNA-treated cells (Fig. 3F). There was almost a 2-fold difference in the number of neurites/cell in C2-siRNA-treated cells compared with nonspecific siRNA-treated cells at 6 days (p < 0.05; Fig. 3G). These observations reveal that the effects of C2-siRNA (decreasing C1C2 expression in differentiated cells) account for the decrease in neurite length and number of neurites/cell.
We tested whether each NM II-C isoform tagged with GFP could rescue NM II-C1C2-depleted cells to the siRNA. Plasmid DNA containing full-length NMHC II-C isoform and GFP was transfected into 24-h post-siRNA-treated cells, and cells were cultured for an additional 72 h in DM. For this rescue experiment, we designed siRNA on the 3′-untranslated region of NMHC II-C mRNA (3′-UTR siRNA), which inhibited both prevailing isoforms of NMHC II-C (C1 and C1C2) and, at the same time, was unable to lower the mRNA coming from the NMHC II-C0-GFP, -C1-GFP, -C2-GFP, or -C1C2-GFP plasmids in cells. The phenotype of GFP-expressing cells was analyzed by phase-contrast and fluorescence microscopy. The loss of major isoform, myosin II-C1C2, expression in Neuro-2a cells leads to shortening of neurite length, whereas in rescue cells expressing NMHC II-C1C2-GFP or -C2-GFP, the loss of endogenous myosin II-C1C2 has little effect on neurite length (Fig. 4A). Fig. 4A shows that, unlike NMHC II-C2-GFP- or NMHC II-C1C2-GFP-transfected cells, which showed neurites and localization of NMHC II-C2-GFP or -C1C2-GFP in neurite at 96 h, NMHC II-C0-GFP- or NMHC II-C1-GFP-transfected cells showed almost no neurite. The Fig. 4B immunoblot confirms the absence of NMHC II-C1C2 at 96 h in the 3′-UTR siRNA-treated cells that were subsequently transfected with GFP alone (lane 1, panel 2) and the inability of 3′-UTR siRNA to lower the amount of NMHC II-C-GFPs encoded by NMHC II-C-GFP plasmid DNAs (lanes 2–5, panels 1 and 2). Blots also reveal that an approximately equal amount of each isoforms was expressed in siRNA-treated cells (lanes 2–5, panels 1 and 2). We quantified the effect of NMHC II-C-GFP in II-C knockdown cells (Table 2), considering the cells that possess more than two neurites and fewer than two neurites. Both populations of cells were increased in NMHC II-C2- and NMHC II-C1C2-transfected cells. Only 5–7% of GFP-positive cells showed more than two neurites in only GFP plasmid-transfected cells. In contrast, around 27% of NMHC II-C1C2-GFP- or NMHC II-C2-GFP-positive cells showed more than two neurites compared with 5–8% of NMHC II-C0-GFP- or NMHC II-C1-GFP-positive cells. We also quantified the length of neurites in NMHC II-C-GFP-positive cells. Table 2 shows that GFP alone-positive cells have neurite length around 8 μm. In contrast, NMHC II-C1C2-GFP- or NMHC II-C2-GFP-positive cells showed neurites of around 40 μm compared with NMHC II-C0-GFP and NMHC II-C1-GFP, which showed neurite length around 7 μm.
FIGURE 4.
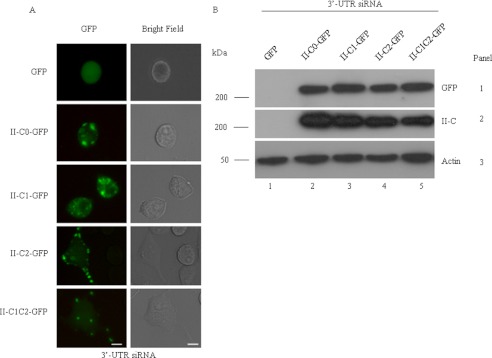
Rescuing the siRNA-induced decrease in neurite length using GFP NMHC II-Cs. A, GFP-tagged NMHC II constructs were transfected into the NMHC II-C siRNA-treated cells, and the cells were cultured for an additional 72 h in DM. Green fluorescence (left column) and the corresponding bright field (right column) images were captured at 72 h postdifferentiation. B, immunoblot of cell lysates with antibody against GFP (top) and NMHC II-C (middle). Note that the transfection efficiency of each isoform was approximately the same and that siRNA against the 3′-UTR suppresses endogenous expression of NMHC II-C but not exogenous expression. Actin was used as a loading control. Scale bar, 20 μm.
TABLE 2.
Percentage of GFP-positive cells having neurites and their average neurite lengths
Plasmid DNA containing GFP-tagged NMHC II-C isoform was transfected into 24-h post-siRNA-treated cells. After 72 h of post-DNA transfection, GFP-positive cells were used for quantification. Data are represented as means ± S.E. from three independent experiments. n > 100 cells from each experiment.
| GFP alone | NMHC II-C0-GFP | NMHC II-C1-GFP | NMHC II-C2-GFP | NMHC II-C1C2-GFP | |
|---|---|---|---|---|---|
| Round cells (%) | 90 ± 2.1 | 85.2 ± 2 | 86.1 ± 2 | 44.4 ± 1.9 | 35.7 ± 1.95 |
| Cells with <2 neurites (%) | 3.3 ± 1 | 7.4 ± 1.6 | 8.4 ± 1 | 29.6 ± 3 | 38.1 ± 2.15 |
| Cells with >2 neurites (%) | 6.7 ± 1 | 7.4 ± 1.1 | 5.5 ± 0.9 | 26 ± 1.95 | 26.2 ± 2.1 |
| Average neurite length (μm) | 7.35 ± 1.2 | 6.32 ± 0.9 | 6.85 ± 1.5 | 35.57 ± 3.2 | 48.43 ± 4.2 |
We next separately inhibited the expression of NMHC II-C1 by >90%, using an siRNA spanning the neucleotide region from 1904 to 1922 of NMHC II-C mRNA (Table 1 and supplemental Fig. S4, A–D), and that inhibition resulted in failure of cytokinesis in Neuro-2a cells, similar to a previous finding in human A549 cells (30). C1-siRNA-treated cells showed normal neuritogenesis but incomplete cytokinesis (supplemental Movie 1 and Fig. S4, B and E). Quantification revealed that nearly 14% of cells failed in cytokinesis, 58% of cells showed normal neuritogenesis, and almost 28% cells were round shaped in C1-siRNA-treated Neuro-2a cells (supplemental Fig. S4F). These data suggest that the C2 insert-containing isoforms NM II-C2 and NM II-C1C2 but not the non-C2 insert-containing isoforms NM II-C0 and NM II-C1 are involved in neuritogenesis.
Inhibition of NMHC II-C1C2 Interferes with a Late Stage of Neuritogenesis in Neuro-2a Cells
Shortening of neurite length and decrease in neurite number by C2-siRNA suggested that the C2 insert-containing isoform plays a role in neuritogenesis. Microscopy detected that a large percentage of the cells treated with C2 exon-specific siRNA rounded up or had one or two neurites compared with control cells treated with nonspecific siRNA. This suggested that NMHC II-C1C2 knockdown cells either lost neurite adhesion or increased retrograde flow or both. To analyze neuritogenesis in detail, time lapse imaging over a 48-h period at 5-min intervals was performed after the addition of DM to cells that had been previously treated with siRNA for 24 h.
For this experiment, we tracked cells' neurites (n > 10 cells) for each siRNA treatment. We made comparisons of neurites in nonspecific and C2-siRNA-treated cells that extended and/or then retracted neurites to make sure that we were measuring neuritogenesis. Because an increase or decrease of neurite length was associated with either neurite outgrowth, retrograde, or cell body movement or a combination of factors, we measured the CNL/min during the process of neuritogenesis. CNL/min was calculated by dividing the change of distance by a neurite by the time taken for such change. Fig. 5, A and C, and supplemental Movie 2 show the pattern of CNL/min of a neurite in a control cell treated with nonspecific siRNA. CNL/min was variable and followed a cyclic pattern with an amplitude value between +2.25 and −3.21 μm/min during the early stage of neuritogenesis until 8–10 h, when the cyclic amplitude was reduced to +1.9 to −1.2 μm/min during the later stage of neuritogenesis. A positive value indicates an increase of neurite length, and a negative value indicates shortening of neurite length. In contrast to neurite of nonspecific siRNA-treated cells, neurite of C2-siRNA-treated cells (Fig. 5, B and D, and supplemental Movie 3) showed that CNL/min followed a cyclic pattern with amplitude value from +1.7 to −1.13 μm/min, but after 8–10 h of neuritogenesis, the amplitude values were reduced to +0.26 and to −0.3 μm/min, respectively. We measured net CNL/min (average of all positive values plus average of all negative values) of a total of >10 neurites before and after the 8–10-h period during neuritogenesis and found that neurites of nonspecific siRNA-treated cells increase their length at 0.217 ± 0.04 and 0.117 ± 0.007 μm/min during the early and later stage of neuritogenesis, respectively, whereas neurites of C2-siRNA-treated cells increase their length at 0.16 ± 0.003 μm/min during the early phase but reduce their length at −0.06 ± 0.01 μm/min during the later stage of neuritogenesis (Fig. 5, E and F). The difference of net CNL/min between nonspecific siRNA and C2-siRNA was statistically significant at the later stage of neuritogenesis.
FIGURE 5.

NM II-C1C2-depleted cells show shortening of neurite length. Time lapse images of Neuro-2a cells during differentiation, which were previously treated with nonspecific (A) or C2-siRNA (B). Note that the C2-siRNA-treated cell shows an increase in neurite length, but after 8–10 h of neuritogenesis, the neurite starts having frequent periods of shortening and shows no growth until 28 h of neuritogenesis, whereas control cells treated with nonspecific siRNA show no shortening of neurite length. The arrows indicate the cells that were tracked, and the arrowheads show neurites for which changes of length were measured. C and D, change in neurite length/min over the 28-h period of neuritogenesis of nonspecific siRNA- and C2-siRNA-treated cells. Note that the amplitude value in control neurites is reduced with time, and neurites of C2-siRNA-treated cells show a net average negative value of CNL/min between 8 and 28 h of neuritogenesis. E and F, quantification of net average CNL/min value in nonspecific siRNA- and C2-siRNA treated neurites during initial and later stage of neuritogenesis. Data are represented as means ± S.E. (error bars). *, p < 0.05, not significant (NS) versus C2-siRNA; n > 10 neurites from each treatment; h, hours; scale bar, 10 μm.
In our culture system, neuritogenesis followed a well defined stereotyped program: stage 1, within a few h (0–2 h after changing to DM) of neuritogenesis, most cells started sprouting but were devoid of neurites; stage 2, within 2–4 h of neuritogenesis, neurons developed neurites and, depending on the number of neurites, cells' morphology could be divided into groups like unipolar, bipolar, and multipolar but none with established axon; stage 3, after 4 h of neuritogenesis, neurites started to grow and form axons, and the remaining neurites could not extend during this stage, consistent with a similar observation by de Lima et al. (37). In control cells treated with nonspecific siRNA, we found that stage 1 and stage 2 were interchangeable (Fig. 6A), and in stage 2 polarity changed reversibly, bipolar to multipolar and vice versa. In stage 3, neurons changed axonal position, meaning the axon became a neurite, and another neurite became an axon, which we termed as stage 3A (Fig. 6B). We found that Neuro-2a cells treated with nonspecific siRNA showed an increase in neurite length at a net rate of 0.217 μm/min and reached a length 112 μm within 10 h (Fig. 5E). Both a movie (supplemental Movie 2) and images (Fig. 5A) show that nonspecific siRNA-treated cell bodies and neurites undergo retrograde and forward movement even after 24 h.
FIGURE 6.

Early and later stage of neuritogenesis. A, images of a Neuro-2a cell at 5-min intervals during the early stage of neuritogenesis. The arrowhead indicates the neurite that retracts to the cell body after 10 min. B, images of Neuro-2a cells at the later stage of neuritogenesis, when neurites become axons. The numeric value indicates the number of the neurite, and the arrow indicates the neurite that is an axon. After 20 min of neuritogenesis, neurite 1, which was an axon, becomes a neurite, whereas neurite 2 becomes an axon. Scale bar, 10 μm.
C2 exon-specific siRNA-treated Neuro-2a cells (n > 14) also formed neurites and reached the same maximum length as control cell neurites (Fig. 5B). Unlike control cells, the neurites started shortening at a rate of −0.06 ± 0.01 μm/min after 8–10 h (Fig. 5F), the time point when expression of NM II-C1C2 reached a maximum level in control cells (Fig. 2, panel 2). 28 h after neuritogenesis, no neurite was seen, and cells became round. Thus, adhesion of the cell body and retrograde flow depend on myosin II-C1C2, which is the major isoform of NM II-C, during neuritogenesis. Because no conversion of stage 3 to 3A was involved (rather stage 3 returns to stage 1) in NMHC II-C1C2 knockdown cells, NM II-C1C2, but not NM II-C1, may be involved in maintaining the stability of stage 3 and/or conversion from stage 3 to 3A.
Localization of Myosin II-C1C2 during Neuritogenesis
Time lapse imaging analysis established that NM II-C1C2 may play a role in the stability of stage 3 of neuritogenesis, which prompted us to study the localization of NMHC II-C1C2 in Neuro-2a cells during neuritogenesis. Supplemental Fig. S5 shows that NMHC II-C1C2 initially localizes at sprouts at 24 h and then in the neurite and neurite tip at 72 h postdifferentiation. Other isoforms (NM II-C1, II-A, and II-B) are distributed throughout the cell body and neurites of Neuro-2a cells during neuritogenesis (supplemental Fig. S5). This observation suggests that the C2 insert is responsible for the observed localization pattern for NM II-C1C2.
It had been reported that blocking of myosin activity by pharmacological drugs modulates integrin and the extracellular matrix (38). Because β1-integrin is involved in adhesion of neuronal cells during neuritogenesis (39–41), we were interested to see which isoform of NM II can interact with β1-integrin. The immunoblot (Fig. 7A) shows that the β1-integrin antibody (lane 2), but not mouse IgG (lane 1), co-immunoprecipitates NM II-C1C2, as detected by the C2-specific antibody. A cell lysate of Neuro-2a cells transfected with NMHC II-C1C2-GFP (lane 3) served as a positive control for the C2-specific antibody. Similarly, the immunoblot (Fig. 7B) shows that immunoprecipitation with the C2 insert antibody (lane 2) but not with rabbit IgG (lane 1) contains β1-integrin, as detected by the β1-integrin antibody. Interestingly, we could not detect NM II-A or II-B in the immunoprecipitate with β1-integrin using NMHC II-A- or NMHC II-B-specific antibodies (Fig. 7C). Fig. 7D shows that both at days 3 and 6 postdifferentiation, NM II-C1C2 colocalizes with β1-integrin in 6 days, suggesting that NM II-C1C2 may play a role in the adhesion of neurites to the substratum as the neurites mature during the later stages of neuritogenesis.
FIGURE 7.

β1-integrin colocalizes and co-precipitates with NM II-C1C2 in differentiated Neuro-2a cells. After 6 days of differentiation, a cell lysate of Neuro-2a cells was used for immunoprecipitation (IP) with β1-integrin or C2 insert-specific antibody. A, immunoprecipitate of β1-integrin antibody was subjected to immunoblot (WB) with C2 insert-specific antibody (lane 2); B, the reverse experiment. Mouse IgG and rabbit IgG were used as negative controls for immunoprecipitation with β1-integrin and C2 insert-specific antibodies, respectively (lane 1). NMHC II-C2-GFP-transfected Neuro-2a cell lysate was used as a positive control for immunoblots with C2 insert-specific antibody (lane 3). C, immunoprecipitate of β1-integrin antibody was subjected to immunoblot with antibodies against NMHC II-A and II-B. Note that both NM II-A and II-B are detectable in the supernatant, not in the pellet. D, colocalization of β1-integrin and NM II-C1C2 in neurites of Neuro-2a cells. Three and 6 days after differentiation, Neuro-2a cells were co-stained for β1-integrin (green, panel 3) and C2 insert (red, panel 2). DAPI was used to stain DNA (blue, panel 1). A merged image is shown in panel 4. The yellow color shown with arrows in panel 5 (magnified image of inset in panel 4) indicates the colocalization of β1-integrin and NM II-C1C2. Scale bar, 10 μm.
Shortening of Filopodial Length
Filopodia play a significant role in neurite protrusion (42, 43). We used actin staining to visualize filopodia following inhibition of NM II-C1C2 protein in Neuro-2a cells during differentiation. Fig. 8A shows that NM II-C1C2 depletion greatly inhibited the number of filopodia across the neuronal shaft and cell body. We quantified the number of filopodia/cell >5 μm in length and spread along the leading edge, cell body, and neuronal shaft, between nonspecific siRNA-treated and C2-siRNA-treated cells (Fig. 8B). Nonspecific siRNA-treated cells showed 28.3 ± 2.9 filopodia/cell, whereas C2-siRNA-treated cells showed only 3.1 ± 1.2. We also used filopodia marker fascin and VASP staining and found decreased number of both lateral and dorsal filopodia in C2-siRNA-treated cells compared with nonspecific siRNA-treated cells (supplemental Fig. S6, C and E). Inhibition of NMHC II-C1C2 expression did not interfere with the expression of either fascin or VASP (supplemental Fig. S6, A and B). Interestingly, Ectopic expression of GFP-tagged NMHC II-C1C2 rescued the decreased number of filopodia in NM II-C1C2 knockdown Neuro-2a cells (supplemental Fig. S6D). Taken together, our data may suggest that integrin-myosin-dependent stable adhesion complexes in neurites are required for filopodia formation.
FIGURE 8.

Depletion of NM II-C1C2 reduces filopodia frequency in differentiated Neuro-2a cells. A, phalloidin staining to indicate actin localization (green) in nonspecific siRNA-treated (top row) and C2-siRNA-treated (bottom row) Neuro-2a cells after 3 days of differentiation. DAPI (blue) was used to stain nuclei. Note that C2-siRNA reduces the expression of NM II-C1C2 (red) in C2-siRNA-treated cells. B, filopodia frequency in cells transfected with nonspecific and C2-siRNA. Scale bar, 10 μm. *, statistical significance of nonspecific siRNA-treated versus C2-siRNA-treated cells (p < 0.05, n > 25 cells from each group). Error bars, S.E. DIC, differential interference contrast.
DISCUSSION
In this study, we uncovered essential requirements for NM II-C1C2 during neuritogenesis. We found that inhibition of NM II-C1C2 expression caused a decreased outgrowth of neurites and reduced the number of filopodia in differentiated Neuro-2a cells.
Focal adhesion complex composition is modulated by myosin II activity (44), and changes in focal adhesion complex composition are necessary to modulate integrin-mediated cell signaling for differentiation. Of note, during differentiation of Neuro-2a cells, the composition of NM II-C has also been remarkably changed from a myosin light chain phosphorylation-dependent NM II-C1 to a phosphorylation-independent NM II-C1C2 isoform. Switching of these isoforms arises due to alternative splicing that is regulated during differentiation. Furthermore, we had also analyzed the splicing of NM II-B and found that only the B1 and not the B2 insert is present in the heavy chain of NM II-B in these cells. Expression of B1 is very low, probably 10% of total NM II-B, and remains essentially unchanged during neuritogenesis (supplemental Fig. S2).
Integrin-mediated focal adhesions play an important role in cell adhesion, cytoskeleton reorganization, and the signaling that controls cell division, differentiation, and migration. Recently, it has been documented that during cell migration, the composition of focal adhesion complexes changes in response to tension during focal adhesion complex maturation from nascent to mature and is modulated by nonmuscle myosin II activity in human foreskin fibroblast cells, HFF1 (44). There are two hypotheses for how NM II mediates adhesion maturation. NM II bundles actin filaments, and as a consequence, adhesion proteins are brought together and clustered. This event increases molecular interaction between adhesion proteins, resulting in increased integrin avidity for ligand and signaling. In the second hypothesis, NM II generates force that induces conformational changes in adhesion-associated molecules and non-mechanosensitive signaling molecules (45). Although our data do not favor one hypothesis over the other, our results do show evidence of a possible interaction between β1-integrin and the C2 insert-containing isoform of NM II-C under the low serum/DMSO differentiation conditions. Our results allow us to hypothesize that integrin-NM II-C1C2-dependent stable adhesion complexes in neurites can help the dendritic network of actin to reorganize into bundles of long filaments. Activities of Ena/VASP (46), fascin (47), formin (48), Arp2/3 complex, and Rho GTPases (RhoA-kinase) (49–51) may be responsible for this process. This stable adhesion complex may be involved in maintaining filopodial length. We hypothesize that punctate localization of NM II-C1C2 in the neurite (supplemental Fig. S5) may be responsible for generating stable adhesion by making complexes with integrin that stabilize the neurites during the later stage of neuritogenesis. Rosado et al. (52) have recently shown that NM II-A associates with the cytoplasmic domain of α4 subunit of the α4β1-integrin and regulates cell migration in CHO cells. Although it remains unclear whether myosin and integrin association is due to direct binding or occurs through other proteins, both studies support the notion that interaction between NM II and integrin is involved in cell-matrix adhesion.
Inhibition of C2 insert-containing isoforms does not interfere with neurite initiation, implying that NM II-C1C2 may not be required for symmetry breaking of neurospheres, although exogenous GFP-tagged NM II-C1C2 could localize to the initiation site, the place where the neuroblast sphere would be broken to make the first neurite (supplemental Fig. S5). It is important to note that neurons having lowered amounts of NM II-C1C2 do form axons and extend to a maximum length but are not stabilized for long periods of time. We hypothesize that neurite growth displays biphasic speed (retrograde phase with a negative CNL value and protrusion phase with a positive value) during neuritogenesis. The neurite final length is dependent on the relative duration of the two phases. At a later stage of neuritogenesis, neurites of C2-siRNA-treated Neuro-2a cells pass through a retrograde phase, which leads to shortening of their length.
NM II-C knock-out mice show no obvious defect in the brain (32). This difference from our in vitro result may reflect the complexity of the mammalian brain. Possibly, other isoforms of NM II-C compensate for the loss of NM II-C1C2, or the cells are exposed to external cues that bypass the requirement for NM II-C1C2. All isoforms of NM II, including possible alternatively spliced isoforms of NM II-B and NM II-C, are found in neuronal cells (31, 33, 43), giving rise to complexity as to whether these isoforms have different or redundant functions in neuronal cells. Previously, NM II-C was shown to be involved in multitasking in neuronal cells (33). Neurite outgrowth, adhesion, retraction, and cell spreading are affected by the NM II-C isoform. NM II-C shares a functional role with NM II-B in neurite outgrowth and with NM II-A and II-B in adhesion but has unique features distinct from either NM II-A or NM II-B in cell spreading and in neurite retraction. The previous study did not analyze the isoform specificity of NM II-C. Our study supports the idea that the C2 insert-containing isoform, NM II-C1C2, is involved in neuritogenesis and that the C1 insert-containing isoform, NM II-C1, participates in cytokinesis of neuronal cells.
We used DMSO/low serum to induce neuritogenesis, in which alternative splicing of NMHC II-C, but not of NMHC II-B or II-A, occurred at loop 2 of its motor domain. Interestingly, we could not detect the expression of the C2 insert when Neuro-2a cells were induced to differentiate in the presence of retinoic acid or nerve growth factor.3 Previously, Seidman et al. (35) have shown that DMSO/low serum induced the expression of the angiotensin type 2 receptor, AT2, in neuroblastoma cells, NG108-15. The brain AT system has been shown to be involved in learning and memory as well as growth and development (53–56). We are now further investigating the importance of the C2 insert-containing isoforms of NM II-C in this physiological process.
Acknowledgments
We thank Dr. Robert S. Adelstein (NHLBI, National Institutes of Health) for kind gifts of antibodies and plasmid DNAs and Drs. Debi P. Sarkar and Pijush K. Das and their groups for reagents and helpful discussions. We also thank Drs. Adelstein, Mary Anne Conti, Kaustuv Datta, and Malancha Ta for valuable comments on the manuscript.
This work was supported by the Indian Association for the Cultivation of Science and the Department of Biotechnology, Government of India.

This article contains supplemental Figs. S1–S6 and Movies 1–3.
S. Saha, S. K. Dey, A. Biswas, P. Das, M. R. Das, and S. S. Jana, unpublished observation.
- NM II
- nonmuscle myosin II
- NMHC
- nonmuscle myosin heavy chain
- DM
- differentiation medium
- RLC
- regulatory light chain
- CNL
- change of neurite length.
REFERENCES
- 1. da Silva J. S., Dotti C. G. (2002) Breaking the neuronal sphere. Regulation of the actin cytoskeleton in neuritogenesis. Nat. Rev. Neurosci. 3, 694–704 [DOI] [PubMed] [Google Scholar]
- 2. Ahmad F. J., Hughey J., Wittmann T., Hyman A., Greaser M., Baas P. W. (2000) Motor proteins regulate force interactions between microtubules and microfilaments in the axon. Nat. Cell Biol. 2, 276–280 [DOI] [PubMed] [Google Scholar]
- 3. Kaether C., Skehel P., Dotti C. G. (2000) Axonal membrane proteins are transported in distinct carriers. A two-color video microscopy study in cultured hippocampal neurons. Mol. Biol. Cell 11, 1213–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou F.-Q., Waterman-Storer C. M., Cohan C. S. (2002) Focal loss of actin bundles causes microtubule redistribution and growth cone turning. J. Cell Biol. 157, 839–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. He M., Zhang Z.-H., Guan C.-B., Xia D., Yuan X.-B. (2010) Leading tip drives soma translocation via forward F-actin flow during neuronal migration. J. Neurosci. 30, 10885–10898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rösner H., Möller W., Wassermann T., Mihatsch J., Blum M. (2007) Attenuation of actinomyosinII contractile activity in growth cones accelerates filopodia-guided and microtubule-based neurite elongation. Brain Res. 1176, 1–10 [DOI] [PubMed] [Google Scholar]
- 7. Brown M. E., Bridgman P. C. (2003) Retrograde flow rate is increased in growth cones from myosin IIB knockout mice. J. Cell Sci. 116, 1087–1094 [DOI] [PubMed] [Google Scholar]
- 8. Turney S. G., Bridgman P. C. (2005) Laminin stimulates and guides axonal outgrowth via growth cone myosin II activity. Nat. Neurosci. 8, 717–719 [DOI] [PubMed] [Google Scholar]
- 9. Wylie S. R., Chantler P. D. (2001) Separate but linked functions of conventional myosins modulate adhesion and neurite outgrowth. Nat. Cell Biol. 3, 88–92 [DOI] [PubMed] [Google Scholar]
- 10. Wylie S. R., Chantler P. D. (2003) Myosin IIA drives neurite retraction. Mol. Biol. Cell 14, 4654–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wylie S. R., Wu P. J., Patel H., Chantler P. D. (1998) A conventional myosin motor drives neurite outgrowth. Proc. Natl. Acad. Sci. U.S.A. 95, 12967–12972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Golomb E., Ma X., Jana S. S., Preston Y. A., Kawamoto S., Shoham N. G., Goldin E., Conti M. A., Sellers J. R., Adelstein R. S. (2004) Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J. Biol. Chem. 279, 2800–2808 [DOI] [PubMed] [Google Scholar]
- 13. Itoh K., Adelstein R. S. (1995) Neuronal cell expression of inserted isoforms of vertebrate nonmuscle myosin heavy chain II-B. J. Biol. Chem. 270, 14533–14540 [DOI] [PubMed] [Google Scholar]
- 14. Ma X., Kawamoto S., Uribe J., Adelstein R. S. (2006) Function of the neuron-specific alternatively spliced isoforms of nonmuscle myosin II-B during mouse brain development. Mol. Biol. Cell 17, 2138–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Breckenridge M. T., Dulyaninova N. G., Egelhoff T. T. (2009) Multiple regulatory steps control mammalian nonmuscle myosin II assembly in live cells. Mol. Biol. Cell 20, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goeckeler Z. M., Bridgman P. C., Wysolmerski R. B. (2008) Nonmuscle myosin II is responsible for maintaining endothelial cell basal tone and stress fiber integrity. Am. J. Physiol. Cell Physiol. 295, C994–C1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smutny M., Cox H. L., Leerberg J. M., Kovacs E. M., Conti M. A., Ferguson C., Hamilton N. A., Parton R. G., Adelstein R. S., Yap A. S. (2010) Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat. Cell Biol. 12, 696–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Betapudi V., Licate L. S., Egelhoff T. T. (2006) Distinct roles of nonmuscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer Res. 66, 4725–4733 [DOI] [PubMed] [Google Scholar]
- 19. Glotzer M. (2005) The molecular requirements for cytokinesis. Science 307, 1735–1739 [DOI] [PubMed] [Google Scholar]
- 20. Ivanov A. I., Bachar M., Babbin B. A., Adelstein R. S., Nusrat A., Parkos C. A. (2007) A unique role for nonmuscle myosin heavy chain IIA in regulation of epithelial apical junctions. PLoS One 2, e658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leal A., Endele S., Stengel C., Huehne K., Loetterle J., Barrantes R., Winterpacht A., Rautenstrauss B. (2003) A novel myosin heavy chain gene in human chromosome 19q13.3. Gene 312, 165–171 [DOI] [PubMed] [Google Scholar]
- 22. Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y., Kaibuchi K. (1996) Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271, 20246–20249 [DOI] [PubMed] [Google Scholar]
- 23. Hartshorne D. J., Ito M., Erdödi F. (2004) Role of protein phosphatase type 1 in contractile functions. Myosin phosphatase. J. Biol. Chem. 279, 37211–37214 [DOI] [PubMed] [Google Scholar]
- 24. Kamm K. E., Stull J. T. (2001) Dedicated myosin light chain kinases with diverse cellular functions. J. Biol. Chem. 276, 4527–4530 [DOI] [PubMed] [Google Scholar]
- 25. Matsumura F. (2005) Regulation of myosin II during cytokinesis in higher eukaryotes. Trends Cell Biol. 15, 371–377 [DOI] [PubMed] [Google Scholar]
- 26. Sandquist J. C., Swenson K. I., Demali K. A., Burridge K., Means A. R. (2006) Rho kinase differentially regulates phosphorylation of nonmuscle myosin II isoforms A and B during cell rounding and migration. J. Biol. Chem. 281, 35873–35883 [DOI] [PubMed] [Google Scholar]
- 27. Watanabe T., Hosoya H., Yonemura S. (2007) Regulation of myosin II dynamics by phosphorylation and dephosphorylation of its light chain in epithelial cells. Mol. Biol. Cell 18, 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adelstein R. S., Conti M. A. (1975) Phosphorylation of platelet myosin increases actin-activated myosin ATPase activity. Nature 256, 597–598 [DOI] [PubMed] [Google Scholar]
- 29. Heissler S. M., Manstein D. J. (2011) Comparative kinetic and functional characterization of the motor domains of human nonmuscle myosin-2C isoforms. J. Biol. Chem. 286, 21191–21202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jana S. S., Kawamoto S., Adelstein R. S. (2006) A specific isoform of nonmuscle myosin II-C is required for cytokinesis in a tumor cell line. J. Biol. Chem. 281, 24662–24670 [DOI] [PubMed] [Google Scholar]
- 31. Jana S. S., Kim K.-Y., Mao J., Kawamoto S., Sellers J. R., Adelstein R. S. (2009) An alternatively spliced isoform of non-muscle myosin II-C is not regulated by myosin light chain phosphorylation. J. Biol. Chem. 284, 11563–11571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma X., Jana S. S., Conti M. A., Kawamoto S., Claycomb W. C., Adelstein R. S. (2010) Ablation of nonmuscle myosin II-B and II-C reveals a role for nonmuscle myosin II in cardiac myocyte karyokinesis. Mol. Biol. Cell 21, 3952–3962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wylie S. R., Chantler P. D. (2008) Myosin IIC. A third molecular motor driving neuronal dynamics. Mol. Biol. Cell 19, 3956–3968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 35. Seidman K. J., Barsuk J. H., Johnson R. F., Weyhenmeyer J. A. (1996) Differentiation of NG108-15 neuroblastoma cells by serum starvation or dimethyl sulfoxide results in marked differences in angiotensin II receptor subtype expression. J. Neurochem. 66, 1011–1018 [DOI] [PubMed] [Google Scholar]
- 36. Kim J. H., Adelstein R. S. (2011) LPA1-induced migration requires nonmuscle myosin II light chain phosphorylation in breast cancer cells. J. Cell. Physiol. 226, 2881–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Lima A. D., Merten M. D., Voigt T. (1997) Neuritic differentiation and synaptogenesis in serum-free neuronal cultures of the rat cerebral cortex. J. Comp. Neurol. 382, 230–246 [DOI] [PubMed] [Google Scholar]
- 38. Chrzanowska-Wodnicka M., Burridge K. (1996) Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133, 1403–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arregui C. O., Carbonetto S., McKerracher L. (1994) Characterization of neural cell adhesion sites. Point contacts are the sites of interaction between integrins and the cytoskeleton in PC12 cells. J. Neurosci. 14, 6967–6977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shigeta M., Shibukawa Y., Ihara H., Miyoshi E., Taniguchi N., Gu J. (2006) β1,4-N-Acetylglucosaminyltransferase III potentiates β1 integrin-mediated neuritogenesis induced by serum deprivation in Neuro2a cells. Glycobiology 16, 564–571 [DOI] [PubMed] [Google Scholar]
- 41. Vicente-Manzanares M., Horwitz A. R. (2010) Myosin light chain mono- and diphosphorylation differentially regulate adhesion and polarity in migrating cells. Biochem. Biophys. Res. Commun. 402, 537–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dent E. W., Kwiatkowski A. V., Mebane L. M., Philippar U., Barzik M., Rubinson D. A., Gupton S., Van Veen J. E., Furman C., Zhang J., Alberts A. S., Mori S., Gertler F. B. (2007) Filopodia are required for cortical neurite initiation. Nat. Cell Biol. 9, 1347–1359 [DOI] [PubMed] [Google Scholar]
- 43. Jang K.-J., Kim M. S., Feltrin D., Jeon N. L., Suh K.-Y., Pertz O. (2010) Two distinct filopodia populations at the growth cone allow to sense nanotopographical extracellular matrix cues to guide neurite outgrowth. PLoS One 5, e15966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuo J.-C., Han X., Hsiao C.-T., Yates J. R., 3rd, Waterman C. M. (2011) Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 13, 383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vicente-Manzanares M., Ma X., Adelstein R. S., Horwitz A. R. (2009) Non-muscle myosin II takes center stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 10, 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kwiatkowski A. V., Rubinson D. A., Dent E. W., Edward van Veen J., Leslie J. D., Zhang J., Mebane L. M., Philippar U., Pinheiro E. M., Burds A. A., Bronson R. T., Mori S., Fässler R., Gertler F. B. (2007) Ena/VASP is required for neuritogenesis in the developing cortex. Neuron 56, 441–455 [DOI] [PubMed] [Google Scholar]
- 47. Adams J. C. (2004) Roles of fascin in cell adhesion and motility. Curr. Opin. Cell Biol. 16, 590–596 [DOI] [PubMed] [Google Scholar]
- 48. Schirenbeck A., Bretschneider T., Arasada R., Schleicher M., Faix J. (2005) The Diaphanous-related formin dDia2 is required for the formation and maintenance of filopodia. Nat. Cell Biol. 7, 619–625 [DOI] [PubMed] [Google Scholar]
- 49. Korobova F., Svitkina T. (2008) Arp2/3 complex is important for filopodia formation, growth cone motility, and neuritogenesis in neuronal cells. Mol. Biol. Cell 19, 1561–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Loudon R. P., Silver L. D., Yee H. F., Jr., Gallo G. (2006) RhoA-kinase and myosin II are required for the maintenance of growth cone polarity and guidance by nerve growth factor. J. Neurobiol. 66, 847–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang Q., Zhang X.-F., Pollard T. D., Forscher P. (2012) Arp2/3 complex-dependent actin networks constrain myosin II function in driving retrograde actin flow. J. Cell Biol. 197, 939–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rivera Rosado L. A., Horn T. A., McGrath S. C., Cotter R. J., Yang J. T. (2011) Association between α4 integrin cytoplasmic tail and non-muscle myosin IIA regulates cell migration. J. Cell Sci. 124, 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jing F., Mogi M., Sakata A., Iwanami J., Tsukuda K., Ohshima K., Min L.-J., Steckelings U. M., Unger T., Dahlöf B., Horiuchi M. (2012) Direct stimulation of angiotensin II type 2 receptor enhances spatial memory. J. Cereb. Blood Flow Metab. 32, 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Phillips M. I., de Oliveira E. M. (2008) Brain renin angiotensin in disease. J. Mol. Med. 86, 715–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vervoort V. S., Beachem M. A., Edwards P. S., Ladd S., Miller K. E., de Mollerat X., Clarkson K., DuPont B., Schwartz C. E., Stevenson R. E., Boyd E., Srivastava A. K. (2002) AGTR2 mutations in X-linked mental retardation. Science 296, 2401–2403 [DOI] [PubMed] [Google Scholar]
- 56. Wright J. W., Reichert J. R., Davis C. J., Harding J. W. (2002) Neural plasticity and the brain renin-angiotensin system. Neurosci. Biobehav. Rev. 26, 529–552 [DOI] [PubMed] [Google Scholar]