

Background: The dioxin receptor (AhR) regulates cell migration and has a role in TGFβ activation.
Results: AhR expression inhibits basal and TGFβ-induced epithelial-to-mesenchymal transition (EMT).
Conclusion: AhR has an intrinsic role in EMT and cross talks with TGFβ.
Significance: The involvement of AhR in EMT can help explain its functions in organ homeostasis and tumor progression.
Keywords: Aryl Hydrocarbon Receptor, Cell Migration, Epithelial to Mesenchymal Transition, Keratinocytes, Transforming Growth Factor β (TGFβ)
Abstract
Recent studies have emphasized the role of the dioxin receptor (AhR) in maintaining cell morphology, adhesion, and migration. These novel AhR functions depend on the cell phenotype, and although AhR expression maintains mesenchymal fibroblasts migration, it inhibits keratinocytes motility. These observations prompted us to investigate whether AhR modulates the epithelial-to-mesenchymal transition (EMT). For this, we have used primary AhR+/+ and AhR−/− keratinocytes and NMuMG cells engineered to knock down AhR levels (sh-AhR) or to express a constitutively active receptor (CA-AhR). Both AhR−/− keratinocytes and sh-AhR NMuMG cells had increased migration, reduced levels of epithelial markers E-cadherin and β-catenin, and increased expression of mesenchymal markers Snail, Slug/Snai2, vimentin, fibronectin, and α-smooth muscle actin. Consistently, AhR+/+ and CA-AhR NMuMG cells had reduced migration and enhanced expression of epithelial markers. AhR activation by the agonist FICZ (6-formylindolo[3,2-b]carbazole) inhibited NMuMG migration, whereas the antagonist α-naphthoflavone induced migration as did AhR knockdown. Exogenous TGFβ exacerbated the promigratory mesenchymal phenotype in both AhR-expressing and AhR-depleted cells, although the effects on the latter were more pronounced. Rescuing AhR expression in sh-AhR cells reduced Snail and Slug/Snai2 levels and cell migration and restored E-cadherin levels. Interference of AhR in human HaCaT cells further supported its role in EMT. Interestingly, co-immunoprecipitation and immunofluorescence assays showed that AhR associates in common protein complexes with E-cadherin and β-catenin, suggesting the implication of AhR in cell-cell adhesion. Thus, basal or TGFβ-induced AhR down-modulation could be relevant in the acquisition of a motile EMT phenotype in both normal and transformed epithelial cells.
Introduction
The aryl hydrocarbon receptor (AhR)3 is a basic-helix-loop-helix (bHLH) transcription factor (1) well known for its relevant role in xenobiotic-induced toxicity and carcinogenesis and, more recently, for its implication in different cellular processes including proliferation, differentiation, cell adhesion, and migration and organ homeostasis (2–6). Cell morphology, adhesion, and migration are essential cell properties requiring endogenous AhR activity. AhR exerts a differential effect on cell motility depending on the mesenchymal, epithelial, or endothelial phenotype of the target cell. Thus, immortalized and embryonic primary murine fibroblasts lacking AhR have spread morphology and impaired adhesion and migration (7–9). Contrary to mesenchymal fibroblasts, AhR knockout in epidermal keratinocytes increases their motility and migration both in vitro and in vivo (10). In additional cell types such as primary mouse endothelial cells (11) and CD4−CD8− thymocytes (4, 12), AhR activation promoted cell migration to newly formed blood vessels and to the spleen, respectively. The fact that AhR depletion increased primary keratinocytes migration and improved wound healing in vivo led us to suggest that AhR could be involved in the epithelial-to-mesenchymal transition (EMT).
EMT is a phenotypic switch that permanently or transiently converts epithelial cells into motile mesenchymal-like cells. During this process, epithelial cells suffer a spectrum of changes that affect their adhesion to neighboring cells and to the substratum, their migration, and their normal functioning (13). EMT is essential during embryonic development and in tissue repair, although a large body of evidence indicates that it also contributes to pathology (13–15). Because EMT enables epithelial cells with migration and invasion capabilities, it is generally accepted that it contributes to the early stages of tumor metastasis (15, 16). Among the EMT features that are conserved in most epithelial cell types are the repression of the adherents junctions protein E-cadherin (E-Cad), the up-regulation of mesenchymal markers vimentin, fibronectin, and N-cadherin (N-Cad), and the change toward a mesenchymal-like morphology (13, 17, 18). Several transcription factors promote EMT through the down-regulation of E-Cad (13, 15), and a central role has been given to members of the Snail family of proteins (Snail and Slug/Snai2), which repress E-Cad by binding to an E-box located in its promoter (19–21). Interestingly, Snail and Slug/Snai2 modulate common as well as specific gene regulatory pathways that likely differentiate their contribution to cancer progression and dissemination (22). An additional inducer of EMT is the extracellular cytokine transforming growth factor β (TGFβ), which can be produced and secreted by tumor cells or by the stroma. TGFβ induces EMT and cancer metastasis (23–25) possibly by promoting the early dissolution of the tight junctions that interconnect epithelial cells (26, 27).
AhR is functionally related to TGFβ in different cell types and in vivo. Several studies have shown that AhR-null fibroblasts have increased expression and activation of TGFβ that presumably contribute to their lower proliferation ability (28–31). In addition, TGFβ secreted by the stroma was able to increase the migration efficiency of primary AhR−/− keratinocytes in vitro and in vivo (10). Interestingly, TGFβ exerts cell type-specific effects on AhR by inhibiting receptor expression and activation in A549 lung cancer cells while enhancing receptor function in HepG2 hepatoma cells (32, 33). Thus, it is likely that AhR and TGFβ could cross-talk during EMT.
In this study we have investigated the role of AhR in EMT under both basal and TGFβ-induced conditions with the aim to determine whether or not AhR expression restrains the acquisition of a migratory EMT phenotype in epithelial cells. Thus, we have used primary keratinocytes from AhR+/+ and AhR−/− mice and NMuMG cells engineered to have either depleted or enhanced AhR activity to investigate the role of AhR in EMT. We have found that AhR knockdown by itself triggers an EMT-like phenotype characterized by changes in epithelial and mesenchymal markers and by increased cell migration. Moreover, AhR deficiency seems to cooperate with TGFβ to enhance the EMT process. These results support a mechanism by which AhR is a negative regulator of EMT and suggest that metastasis of TGFβ-responsive tumors could be exacerbated if they undergo AhR down-modulation.
EXPERIMENTAL PROCEDURES
Primary Keratinocytes and Cell Lines
Primary keratinocytes were obtained from newborn AhR+/+ and AhR−/− mice at 2–3 days of age. Pups were sequentially washed in povidone solution, sterile water, and 70% ethanol in PBS. Legs and tail were removed, and the complete skin was dissected using forceps. The skins were floated dermis side down in 0.25% trypsin for 16–18 h at 4 °C. Next, the epidermis was separated from the dermis and minced in 2–3 ml/mouse of plating medium (minimum essential medium Eagle containing gentamycin and 4% FBS pretreated with Chelex and 0.2 mm Ca2+). Tissues were digested by gentle incubation for 45 min at 4 °C and filtered through a 140-μm mesh to remove aggregates and undigested material. The resulting cell suspension was seeded at 2 × 106 cells in 60-mm culture plates pretreated with 5 μg/ml collagen or 15 μg/ml fibronectin. After 24 h, keratinocytes were washed with PBS and grown in CnT-0.7 culture medium (CELLnTEC) to promote proliferation and to inhibit differentiation. NMuMG mouse epithelial cells were grown in DMEM containing 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mm l-glutamine, and 10 μg/ml insulin at 37 °C in a 5% CO2 atmosphere. Human HaCaT and Phoenix packing cells were cultured in the above medium without added insulin at 37 °C and 5% CO2 atmosphere. Cell lines were trypsinized and passaged at 1:3 dilution when they reached 70–80% confluence.
Antibodies, Reagents, Expression Vectors, and Treatments
Antibodies against the following proteins were used: β-catenin (BD Biosciences); E-cadherin (Calbiochem); AhR (Immunostep and Biomol); TGFβ (R&D Systems); p-Smad2 (Cell Signaling); Slug/Snai2 (Santa Cruz); N-cadherin (Invitrogen); fibronectin (Chemicon), vimentin, α-smooth muscle actin, and β-actin (Sigma). The AhR agonist 6-formylindolo[3,2-b]carbazole (FICZ) was from Enzo, and the AhR antagonist α-naphthoflavone (α-naph) was from Sigma. The pharmacological inhibitor of the TGFβ pathway SB431542 was from Selleckchem. Rhodamine-phalloidin was from Invitrogen. Matrigel solution was from BD Biosciences. TaqDNA polymerase was from Ecogen. iScript reverse transcription supermix and SYBR Green master mix were obtained from Bio-Rad. Small hairpin RNA was from Sigma. Small interfering RNA for AhR and scrambled siRNA were synthesized by Dharmacon. The constitutively active form of the AhR (CA-AhR) was produced from the wild type mouse receptor by deleting the minimal PAS-B motif (amino acids 288–421) without altering the N-terminal half of the binding domain (PAS-A). This constitutively active receptor heterodimerizes with ARNT and has intrinsic transcriptional activity in a ligand-independent manner (34). Recombinant human TGFβ (Sigma) was added to the cultures at 10 ng/ml (primary keratinocytes and HaCaT cells) or 5 ng/ml (NMuMG cells). Control cultures were treated with the same volume of solvent (PBS).
Retroviral Transduction
NMuMG cells were stably transduced with expression vectors containing a small hairpin RNA for AhR (sh-AhR) or a constitutively active form of the protein (CA-AhR) as described (Stanford University Medical Center). In brief, constructs LMP-sh-AhR, pBABE-CA-AhR, or the empty vectors pBABE+LMP were transfected by calcium phosphate precipitation in Phoenix cells, and virus production was allowed for 48 h. NMuMG cells were exposed overnight to the viral supernatants, and 48 h later selection was started with 1 μg/ml puromycin for 14 days. Individual clones surviving selection were isolated by cell sorting and then analyzed for AhR expression by immunoblotting or for the Cyp1a1 AhR target gene by qRT-PCR.
Transient Transfection and RNA Interference
NMuMG cells were transiently transfected using the Turbofect reagent (Fermentas). Briefly, the DNA was incubated for 15 min at room temperature with Turbofect, and the mix was added to the cells in complete medium. After 24 h-48 h of transfection, cultures were processed and analyzed. HaCaT cells were transfected with small interfering RNAs for AhR (si-AhR) or scrambled siRNA by nucleoporation using a MicroPorator MP-100 (Digital-Bio). In brief, cells were trypsinized, washed, and resuspended in transfection solution. si-AhR or scrambled siRNA were added at 50 nm to the cells, and nucleofection was performed with a single pulse of 1400 V for 30 ms. After 24 h, cells were transferred to fresh complete medium, and culture was continued for an additional 48 h.
Cellular Morphology
Cell area and circularity of AhR+/+ and AhR−/− keratinocytes and NMuMG cells were determined by blinded analysis in different random fields for each culture as indicated (7). Cellular contour and axis were analyzed using the ImageJ software (Version 1.45S). Significance of the data was analyzed as indicated below (“Statistical Analyses”).
Trans-epithelial Resistance (TER)
Cells seeded on transwell filter inserts (BD Biosciences) of 0.4-μm pore size were allowed to reach confluence. A volume of 2 ml of medium was added to the inner insert chamber and 4 ml to the outer chamber, with daily replacement of fresh medium. TER was measured every day using the Millicell Electrical Resistance System according to the manufacturer's instructions.
Clonogenic Assays
Clonogenic assays in two dimensions (2-D) cultures were done by platting 5 × 102 or 103 sh-AhR or CA-AhR NMuMG cells in plain tissue culture dishes. Empty vector-transduced (EV) NMuMG cells were used as controls. After 5 days of culture, medium was removed, and clones were washed in PBS, stained for 10 min with 0.5% (w/v) crystal violet, and counted. Clone formation in three dimensions (3-D) was analyzed by growing each cell line on Matrigel as described (11). Briefly, 5 × 103 sh-AhR, CA-AhR, or EV NMuMG cells were seeded on Matrigel plugs for 3 days in complete medium. Clones formed were counted and analyzed for cell spreading using the ImageJ software (Version 1.45S).
Cell Migration
Cell migration was analyzed using wound healing experiments as previously reported (9, 10).
Immunofluorescence and Rhodamine-phalloidin
Fluorescence analysis was done using a Fluoview 1000 confocal microscope (Olympus) on cultures fixed for 20 min at room temperature in 4% paraformaldehyde (Polysciences, Inc). Secondary antibodies labeled with Alexa 488 for E-cadherin (rat monoclonal ECCd2), β-catenin (mouse monoclonal), Snail (mouse monoclonal), and Slug/Snai2 (goat polyclonal) or Alexa 633 for AhR (rabbit polyclonal) were used. DAPI staining served to label cell nuclei. Cells stained with only secondary antibody were used as negative controls. To label actin stress fibers, cells were also incubated with rhodamine-phalloidin as indicated (9). Fluorescence distribution analysis was performed using the FV1000 software (Olympus).
Immunoprecipitation and Immunoblotting
Immunoprecipitation from NMuMG cells was performed essentially as described (35, 36). Briefly, cells were lysed, and 1 mg of total cellular protein was precleared by incubation with protein A/G plus-agarose beads. Extracts were then mixed with 2 μg of the corresponding specific antibody and fresh protein A/G-Sepharose beads. After overnight incubation at 4 °C, beads were washed twice with buffer A (20 mm Tris-HCl pH 7.4, 50 mm NaCl, 1% Nonidet P-40, 10 mm EDTA, 1 mm sodium orthovanadate, 50 mm NaF, 0.5 mm PMSF, and 4 mg/ml Complete protease inhibitor mixture) and buffer B (25 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 1 mm EDTA). Washed beads were then used for immunoblotting. SDS-PAGE and immunoblotting were performed as described (9, 31).
Reverse Transcription and Real-time PCR
Total RNA was isolated from primary keratinocytes, NMuMG, and HaCaT cells using the RNeasy kit (Qiagen). Reverse transcription was performed using random hexamers and iScript reverse transcription super mix (Bio-Rad). Real-time PCR was done to quantify the levels of human and mouse Snail and Slug/Snai2 and of mouse Cyp1a1 mRNAs. Gapdh mRNA was used to normalize individual gene expression (ΔCt). Reactions were done using SYBR Green I/QTaq DNA polymerase mix on an iCycler equipment (Bio-Rad) as described (37). 2−ΔΔCt was used to calculate Gapdh-normalized gene expression with respect to the control or untreated conditions. Primer sequences used in this study are indicated in supplemental Table 1.
Statistical Analyses
Data are shown as the mean ± S.D. Statistical comparison between experimental conditions was done using GraphPad Prism 4.0 software (GraphPad). Student's t test or analysis of variance were applied. Experiments were done in duplicate or triplicate in two or three cultures of each cell line.
RESULTS
AhR Knockdown Induces EMT under Basal Cell Conditions
To analyze the role of AhR in EMT under normal cellular conditions, we have used primary keratinocytes from AhR+/+ and AhR−/− mouse pups and NMuMG epithelial cells engineered by retroviral transduction to encode a small hairpin RNA for AhR (sh-AhR) or a constitutively active AhR receptor (CA-AhR). NMuMG cells we also transduced with an empty retrovirus to account for the basal AhR expression. After antibiotic selection, clones carrying each of those constructs were isolated. Immunoblotting analysis with an AhR-specific antibody revealed a marked knockdown of the endogenous AhR expression in the sh-AhR cell line (supplemental Fig. S1A). Constitutively active AhR was ectopically expressed in CA-AhR cells, and it was functional as determined by the increase in mRNA expression observed for its canonical target gene Cyp1a1 (supplemental Fig. S1B).
EMT is generally followed by a change in morphology toward a mesenchymal-like phenotype in parallel with a switch in the pattern of expression of cell adhesion molecules and their regulators. AhR depletion significantly reduced the circularity of NMuMG cells, inducing a more elongated morphology, whereas AhR over-activation further stressed their circular form (Fig. 1A). AhR has a role in that phenotype as primary keratinocytes from AhR-null mice were significantly more elongated than their AhR+/+ counterparts (Fig. 1B). Mesenchymal cells usually have more relaxed cell-cell interactions than epithelial cells when grown at high confluence, thus producing less compact monolayers. The TER of sh-AhR confluent cultures was significantly lower than that of wild type (EV) or CA-AhR NMuMG cultures (Fig. 1C), supporting that reduction of AhR levels in epithelial cells favors a more mesenchymal phenotype.
FIGURE 1.

AhR knockdown alters the morphology of NMuMG cells and of primary mouse keratinocytes. A, wild type (EV), sh-AhR, and CA-AhR cell lines were grown in plain tissue culture dishes, and their morphology was determined by measuring the circularity factor (4π × area/perimeter2). n.s., not significant. B, cell morphology of wild type (AhR+/+) and AhR-null (AhR−/−) primary keratinocytes was also determined as indicated above. C, TER of each NMuMG cell line was measured in confluent cultures grown in transwell filter inserts as indicated under “Experimental Procedures.” Determinations were done in triplicate in at least three independent cultures of each cell line. Data are shown as mean ± S.D. Bar, 200 μm.
The acquisition of an EMT phenotype implies a reduction in the expression of epithelial markers such as E-Cad and a gain in mesenchymal molecules like fibronectin, N-cadherin, and vimentin. sh-AhR NMuMG cells had a significant reduction in E-Cad and β-Cat protein levels and an increase in fibronectin (Fig. 2A). CA-AhR cells, on the contrary, increased their E-Cad protein content by about 3-fold and reduced vimentin and fibronectin expression (Fig. 2A). Immunofluorescence experiments showed that AhR depletion reduced the cellular content of E-Cad and β-Cat and altered their location at the plasma membrane while increasing the levels of F-actin fibers, suggesting that low AhR expression could destabilize cell-cell interactions in epithelial cells (Fig. 2C). Oppositely, CA-AhR cells concentrated E-Cad and β-Cat at the plasma membrane and had much fewer prominent F-actin fibers (Fig. 2C). Primary AhR−/− keratinocytes had marked reductions in E-Cad and β-Cat and higher contents of vimentin and α-smooth muscle actin as compared with AhR+/+ cells (Fig. 2B), confirming that AhR down-modulation triggers phenotypical changes indicative of EMT. TGFβ is considered a potent EMT inducer in different cell types (24, 25), and we have previously shown that AhR negatively regulates TGFβ activation (29–31). Protein expression analysis revealed that AhR depletion increased TGFβ content in sh-AhR NMuMG cells by close to 2-fold without a significant effect in AhR−/− keratinocytes (Fig. 2, A and B). Yet both cell types had a significant increase in the activation of the TGFβ pathway based on the phosphorylation of pSmad2 (Fig. 2, A and B), indicating that TGFβ-dependent signaling could mediate the pro-EMT effects induced by reduced AhR expression. To further analyze this possibility, we treated sh-AhR NMuMG cells with the TGFβ-RI inhibitor SB431542. SB431542 was effective in blocking TGFβ-dependent signaling in this cell line, as determined by a significant reduction in Smad2 phosphorylation (Fig. 2D). SB431542 increased E-Cad expression (Fig. 2D), suggesting that enhanced TGFβ signaling could contribute to the EMT-like features observed under absent or reduced AhR expression.
FIGURE 2.
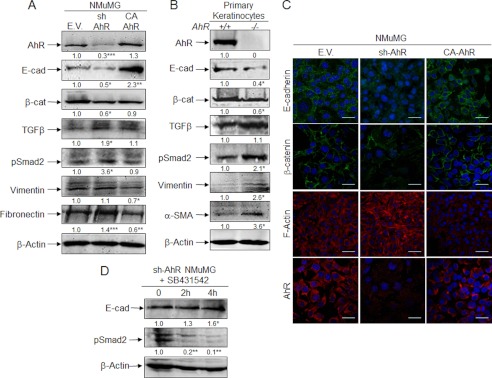
AhR depletion alters the pattern of EMT molecular markers in NMuMG cell lines and in primary keratinocytes. A, total cell extracts from wild type (EV), sh-AhR, and CA-AhR NMuMG cells were analyzed for the expression of relevant EMT markers by immunoblotting. B, total cell extracts from AhR+/+ and AhR−/− primary keratinocytes were also analyzed for EMT markers as above. The expression of β-actin was used to normalize protein levels. C, the expression patterns of E-cad, β-cat, and F-actin were also studied by confocal microscopy in NMuMG cell lines. Cell nuclei were stained with DAPI. D, sh-AhR NMuMG cells were treated with 10 μm TGFβ-RI inhibitor SB431542, and the expression of E-Cad and pSmad2 was analyzed by immunoblotting. β-Actin was used to normalize protein levels. Experiments were done in duplicate in at least three independent cultures. Numbers under the figures stand for the expression levels relative to the wild type condition (EV or AhR+/+). Bar, 50 μm. Statistical significance: *, p < 0.05; **, p < 0.001; ***, p < 0.0001.
Because E-Cad is regulated by transcription factors Snail and Slug/Snai2 (20, 21), we next investigated if their expression was AhR-dependent. As shown in Fig. 3A, AhR knockdown significantly increased Slug/Snai2 mRNA expression in NMuMG cells (upper panel) and in primary keratinocytes (lower panel); Snail expression was largely increased in primary cells (lower panel) but only marginally in NMuMG cells (upper panel). Consistently, constitutive AhR activation diminished Slug/Snai2 and Snail mRNA levels in NMuMG cells (Fig. 3A). Immunocytochemical analysis showed that Slug/Snai2 and Snail were barely detectable in wild type and CA-AhR NMuMG cells but significantly expressed in sh-AhR cells (Fig. 3B). These data support that AhR represses Slug/Snai2 and Snail and maintains the expression and localization of E-Cad at the plasma membrane.
FIGURE 3.
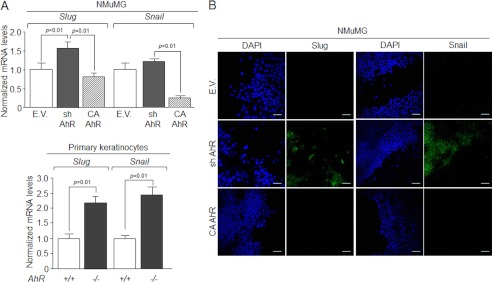
AhR modulates the expression of EMT regulators Snail and Slug/Snai2. A, Snail and Slug/Snai2 mRNA expression was determined by qRT-PCR in total RNA isolated from wild type (EV), sh-AhR, and CA-AhR NMuMG cell lines (upper) or primary AhR+/+ and AhR−/− keratinocytes (lower). mRNA levels were normalized by the expression of Gapdh and referred to the expression of control cells (EV or AhR+/+). B, the expression patterns of Snail and Slug/Snai2 proteins were analyzed by confocal microscopy in NMuMG cell lines. Cell nuclei were stained with DAPI. The experiment was done in triplicate in at least three cultures of each genotype. Data are shown as the mean ± S.D. Bar, 100 μm.
We then explored if the mesenchymal-like status of sh-AhR NMuMG cells affected their clonogenic and migratory potentials. Experiments in two dimensions (2-D) cultures revealed that AhR knockdown produced a larger number of clones that were significantly more spread and that contained mesenchymal-like cells readily moving out from the periphery (Fig. 4A). sh-AhR NMuMG cells grown in three dimensions (3-D) cultures also formed more and larger clones than wild type (EV) and CA-AhR cells (Fig. 4B). The EMT generally induces a pro-migratory phenotype (15) that can be associated to the clonogenic potential of the cell. Wound healing experiments indicated that sh-AhR NMuMG cells had a moderate increase in migration with respect to wild type (EV) cells, unlike CA-AhR cells, which had a significantly reduced motility as compared with both EV and sh-AhR cells (Fig. 5A). Likewise, primary AhR−/− keratinocytes had increased migration rates with respect to AhR+/+ cells (Fig. 5B). Thus, the EMT process induced by AhR depletion concurs with enhanced migration and increased clonogenic potential.
FIGURE 4.

AhR down-modulation in NMuMG cells increases their clonogenicity. A, wild type (EV), sh-AhR, and CA-AhR NMuMG cell lines were grown in plain tissue culture dishes at low density (2-D), and the clones formed were counted and analyzed for their morphology and spreading. B, the clonogenicity and invasive potential of NMuMG cell lines were also analyzed in Matrigel-treated culture dishes (3-D). Clone number was determined using the ImageJ software (Version 1.45S). The experiments were done in duplicate in three cultures of each genotype. Data are shown as the mean ± S.D. Bar, 200 μm.
FIGURE 5.

AhR expression modulates cell migration in epithelial cells. Wild type (EV), sh-AhR, and CA-AhR NMuMG cells (A) or primary AhR+/+ and AhR−/− keratinocytes (B) were grown to confluence, and their ability to migrate was analyzed in wound healing assays after 48 h. The experiments were done in triplicate in three independent cultures of each genotype. Data are shown as the mean ± S.D. Bar, 200 μm. A.U., arbitrary units.
Pharmacological Modulation of AhR Affects EMT Markers and Epithelial Cell Migration
These results suggested that non-toxic (e.g. potentially endogenous) molecules known to regulate AhR could be potential EMT modulators. To address this issue, we have investigated the effects of the AhR antagonist α-naphthoflavone (38) and of the potential AhR endogenous ligand derived from tryptophan FICZ (39, 40) on relevant EMT parameters (Fig. 6). FICZ was an efficient AhR agonist in NMuMG cells markedly inducing Cyp1a1 mRNA expression (Fig. 6A). Although its effects on fibronectin protein levels were only slight, FICZ significantly increased E-Cad levels (Fig. 6B) and inhibited cell migration in wound healing assays (Fig. 6C). Treatment of NMuMG cells with the antagonist α-naph, on the contrary, reduced AhR levels and increased fibronectin expression while diminishing E-Cad amounts (Fig. 6D). Regarding its effects on migration, α-naph increased the ability of NMuMG cells to close wounds and to spread out at the margin of the wounds (Fig. 6E and insets). These results support our hypothesis that increasing AhR activity by an endogenous ligand inhibits EMT-like processes, whereas blockade of AhR enhances EMT properties in epithelial cells.
FIGURE 6.

Pharmacological modulation of AhR alters EMT markers and epithelial cell migration. A, wild type NMuMG cells were treated with the AhR endogenous agonist FICZ (5 nm) for 16 h, and Cyp1a1 mRNA expression was analyzed by real-time RT-PCR. The expression of Gapdh was used to normalize gene expression. B, protein expression of fibronectin and E-Cad was analyzed at different times of FICZ treatment by immunoblotting. β-Actin was used as normalization control. C, cells were treated with FICZ for 48 h and then used to analyze migration in wound healing assays. A.U., arbitrary units. D, wild type NMuMG cells were exposed to the AhR antagonist α-naph (50 μm) for the indicated times. Protein levels of fibronectin, E-Cad, and AhR were determined by immunoblotting. β-Actin was used as normalization control. E, cell migration in wound healing assays was also analyzed in cells treated with α-naph for 48 h. Inset, higher magnification of cells at the leading edge of migration. The experiments were done in duplicate in three independent cultures of each genotype. Data are shown as the mean ± S.D. Bar, 200 μm. Statistical significance: *, p < 0.05; **, p < 0.001; ***, p < 0.0001.
AhR Knockdown Has Synergistic Effects with TGFβ in EMT
Considering the results above, we decided to address how knockdown or constitutive activation of AhR affected cell response to exogenous TGFβ. In wild type NMuMG cells, TGFβ induced a mesenchymal-like morphology that was exacerbated in sh-AhR but not in CA-AhR cells (Fig. 7A). Immunoblot analysis of EMT markers confirmed the lower basal levels of E-Cad and the higher amounts of fibronectin in sh-AhR cells (Fig. 7, B–D). TGFβ treatment reduced E-Cad to almost undetectable levels in sh-AhR cells, although a significant reduction was also observed in CA-AhR cells (Fig. 7, B and D). Fibronectin content was enhanced by 48 and 72 h of TGFβ treatment in all cell lines (Fig. 7, B and C). Interestingly, TGFβ reduced AhR protein levels in all three cell lines, particularly in EV and CA-AhR cells (Fig. 7E). Primary keratinocytes from AhR-null mice treated with TGFβ grew as more elongated cells with loosen cell-cell interactions (Fig. 7F). E-Cad protein levels were adversely affected by TGFβ in both primary cell lines, reaching very low levels in AhR−/− cells (Fig. 7, G and I). α-Smooth muscle actin was overexpressed in AhR−/− cells and remained unchanged after TGFβ treatment (Fig. 7G); fibronectin expression, on the other hand, increased upon TGFβ addition in AhR-null keratinocytes (Fig. 7, G and H). Similarly to NMuMG cells, TGFβ also reduced AhR protein amounts in AhR+/+ cells (Fig. 7, G and J). Confocal microscopy was used to analyze by immunocytochemistry the pattern of EMT regulators in NMuMG cells and in primary keratinocytes exposed to TGFβ (Fig. 8). E-Cad staining was reduced and delocalized from the plasma membrane in sh-AhR NMuMG cells, and such a pattern was stressed by 48 h treatment with TGFβ (Fig. 8A). The well defined membrane location of E-Cad was disturbed by the cytokine in wild type (EV) and CA-AhR cells, although to a lesser degree than in AhR-depleted cells. F-actin was enhanced by TGFβ to a similar extent in each cell line despite its higher basal levels in sh-AhR cells (Fig. 8A). Primary keratinocytes responded alike to TGFβ, delocalizing E-Cad from the plasma membrane and increasing F-actin fibers content, although as for NMuMG cells, both effects were more apparent in AhR−/− than in AhR+/+ cells (Fig. 8B). We subsequently examined how AhR expression modulates the effects of TGFβ on Snail and Slug/Snai2. In agreement with the effects seen on E-Cad, TGFβ increased Snail mRNA levels in sh-AhR, wild type (EV) and CA-AhR NMuMG cells (Fig. 9A, left panel). Despite its higher basal levels in sh-AhR cells, Slug/Snai2 was not significantly induced by TGFβ treatment in either cell line (Fig. 9A, right panel). Similar results were also found for primary keratinocytes, in which TGFβ increased their Snail mRNA levels (with a delayed kinetics in AhR−/− with respect to AhR+/+ cells) without significantly affecting Slug/Snai2 mRNA (Fig. 9B). Wound healing assays revealed no significant differences in migration between NMuMG cell lines by TGFβ treatment (Fig. 10A), whereas TGFβ markedly enhanced migration rates in AhR knockout primary keratinocytes with respect to wild type cells (Fig. 10B). Altogether, these results suggest that AhR expression counteracts the EMT-like phenotype induced by TGFβ.
FIGURE 7.

NMuMG cell lines and primary keratinocytes respond to TGFβ-induced EMT. Wild type (EV), sh-AhR, and CA-AhR NMuMG cell lines (A) or primary AhR+/+ and AhR−/− keratinocytes (F) were left untreated (control) or treated with TGFβ for 48 h or 72 h, and their morphology was analyzed. Protein expression for molecular EMT markers was analyzed in NMuMG cell lines (B) or primary keratinocyte cultures (G) exposed to TGFβ. Quantification of E-Cad (D and I), fibronectin (C and H) and AhR (E and J) protein expression in NMuMG cells and primary keratinocytes is shown, respectively. The expression of β-actin was used to normalize protein levels. Determinations were done in duplicate of two different cultures. Data are shown as the mean ± S.D. Bar, 100 μm.
FIGURE 8.

TGFβ alters the pattern of E-cad and F-actin in NMuMG cell lines and primary keratinocytes. Wild type (EV), sh-AhR, and CA-AhR NMuMG cell lines (A) or primary AhR+/+ and AhR−/− keratinocytes (B) were left untreated (control) or treated with TGFβ for 48 h, and the expression patterns of E-cad and F-actin were analyzed by confocal microscopy. Duplicate determinations were performed in three different cultures of each genotype. Bar, 50 μm.
FIGURE 9.

TGFβ increases the expression of Snail and Slug/Snai2 in NMuMG cell lines and primary keratinocytes. Wild type (EV), sh-AhR, and CA-AhR NMuMG cell lines (A) or primary AhR+/+ and AhR−/− keratinocytes (B) were left untreated (control) or treated with TGFβ for 48 h or 72 h, and the mRNA expression of Snail and Slug/Snai2 was quantified by qRT-PCR. mRNA levels were normalized by the expression of Gapdh and referred to the expression of untreated control cells (EV or AhR+/+). Three different cultures of each genotype were analyzed in triplicate experiments. Data are shown as the mean ± S.D.
FIGURE 10.

Effect of TGFβ on the migratory potential of NMuMG cell lines and primary keratinocytes. Wound healing experiments were performed in wild type (EV), sh-AhR, and CA-AhR cell lines (A) or in primary AhR+/+ and AhR−/− keratinocytes (B) treated with TGFβ for 48 h. Cell migration was quantified using the ImageJ software (Version 1.45S). The experiments were done in triplicate in at least three different cultures of each genotype (three wounds per experiment). Data are shown as the mean ± S.D. Bar, 200 μm. A.U., arbitrary units.
The antagonism between TGFβ and AhR in EMT was also investigated in the human HaCaT cell line. TGFβ altered EMT markers, promoting a mesenchymal-like morphology with a reduction in E-Cad levels and an increase in fibronectin and F-actin contents (Fig. 11A). Interestingly, TGFβ reduced AhR levels in HaCaT cells (Fig. 11A, right panel), which is consistent with the effects found in NMuMG and primary keratinocytes. TGFβ increased Snail and Slug/Snai2 mRNA levels in basal HaCaT cells, whereas transient AhR knockdown (si-AhR) slightly enhanced Snail without a significant change in Slug/Snai2 expression (Fig. 11B). TGFβ treatment of si-AhR cells markedly increased both Snail and Slug/Snai2 mRNAs (Fig. 11B). Because TGFβ reduced AhR levels in HaCaT cells (Fig. 11A, right panel), it is likely that the cytokine acts synergistically with AhR knockdown in inducing EMT. Indeed, AhR depletion by si-AhR not only reduced E-Cad protein levels in basal HaCaT cells but also enhanced the effects of TGFβ (Fig. 11C).
FIGURE 11.

TGFβ induces EMT in HaCaT cells and alters AhR expression. A, human HaCaT cells were left untreated or treated with TGFβ for 48 h or 72 h, and the expression of E-cad and F-actin was determined by confocal microscopy (left panel). Protein expression of E-cad, fibronectin, and AhR was also determined by immunoblotting using specific antibodies (right panel). B, the effects of TGFβ, a siRNA against AhR (si-AhR), or both on Snail and Slug/Snai2 mRNA expression were analyzed by qRT-PCR. mRNA levels were normalized by the expression of Gapdh and referred to the expression of control cells (EV). C, the effects of TGFβ on the expression of EMT markers in the absence or presence of a si-AhR were also analyzed by immunoblotting. The expression of β-actin was used to normalize protein levels. Three experiments were performed in at least three independent HaCaT cultures. Data are shown as mean ± S.D. Bar, 50 μm. Statistical significance: * p < 0.05, **p < 0.001, ***p < 0.0001.
The causal role of AhR in EMT was analyzed by rescue experiments in which the CA-AhR construct was transfected in sh-AhR NMuMG cells (Fig. 12). CA-AhR efficiently restored AhR expression and activity in sh-AhR cells as determined by immunoblotting and qRT-PCR for the canonical target gene Cyp1a1 (Fig. 12A). Although under these conditions E-Cad was only slightly modified, CA-AhR significantly reduced fibronectin levels (Fig. 12B) and Snail and Slug/Snai2 mRNA expression (Fig. 12C). The apparent mild increase in E-Cad observed under Snail and Slug/Snai2 repression in CA-AhR-rescued cells could derive from slow accumulation kinetics not fully addressed in the 48-h time frame of the experiment. Yet in vitro wound healing showed that CA-AhR was able to reduce the migration potential of sh-AhR cells (Fig. 12D). Therefore, AhR re-expression can partially reestablish EMT markers and impair the migration of cells undergoing EMT by AhR down-modulation.
FIGURE 12.

Rescue of AhR expression in AhR knockdown NMuMG cells reduces the EMT markers and inhibits cell migration. sh-AhR NMuMG cells were transiently transfected with the CA-AhR construct (A), and the expression of E-cad and fibronectin was analyzed by immunoblotting (B). The expression of β-actin was used to normalize protein levels. C, the effects of CA-AhR on the mRNA levels of Snail and Slug/Snai2 in sh-AhR NMuMG cells were analyzed by qRT-PCR. mRNA levels were normalized by the expression of Gapdh and referred to the expression of control cells (sh-AhR). D, sh-AhR NMuMG cells transfected with CA-AhR were treated with TGFβ and studied for cell migration using would healing assays. The experiments were done in duplicate in at least three independent transfections. Data are shown as the mean ± S.D. Bar, 200 μm. Statistical significance: **, p < 0.001. A.U., arbitrary units.
AhR Associates to E-Cad and β-Cat in NMuMG Epithelial Cells
The mechanisms by which AhR participates in signaling pathways controlling EMT are practically unknown. One possibility is that AhR interacts with the function of molecules maintaining cell-cell adhesions such as E-Cad and β-Cat. Co-immunoprecipitation experiments for AhR showed that it was associated in a common protein complex with both E-Cad and β-Cat in wild type (EV) and CA-AhR NMuMG cells (Fig. 13A). Consistently, E-Cad was also able to co-immunoprecipitate with AhR in both cell lines (Fig. 13B). For β-Cat, co-immunoprecipitation with AhR was more evident in CA-AhR than in wild type (EV) cells (Fig. 13C), although the presence of AhR in the immunoprecipitates was reproducibly observed in the latter. The plausible association of AhR in a common protein complex with E-Cad and β-Cat gained additional support by confocal microscopy analysis. As shown in Fig. 13D, AhR co-localized with both E-Cad and β-Cat at the periphery of NMuMG cells (yellow color in merge panels). Thus, one likely mechanism for AhR to modulate EMT is through interaction with proteins preserving epithelial integrity and cell-cell interactions. In addition, the fact that the constitutively active AhR did not show an increased association to E-Cad suggests that AhR could modulate additional signaling pathways unrelated to its intrinsic transcriptional activity.
FIGURE 13.

AhR associates in a common protein complex with E-cad and β-cat in NMuMG cells. A, AhR was immunoprecipitated (IP) in wild type (EV), sh-AhR, and CA-AhR NMuMG cell lines, and the amounts of associated E-cad and β-cat were determined by immunoblotting using specific antibodies. AhR association to E-cad (B) and β-cat (C) was also determined in E-cad and β-cat immunoprecipitates, respectively. Total cell lysates are shown on the right side of panels A, B, and C. D, the pattern of association of AhR to E-cad and β-cat was analyzed by confocal microscopy. Merge for each pair of proteins is shown on the right panel. Determinations were done in triplicate in three independent cultures of each genotype. Bar, 30 μm.
DISCUSSION
The interest in understanding the physiological functions of AhR has significantly increased as recent studies have anticipated an important and perhaps causal role of this receptor in reproduction and organ homeostasis (6). In addition to the control of cell cycle and cell proliferation (28, 41, 42), cell adhesion and migration are recognized as important cellular functions requiring AhR activity (43). This assumption is partially based on in vitro studies using MCF-7 breast tumor cells (44–46), thymocytes (4, 12, 47), and hematopoietic stem cells (48) treated with the potent exogenous AhR ligand dioxin. AhR is also involved in cell adhesion and migration under physiological conditions through cell type-specific mechanisms (6, 49). An important observation is that although the lack of AhR alters the morphology and compromises the adhesion and migration of mesenchymal fibroblasts (7, 9), its deficiency enhances keratinocyte motility and epithelial wound healing in vivo (10). From these studies it can be proposed that the variations in AhR levels, which presumably take place during development and disease, might contribute to altered cell migration. Closely related to such a scenario is the epithelial-to-mesenchymal transition that provides epithelial cells with some, but generally not all, mesenchymal characteristics of a migratory cell (13, 15, 50).
In this study we have decided to investigate if the cellular levels of AhR are enough to block EMT and if this receptor antagonizes the effects of TGFβ. The latter hypothesis is based on previous studies revealing a cross-regulation between AhR and TGFβ in epithelial cells (10, 32) and mesenchymal fibroblasts (28, 30, 31).
Cell morphology is closely related and, in a large part dependent, on cell-cell and cell-substratum interactions (51). As a result, changes in cell attachment largely influence cell migration and can be relevant in processes such as EMT (52). AhR expression affects epithelial cell morphology as receptor depletion in NMuMG cells induced a mesenchymal-like phenotype that was AhR-dependent because it was also observed in primary AhR−/− keratinocytes but not in NMuMG cells having a constitutively active receptor. The mesenchymal-like morphology of AhR knockout/knockdown cells apparently reduced the strength of their cell-cell interactions as revealed by their lower trans-epithelial resistance when grown at confluence. Based on these results, we considered the possibility that AhR down-modulation could alter the expression of proteins maintaining epithelial identity. In AhR-interfered NMuMG cells and primary AhR-null keratinocytes, epithelial markers E-Cad and β-Cat were significantly reduced, whereas mesenchymal markers fibronectin, vimentin, α-smooth muscle actin, and F-actin were increased, suggesting that AhR expression could impair EMT under normal cellular conditions and in the absence of exogenous stimuli. Additional sets of data strengthen this conclusion. First, a constitutively active CA-AhR was able to rescue epithelial markers and to inhibit cell migration in AhR-depleted cells, showing that AhR has a causal role in the EMT phenotype. Second, AhR appears to be associated to and to co-localize with E-Cad and β-Cat in the periphery of NMuMG cells, indicating that AhR deficiency could enhance EMT-dependent cell migration through altered cell-cell adhesion. In agreement with these results, early studies showed that AhR becomes induced in 10T1/2 fibroblasts after de-assembling of cell-cell contacts (53). Finally, the increased migration of AhR knockdown cells is consistent with the more efficient wound healing found in AhR-null mice due to increased keratinocyte migration (10). Importantly, the fact that AhR inhibition by the antagonist α-naph switched E-Cad, fibronectin, and migration to a pattern similar to that of both AhR−/− and sh-NMuMG cells, strongly support that AhR depletion favors an EMT-like phenotype. Moreover, the effects induced by the potential endogenous AhR ligand FICZ not only indicate that AhR expression could impair EMT but also offer the possibility to block EMT by pharmacological modulation of AhR activity.
Interestingly, although AhR knockout/knockdown homogeneously affects most EMT markers, constitutive AhR activation produces a more variable phenotype that significantly alters such markers as E-Cad and fibronectin but that does not significantly influence cell morphology, TER, or β-Cat levels. Although more extensive studies need to be done, it is probable that AhR functionally interacts with different signaling pathways requiring increased transcriptional activity of the receptor. Alternatively, wild type AhR activity could suffice to repress certain EMT markers, thus attenuating the potential effects of constitutive AhR activation.
It is generally accepted that the initial steps in EMT require the induction of the E-Cad transcriptional repressors Snail and/or Slug/Snai2 (19–21, 54). The reduced levels of E-Cad in AhR-depleted cells was accompanied by a significant increase in Snail and Slug/Snai2 expression, further supporting that a lack of AhR induces an EMT-like phenotype. Indeed, AhR deficiency increased clonogenicity, spreading and migration of epithelial cells, demonstrating that AhR is functionally relevant in EMT. The link between AhR and Slug/Snai2 appears likely as we have found that both proteins cooperate to repress the expression of genes harboring a novel murine SINE B1-X35S retrotransposon in their promoters (55). In addition, treatment of rat mammary cells with the tumor promoter DMBA (7,12-dimethylbenz(a)anthracene) induced EMT by activating c-Rel/CK2-dependent signaling and the downstream target proteins AhR and Slug/Snai2 (56). Although this study suggests that Slug/Snai2 can be a putative AhR target gene, our data clearly indicate that steady AhR knockdown significantly increases Slug/Snai2 expression, represses E-Cad, and promotes cell migration, in agreement with an EMT process. Interestingly, Slug/Snai2 was similarly induced in sh-AhR NMuMG and primary AhR−/− keratinocytes, whereas Snail induction was more significant in the latter, suggesting that the two proteins can be differentially affected by cell type-specific AhR expression. In support of such a possibility, we have found that the AhR-dependent B1-X35S retrotransposon has insulator activity when bound to Slug/Snai2 but not to Snail (37), and an additional study in MDCK epithelial cells has shown that Snail and Slug/Snai2 induce common as well as specific genetic programs during EMT (22).
TGFβ addition enhanced EMT features in AhR knockdown cells (sh-AhR NMuMG and primary keratinocytes) with respect to either wild type (EV and AhR+/+ keratinocytes) or constitutively active CA-AhR. This includes the repression of E-Cad and the induction of Snail, Slug/Snai2, fibronectin, and F-actin. Accordingly, TGFβ increased the migration of primary AhR−/− keratinocytes but not of sh-AhR NMuMG cells, perhaps because the addition of exogenous TGFβ to this cell line overrides the stimulatory effect induced by AhR depletion. The cross-talk between AhR and TGFβ in EMT was also observed in HaCaT cells in which AhR down-modulation potentiated the effects of TGFβ on Snail and Slug/Snai2 expression and on E-Cad repression. It has been shown (57) that short term (e.g. 1 h) treatment with the AhR ligand 3-methylcholanthrene of si-AhR-interfered HaCaT cells blocked Slug/Snai2 mRNA expression, a result differing from ours most probably because of the use of an exogenous ligand (versus none) and a much shorter time of treatment (1 versus 48 h). In fact, the increased response to exogenous TGFβ exhibited by AhR-depleted cells could help explain the effects seen under basal cell conditions. Immunoblotting experiments revealed that sh-AhR NMuMG cells and primary AhR−/− keratinocytes had slightly higher TGFβ protein content and markedly increased levels of activated pSmad2, suggesting that their basal EMT-like phenotype is at least partially due to enhanced TGFβ-dependent signaling. This possibility is in agreement with previous reports indicating that AhR negatively regulates TGFβ activation under basal cell conditions in mouse fibroblasts (8, 28, 30, 58), hepatocytes (59), and dermal fibroblasts (10). Therefore, TGFβ, by reducing AhR levels, could have synergistic activity in inducing molecular and phenotypical features of EMT in epithelial cells.
In summary, we propose that AhR deficiency in epithelial cells is enough to trigger morphological and phenotypical changes indicative of EMT, likely because of increased TGFβ signaling. Exogenous TGFβ further intensifies the EMT phenotype induced by AhR deficiency, probably because of its inhibitory role on AhR expression. This cross-talk could be relevant to the study of tumors in which AhR activity presumably varies during metastatic progression of the disease. In this context, several studies suggest that AhR has tumor suppressor activity and that certain human tumors tend to reduce their AHR expression. For instance, AHR expression was lowered by promoter hypermethylation in close to 35% of acute lymphoblastic leukemia patients (60). We have also found using tissue microarrays that AhR levels are significantly reduced in a large fraction of human glioblastomas and melanomas but not in low grade astrocytomas or benign nevi,4 again supporting an inhibitory role for this receptor in certain types of cancers. A recent study analyzing a panel of 947 human tumor cell lines identified AHR as a predictor of drug sensitivity associated with the efficacy of MEK inhibitors in NRAS-mutant lines (61).
Acknowledgments
We are very grateful to Drs. Alberto Muñoz, Xose R. Bustelo, and Jesús M. Paramio for technical assistance with TER measurements, retroviral transduction, and primary keratinocytes cultures, respectively. We also thank Drs. Antonio Garcia de Herreros for the NMuMG cells and the anti-Snail antibody, Yoshiaki Fujii-Kuriyama for the CA-AhR construct, and Miguel Quintanilla for the HaCaT cell line. The technical help of Eva Barrasa is greatly appreciated. Support of the Servicio de Técnicas Aplicadas a las Biociencias of the Universidad de Extremadura is greatly acknowledged.
This work was funded by grants from the Spanish Ministry of Science and Innovation (SAF2008-00462 and BFU2011-22678), the Red Temática de Investigación Cooperativa en Cáncer, Fondo de Investigaciones Sanitarias, Carlos III Institute, Spanish Ministry of Health (RD06/0020/1016 and RD12/0036/0032), and the Junta de Extremadura (GRU10008). All Spanish funding was co-sponsored by the European Union FEDER (Fondo Europeo de Desarrollo Regional) program.

This article contains supplemental Table S1 and Fig. S1.
E. M. Rico-Leo, M. Contador-Troca, I. Catalina-Fernández, A. Gómez-Durán, J. Sáenz-Santamaría, and P. M. Fernandez-Salguero, unpublished observations.
- AhR
- dioxin receptor
- EMT
- epithelial-to-mesenchymal transition
- α-naph
- α-naphthoflavone
- CA-AhR
- constitutively active AhR receptor
- E-Cad and N-Cad
- E-cadherin and N-cadherin, respectively
- β-Cat
- β-catenin
- FICZ
- 6-formylindolo[3,2-b]carbazole
- sh-AhR
- small-hairpin for AhR
- TER
- trans-epithelial resistance
- qRT
- quantitative real-time
- EV
- empty vector.
REFERENCES
- 1. Massari M. E., Murre C. (2000) Helix-loop-helix proteins. Regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 20, 429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abbott B. D., Birnbaum L. S., Perdew G. H. (1995) Developmental expression of two members of a new class of transcription factors. I. Expression of aryl hydrocarbon receptor in the C57BL/6N mouse embryo. Dev. Dyn. 204, 133–143 [DOI] [PubMed] [Google Scholar]
- 3. Barouki R., Coumoul X., Fernandez-Salguero P. M. (2007) The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett. 581, 3608–3615 [DOI] [PubMed] [Google Scholar]
- 4. Esser C., Rannug A., Stockinger B. (2009) The aryl hydrocarbon receptor in immunity. Trends Immunol. 30, 447–454 [DOI] [PubMed] [Google Scholar]
- 5. Furness S. G., Lees M. J., Whitelaw M. L. (2007) The dioxin (aryl hydrocarbon) receptor as a model for adaptive responses of bHLH/PAS transcription factors. FEBS Lett. 581, 3616–3625 [DOI] [PubMed] [Google Scholar]
- 6. Pohjanvirta R. (2012) The AH Receptor in Biology and Toxicology, John Wiley & Sons, New York [Google Scholar]
- 7. Carvajal-Gonzalez J. M., Mulero-Navarro S., Roman A. C., Sauzeau V., Merino J. M., Bustelo X. R., Fernandez-Salguero P. M. (2009) The dioxin receptor regulates the constitutive expression of the vav3 proto-oncogene and modulates cell shape and adhesion. Mol. Biol. Cell 20, 1715–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gomez-Duran A., Carvajal-Gonzalez J. M., Mulero-Navarro S., Santiago-Josefat B., Puga A., Fernandez-Salguero P. M. (2009) Fitting a xenobiotic receptor into cell homeostasis. How the dioxin receptor interacts with TGFβ signaling. Biochem. Pharmacol. 77, 700–712 [DOI] [PubMed] [Google Scholar]
- 9. Mulero-Navarro S., Pozo-Guisado E., Pérez-Mancera P. A., Alvarez-Barrientos A., Catalina-Fernández I., Hernández-Nieto E., Sáenz-Santamaria J., Martínez N., Rojas J. M., Sánchez-García I., Fernández-Salguero P. M. (2005) Immortalized mouse mammary fibroblasts lacking dioxin receptor have impaired tumorigenicity in a subcutaneous mouse xenograft model. J. Biol. Chem. 280, 28731–28741 [DOI] [PubMed] [Google Scholar]
- 10. Carvajal-Gonzalez J. M., Roman A. C., Cerezo-Guisado M. I., Rico-Leo E. M., Martin-Partido G., Fernandez-Salguero P. M. (2009) Loss of dioxin-receptor expression accelerates wound healing in vivo by a mechanism involving TGFβ. J. Cell Sci. 122, 1823–1833 [DOI] [PubMed] [Google Scholar]
- 11. Roman A. C., Carvajal-Gonzalez J. M., Rico-Leo E. M., Fernandez-Salguero P. M. (2009) Dioxin receptor deficiency impairs angiogenesis by a mechanism involving VEGF-A depletion in the endothelium and transforming growth factor-β overexpression in the stroma. J. Biol. Chem. 284, 25135–25148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Temchura V. V., Frericks M., Nacken W., Esser C. (2005) Role of the aryl hydrocarbon receptor in thymocyte emigration in vivo. Eur. J. Immunol. 35, 2738–2747 [DOI] [PubMed] [Google Scholar]
- 13. Thiery J. P., Sleeman J. P. (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142 [DOI] [PubMed] [Google Scholar]
- 14. Radisky D. C. (2005) Epithelial-mesenchymal transition. J. Cell Sci. 118, 4325–4326 [DOI] [PubMed] [Google Scholar]
- 15. Thiery J. P., Acloque H., Huang R. Y., Nieto M. A. (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 [DOI] [PubMed] [Google Scholar]
- 16. Prall F. (2007) Tumour budding in colorectal carcinoma. Histopathology 50, 151–162 [DOI] [PubMed] [Google Scholar]
- 17. Hajra K. M., Fearon E. R. (2002) Cadherin and catenin alterations in human cancer. Genes Chromosomes Cancer 34, 255–268 [DOI] [PubMed] [Google Scholar]
- 18. Huber M. A., Kraut N., Beug H. (2005) Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 17, 548–558 [DOI] [PubMed] [Google Scholar]
- 19. Batlle E., Sancho E., Francí C., Domínguez D., Monfar M., Baulida J., García De Herreros A. (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2, 84–89 [DOI] [PubMed] [Google Scholar]
- 20. Cano A., Pérez-Moreno M. A., Rodrigo I., Locascio A., Blanco M. J., del Barrio M. G., Portillo F., Nieto M. A. (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2, 76–83 [DOI] [PubMed] [Google Scholar]
- 21. Bolós V., Peinado H., Pérez-Moreno M. A., Fraga M. F., Esteller M., Cano A. (2003) The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions. A comparison with Snail and E47 repressors. J. Cell Sci. 116, 499–511 [DOI] [PubMed] [Google Scholar]
- 22. Moreno-Bueno G., Cubillo E., Sarrió D., Peinado H., Rodríguez-Pinilla S. M., Villa S., Bolós V., Jordá M., Fabra A., Portillo F., Palacios J., Cano A. (2006) Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer Res. 66, 9543–9556 [DOI] [PubMed] [Google Scholar]
- 23. Bhowmick N. A., Moses H. L. (2005) Tumor-stroma interactions. Curr. Opin. Genet. Dev. 15, 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fischer A. N., Herrera B., Mikula M., Proell V., Fuchs E., Gotzmann J., Schulte-Hermann R., Beug H., Mikulits W. (2005) Integration of Ras subeffector signaling in TGF-β mediated late stage hepatocarcinogenesis. Carcinogenesis 26, 931–942 [DOI] [PubMed] [Google Scholar]
- 25. Janda E., Lehmann K., Killisch I., Jechlinger M., Herzig M., Downward J., Beug H., Grünert S. (2002) Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis. Dissection of Ras signaling pathways. J. Cell Biol. 156, 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barrios-Rodiles M., Brown K. R., Ozdamar B., Bose R., Liu Z., Donovan R. S., Shinjo F., Liu Y., Dembowy J., Taylor I. W., Luga V., Przulj N., Robinson M., Suzuki H., Hayashizaki Y., Jurisica I., Wrana J. L. (2005) High-throughput mapping of a dynamic signaling network in mammalian cells. Science 307, 1621–1625 [DOI] [PubMed] [Google Scholar]
- 27. Ozdamar B., Bose R., Barrios-Rodiles M., Wang H. R., Zhang Y., Wrana J. L. (2005) Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science 307, 1603–1609 [DOI] [PubMed] [Google Scholar]
- 28. Chang X., Fan Y., Karyala S., Schwemberger S., Tomlinson C. R., Sartor M. A., Puga A. (2007) Ligand-independent regulation of transforming growth factor β1 expression and cell cycle progression by the aryl hydrocarbon receptor. Mol. Cell. Biol. 27, 6127–6139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gomez-Duran A., Ballestar E., Carvajal-Gonzalez J. M., Marlowe J. L., Puga A., Esteller M., Fernandez-Salguero P. M. (2008) Recruitment of CREB1 and histone deacetylase 2 (HDAC2) to the mouse Ltbp-1 promoter regulates its constitutive expression in a dioxin receptor-dependent manner. J. Mol. Biol. 380, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gomez-Duran A., Mulero-Navarro S., Chang X., Fernandez-Salguero P. M. (2006) LTBP-1 blockade in dioxin receptor-null mouse embryo fibroblasts decreases TGF-β activity. Role of extracellular proteases plasmin and elastase. J. Cell. Biochem. 97, 380–392 [DOI] [PubMed] [Google Scholar]
- 31. Santiago-Josefat B., Mulero-Navarro S., Dallas S. L., Fernandez-Salguero P. M. (2004) Overexpression of latent transforming growth factor-β-binding protein 1 (LTBP-1) in dioxin receptor-null mouse embryo fibroblasts. J. Cell Sci. 117, 849–859 [DOI] [PubMed] [Google Scholar]
- 32. Döhr O., Sinning R., Vogel C., Münzel P., Abel J. (1997) Effect of transforming growth factor-β 1 on expression of aryl hydrocarbon receptor and genes of Ah gene battery. Clues for independent down-regulation in A549 cells. Mol. Pharmacol. 51, 703–710 [DOI] [PubMed] [Google Scholar]
- 33. Wolff S., Harper P. A., Wong J. M., Mostert V., Wang Y., Abel J. (2001) Cell-specific regulation of human aryl hydrocarbon receptor expression by transforming growth factor-β (1). Mol. Pharmacol. 59, 716–724 [DOI] [PubMed] [Google Scholar]
- 34. McGuire J., Okamoto K., Whitelaw M. L., Tanaka H., Poellinger L. (2001) Definition of a dioxin receptor mutant that is a constitutive activator of transcription. Delineation of overlapping repression and ligand binding functions within the PAS domain. J. Biol. Chem. 276, 41841–41849 [DOI] [PubMed] [Google Scholar]
- 35. Pozo-Guisado E., Lorenzo-Benayas M. J., Fernández-Salguero P. M. (2004) Resveratrol modulates the phosphoinositide 3-kinase pathway through an estrogen receptor α-dependent mechanism. Relevance in cell proliferation. Int. J. Cancer 109, 167–173 [DOI] [PubMed] [Google Scholar]
- 36. Santiago-Josefat B., Fernandez-Salguero P. M. (2003) Proteasome inhibition induces nuclear translocation of the dioxin receptor through an Sp1 and protein kinase C-dependent pathway. J. Mol. Biol. 333, 249–260 [DOI] [PubMed] [Google Scholar]
- 37. Román A. C., González-Rico F. J., Moltó E., Hernando H., Neto A., Vicente-Garcia C., Ballestar E., Gómez-Skarmeta J. L., Vavrova-Anderson J., White R. J., Montoliu L., Fernández-Salguero P. M. (2011) Dioxin receptor and SLUG transcription factors regulate the insulator activity of B1 SINE retrotransposons via an RNA polymerase switch. Genome Res. 21, 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Merchant M., Krishnan V., Safe S. (1993) Mechanism of action of α-naphthoflavone as an Ah receptor antagonist in MCF-7 human breast cancer cells. Toxicol. Appl. Pharmacol. 120, 179–185 [DOI] [PubMed] [Google Scholar]
- 39. Bergander L., Wahlström N., Alsberg T., Bergman J., Rannug A., Rannug U. (2003) Characterization of in vitro metabolites of the aryl hydrocarbon receptor ligand 6-formylindolo[3,2-b]carbazole by liquid chromatography-mass spectrometry and NMR. Drug Metab. Dispos. 31, 233–241 [DOI] [PubMed] [Google Scholar]
- 40. Wincent E., Amini N., Luecke S., Glatt H., Bergman J., Crescenzi C., Rannug A., Rannug U. (2009) The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J. Biol. Chem. 284, 2690–2696 [DOI] [PubMed] [Google Scholar]
- 41. Marlowe J. L., Puga A. (2005) Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J. Cell. Biochem. 96, 1174–1184 [DOI] [PubMed] [Google Scholar]
- 42. Puga A., Marlowe J., Barnes S., Chang C. Y., Maier A., Tan Z., Kerzee J. K., Chang X., Strobeck M., Knudsen E. S. (2002) Role of the aryl hydrocarbon receptor in cell cycle regulation. Toxicology 181, 171–177 [DOI] [PubMed] [Google Scholar]
- 43. Fernandez-Salguero P. M. (2010) A remarkable new target gene for the dioxin receptor. THE Vav3 proto-oncogene links AhR to adhesion and migration. Cell Adh. Migr. 4, 172–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bui L. C., Tomkiewicz C., Chevallier A., Pierre S., Bats A. S., Mota S., Raingeaud J., Pierre J., Diry M., Transy C., Garlatti M., Barouki R., Coumoul X. (2009) Nedd9/Hef1/Cas-L mediates the effects of environmental pollutants on cell migration and plasticity. Oncogene 28, 3642–3651 [DOI] [PubMed] [Google Scholar]
- 45. Diry M., Tomkiewicz C., Koehle C., Coumoul X., Bock K. W., Barouki R., Transy C. (2006) Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a JNK-dependent mechanism. Oncogene 25, 5570–5574 [DOI] [PubMed] [Google Scholar]
- 46. Seifert A., Rau S., Küllertz G., Fischer B., Santos A. N. (2009) TCDD induces cell migration via NFATc1/ATX-signaling in MCF-7 cells. Toxicol. Lett. 184, 26–32 [DOI] [PubMed] [Google Scholar]
- 47. Frericks M., Burgoon L. D., Zacharewski T. R., Esser C. (2008) Promoter analysis of TCDD-inducible genes in a thymic epithelial cell line indicates the potential for cell-specific transcription factor crosstalk in the AhR response. Toxicol. Appl. Pharmacol. 232, 268–279 [DOI] [PubMed] [Google Scholar]
- 48. Casado F. L., Singh K. P., Gasiewicz T. A. (2011) Aryl hydrocarbon receptor activation in hematopoietic stem/progenitor cells alters cell function and pathway-specific gene modulation reflecting changes in cellular trafficking and migration. Mol. Pharmacol. 80, 673–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roman A. C., Carvajal-Gonzalez J. M., Mulero-Navarro S., Gomez-Duran A., Rico-Leo E., Merino J. M., Fernandez-Salguero P. M. (2012) in The AH receptor in Biology and Toxicology (Pohjanvirta R., ed.) pp. 485–497, John Wiley & Sons, New York [Google Scholar]
- 50. Peinado H., Olmeda D., Cano A. (2007) Snail, Zeb, and bHLH factors in tumour progression. An alliance against the epithelial phenotype? Nat. Rev. Cancer 7, 415–428 [DOI] [PubMed] [Google Scholar]
- 51. Mitra S. K., Hanson D. A., Schlaepfer D. D. (2005) Focal adhesion kinase. In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 [DOI] [PubMed] [Google Scholar]
- 52. Li S., Guan J. L., Chien S. (2005) Biochemistry and biomechanics of cell motility. Annu. Rev. Biomed. Eng. 7, 105–150 [DOI] [PubMed] [Google Scholar]
- 53. Cho Y. C., Zheng W., Jefcoate C. R. (2004) Disruption of cell-cell contact maximally but transiently activates AhR-mediated transcription in 10T1/2 fibroblasts. Toxicol. Appl. Pharmacol. 199, 220–238 [DOI] [PubMed] [Google Scholar]
- 54. Barrallo-Gimeno A., Nieto M. A. (2005) The Snail genes as inducers of cell movement and survival. Implications in development and cancer. Development 132, 3151–3161 [DOI] [PubMed] [Google Scholar]
- 55. Roman A. C., Benitez D. A., Carvajal-Gonzalez J. M., Fernandez-Salguero P. M. (2008) Genome-wide B1 retrotransposon binds the transcription factors dioxin receptor and Slug and regulates gene expression in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 1632–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Belguise K., Guo S., Yang S., Rogers A. E., Seldin D. C., Sherr D. H., Sonenshein G. E. (2007) Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, slug, and an invasive phenotype. Cancer Res. 67, 11742–11750 [DOI] [PubMed] [Google Scholar]
- 57. Ikuta T., Kawajiri K. (2006) Zinc finger transcription factor Slug is a novel target gene of aryl hydrocarbon receptor. Exp. Cell Res. 312, 3585–3594 [DOI] [PubMed] [Google Scholar]
- 58. Elizondo G., Fernandez-Salguero P., Sheikh M. S., Kim G. Y., Fornace A. J., Lee K. S., Gonzalez F. J. (2000) Altered cell cycle control at the G2/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol. Pharmacol. 57, 1056–1063 [PubMed] [Google Scholar]
- 59. Zaher H., Fernandez-Salguero P. M., Letterio J., Sheikh M. S., Fornace A. J., Jr., Roberts A. B., Gonzalez F. J. (1998) The involvement of aryl hydrocarbon receptor in the activation of transforming growth factor-β and apoptosis. Mol. Pharmacol. 54, 313–321 [DOI] [PubMed] [Google Scholar]
- 60. Mulero-Navarro S., Carvajal-Gonzalez J. M., Herranz M., Ballestar E., Fraga M. F., Ropero S., Esteller M., Fernandez-Salguero P. M. (2006) The dioxin receptor is silenced by promoter hypermethylation in human acute lymphoblastic leukemia through inhibition of Sp1 binding. Carcinogenesis 27, 1099–1104 [DOI] [PubMed] [Google Scholar]
- 61. Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. A., Kim S., Wilson C. J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., Morais P., Meltzer J., Korejwa A., Jané-Valbuena J., Mapa F. A., Thibault J., Bric-Furlong E., Raman P., Shipway A., Engels I. H., Cheng J., Yu G. K., Yu J., Aspesi P., Jr., de Silva M., Jagtap K., Jones M. D., Wang L., Hatton C., Palescandolo E., Gupta S., Mahan S., Sougnez C., Onofrio R. C., Liefeld T., MacConaill L., Winckler W., Reich M., Li N., Mesirov J. P., Gabriel S. B., Getz G., Ardlie K., Chan V., Myer V. E., Weber B. L., Porter J., Warmuth M., Finan P., Harris J. L., Meyerson M., Golub T. R., Morrissey M. P., Sellers W. R., Schlegel R., Garraway L. A. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]