

Background: Corin is a membrane serine protease that activates natriuretic peptides in the heart.
Results: The corin mutant R539C identified in hypertensive patients has impaired zymogen activation and altered ectodomain shedding.
Conclusion: The mutation alters corin protein structure and reduces corin activity.
Significance: Human CORIN gene mutations causing impaired corin activity may be an underlying mechanism in hypertension.
Keywords: Genetic Diseases, Hypertension, Peptide Hormones, Protease, Proteolytic Enzymes, Corin, Natriuretic Peptides
Abstract
Corin is a cardiac transmembrane serine protease that regulates blood pressure by activating natriuretic peptides. Corin variants have been associated with African Americans with hypertension and heart disease. Here, we report a new mutation in exon 12 of the CORIN gene identified in a family of patients with hypertension. The mutation resulted in R539C substitution in the Fz2 (Frizzled-2) domain of the corin propeptide region. We expressed and characterized the corin R539C mutant in HEK293 cells. As determined by Western blot analysis, the R539C mutation did not alter corin expression in transfected cells but impaired corin zymogen activation. In a pro-atrial natriuretic peptide processing assay, the corin mutant had reduced activity and exhibited a dominant-negative effect on wild-type corin. In addition, the R539C mutation altered corin ectodomain shedding, producing an alternative ∼75-kDa fragment that was biologically inactive. Using protease inhibitors and the catalytically inactive corin mutant S985A, we showed that the ∼75-kDa fragment was generated by corin autocleavage. We constructed a series of mutants by replacing single or double Arg residues in the corin propeptide and identified Arg-530 in the Fz2 domain as the alternative autocleavage site. Our results show that the corin mutation R539C identified in hypertensive patients impairs corin zymogen activation and causes an alternative autocleavage that reduces corin activity. These data support that human CORIN gene mutations causing impaired corin activity may be an underlying mechanism in hypertension.
Introduction
Hypertension is a common cardiovascular disease, afflicting nearly 30% adults in developed countries (1). Genetic factors are known to influence the onset and severity of the disease. Mutations in the genes involving electrolyte homeostasis have been reported in hypertensive patients (2–5). To date, however, the underlying disease mechanism in the majority of patients with hypertension remains unknown (4).
Atrial natriuretic peptide (ANP)3 plays an important role in regulating salt-water balance and blood pressure (6, 7). Genetic variants or mutations of the genes encoding ANP or its receptor have been associated with hypertension in patients (8–11). Corin is a transmembrane serine protease that converts the ANP precursor (pro-ANP) to mature ANP in cardiac myocytes (6, 12). In mice, lack of corin impairs renal sodium excretion and causes salt-sensitive hypertension and cardiac hypertrophy (13–16). These data support the physiological importance of corin and ANP in maintaining normal blood pressure.
Structurally, corin belongs to the family of type II transmembrane serine proteases that participate in diverse biological processes (17). Mutations in genes encoding these proteases have been reported to cause human diseases. For example, mutations in the TMPRSS3, TMPRSS6, and ST14 genes have been identified in patients with congenital deafness, iron-refractory anemia, and ichthyotic skin disorders, respectively (18–25). The human CORIN gene is located on chromosome 4, which contains 22 exons and spans >200 kb in length (26). CORIN gene variants (T555I/Q568P) have been identified in African Americans with hypertension and heart disease (27, 28). Studies have shown that the variants have reduced natriuretic peptide processing activity in vitro and in vivo (29, 30). Most recently, two CORIN gene mutations have been reported in patients with pregnancy-induced hypertension (31). The data suggest that variants or mutations of the CORIN gene may contribute to hypertensive disease in humans.
To test this hypothesis, we sequenced the CORIN gene in patients with hypertension. Here, we report a novel CORIN gene mutation (R539C) identified in a patient family with hypertension. We characterized the corin mutant R539C in a series of biochemical and cellular experiments to understand how the mutation may alter corin protein structure and function.
EXPERIMENTAL PROCEDURES
Patient Blood Samples
Venous blood samples were from 183 patients with hypertension that was defined by systolic blood pressure >140 mm Hg and/or diastolic blood pressure >90 mm Hg. Additional blood samples were obtained from 229 normal individuals who underwent routine checkups at the hospitals and had no history of hypertension. All individuals were ethnic Han Chinese. The study was approved by the ethic committees at the hospitals. All participants provided written informed consent.
DNA Extraction and Sequencing
Genomic DNA was extracted from peripheral blood cells using the QIAamp DNA blood kit (Qiagen). DNA fragments from all 22 exons and intron-exon boundaries of the CORIN gene were amplified by PCR using specific oligonucleotide primers. PCR products were sequenced directly by an Applied Biosystems 3100 DNA analyzer. Gene variants or mutations identified were verified by independent PCR and DNA sequencing.
Plasmids
Plasmids expressing human WT corin, the S985A mutant (in which the catalytic Ser-985 was replaced with Ala), and the R801A mutant (in which the activation cleavage site Arg-801 was replaced with Ala) were described previously (32, 33). Plasmids expressing corin mutants R539C, R539A, R539C/R480A, R530A/R539C, R539C/R588A, R539C/R597A/R598A, R539C/R619A, R539C/R670A, R539C/S985A, C540A, and R539C/C540A were made by PCR-based site-directed mutagenesis. Recombinant corin proteins encoded by these plasmids contained a C-terminal V5 tag to facilitate protein detection by Western blotting (32, 33).
Transfection, Immunoprecipitation, and Western Blotting
HEK293 cells were grown in DMEM with 10% FBS in humidified incubators at 37 °C with 5% CO2 and 95% air. Plasmids were transfected into HEK293 cells using FuGENE reagents (Roche Diagnostics). Conditioned medium was collected after 48–72 h. Cells were lysed in buffer containing 50 mmol/liter Tris-HCl (pH 8.0), 150 mmol/liter NaCl, 1% (v/v) Triton X-100, and a protease inhibitor mixture (1:100 dilution; Sigma). Immunoprecipitation and Western analysis of corin proteins were done using an anti-V5 tag antibody as described previously (34, 35).
Pro-ANP Processing Assay
Conditioned medium containing recombinant human pro-ANP from a stable HEK293 cell line was added to HEK293 cells expressing WT corin or mutants and incubated at 37 °C for 2 h. Pro-ANP and ANP in the medium were analyzed by immunoprecipitation and Western blotting (34, 35). To test the activity of soluble corin, conditioned medium containing corin fragments from transfected HEK293 cells was collected, concentrated, and incubated with the pro-ANP medium at 37 °C for 2 h. Pro-ANP processing was assessed by immunoprecipitation and Western blotting.
Effect of Protease Inhibitors on Corin Cleavage
To identify proteases involved in corin cleavage, a panel of protease inhibitors, including benzamidine (a trypsin-like serine protease inhibitor; 5 mm), GM6001 (a metalloproteinase inhibitor; 50 μm), TAPI-1 (an ADAM (a disintegrin and metalloproteinase) inhibitor; 50 μm), and N-acetyl-leucyl-leucyl-methionine (a cysteine protease inhibitor; 50 μm), was added to corin-expressing cells in separate wells of culture plates. The conditioned medium was collected over time. Corin fragments in the conditioned medium were analyzed by immunoprecipitation and Western blotting.
Molecular Modeling
Modeling of the corin Fz2 (Frizzled-2) domain was done based on the crystal structure of mouse Frizzled-8 (36). The sequences of mouse Frizzled-8 and the human corin Fz2 domain were aligned using the ClustalW program (37). Three-dimensional models of the human corin Fz2 domain were created using a computer-based homology modeling program (38). The model inspection and image generation were carried out using the PyMOL program.
Statistical Analysis
Data were analyzed using SPSS 12 software (SPSS, Chicago, IL). All data are presented as means ± S.D. Comparisons between two groups were done by Student's t test. Multigroup comparisons were done by analysis of variance. A p value of <0.05 was considered to be statistically significant.
RESULTS
Identification of CORIN Gene Mutation
We sequenced the CORIN gene from 183 patients with hypertension. A C → T heterozygous mutation at nucleotide 1708 in exon 12 was identified in a hypertensive patient (Fig. 1, A and B). This mutation was absent in 229 unrelated healthy controls. Members of the patient family who carried this mutation had higher blood pressure except for a 14-year-old male (IV-3) (Fig. 1A). The mutation caused an Arg-to-Cys substitution at residue 539 in the Fz2 domain (Fig. 2A). In a three-dimensional model of the corin Fz2 domain (Fig. 2, B and C), Arg-539 was located in an α-helix on the domain surface. The model also suggested that the mutant Cys residue was likely to create an alternative disulfide bond with Cys-502 in an adjacent loop (Fig. 2C), which may alter the overall Fz2 domain structure.
FIGURE 1.

CORIN gene mutation in a patient family. A, patient pedigree. Males (squares) and females (circles) with (black) or without (white) the R539C mutant allele are shown. The arrow indicates the proband (P). Slashes through the symbols indicate decreased individuals. Gray indicates the individuals from whom samples were not available. Numbers below the symbols indicate systolic/diastolic blood pressure (mm Hg) in these individuals. B, CORIN gene sequencing data from a normal control (left panel) and the proband (right panel). The C → T heterozygous mutation at nucleotide 1708 causing the R539C change is indicated (right panel).
FIGURE 2.

Location of the R539C mutation in the corin Fz2 domain. A, corin protein domains. TM, transmembrane; SR, scavenger receptor. The catalytic residues His (H), Asp (D), and Ser (S) are shown. The arrow indicates the activation cleavage site at Arg-801–Ile-802. A disulfide bond (S-S) links the propeptide and the protease domain. B, a surface model of the corin Fz2 domain by molecular modeling. Arg-539 is in green. C, location of Arg-539 in a molecular model of the corin Fz2 domain. The R539C mutation may potentially create an alternative disulfide bond between Cys-539 and Cys-502. Magnified areas highlight the residues involved in the WT corin (upper circle) and R539C mutant (lower circle) models. The N (N-term) and C (C-term) termini of the Fz2 domain are indicated. The conserved disulfide bond between Cys-540 and Cys-502 is shown in stick models.
Expression and Functional Analysis of the R539C Mutant
To examine if the R539C mutation altered corin expression and/or function, we made plasmids expressing the R539C and R539A mutants and transfected them into HEK293 cells. WT corin and the activation cleavage site mutant R801A and the catalytic active site mutant S985A were used as controls. On Western blots, WT and mutant corin proteins were found to be expressed at similar levels in transfected cells (Fig. 3A). However, the level of the ∼40-kDa band in the R539C mutant was lower than those in WT corin and mutants R539A and S985A. This band represents the corin protease domain cleaved at the activation site Arg-801 (Fig. 2A). As predicted, the band was absent in the R801A mutant, which abolished the activation cleavage site (Fig. 3A). As estimated by densitometric analysis of the Western blots, the levels of the ∼40-kDa band were 28.0 ± 10.3 and 17.8 ± 8.3%, respectively, of the total WT and R539C corin proteins (p < 0.01) (Fig. 3B). The data indicate that the R539C (but not R539A) mutation impairs corin zymogen activation.
FIGURE 3.

R539C mutation reduces zymogen activation and pro-ANP processing activity. A, Western analysis of lysates from HEK293 cells expressing WT corin and mutants. The cleaved corin protease domain fragment (corin-p) is indicated by the arrow. B, percentage of the activated protease fragment versus corin zymogen protein as estimated by densitometry. Data are means ± S.D. from five independent experiments. C, Western analysis of pro-ANP processing activities of corin mutants. D, quantitative data of pro-ANP processing as estimated by densitometry. Data are means ± S.D. from seven independent experiments. **, p < 0.01 versus WT corin or R539A. E, protein expression (upper panel) and pro-ANP processing activity (lower panel) of corin mutants C540A and R539C/C540A. WT corin and mutant R801A were used as positive and negative controls, respectively. F, quantitative data (means ± S.D., n = 5) of pro-ANP processing by WT corin and mutants C540A and R539C/C540A. **, p < 0.01 versus the indicated groups.
We next tested the activity of corin mutants in a pro-ANP processing assay. The activity of the R539C (but not R539A) mutant was significantly reduced compared with that of WT corin (Fig. 3C). Two negative controls (mutants R801A and S985A) had little activity (Fig. 3C). As estimated by densitometric analysis of the Western blots, the R539C mutant had 59.8 ± 16.0% activity compared with WT corin (p < 0.01) (Fig. 3D).
Based on the molecular model (Fig. 2C), R539C is likely to interfere with the disulfide bond between Cys-540 and Cys-502. Consistent with this hypothesis, mutant C540A, which was expected to disrupt the disulfide bond, had markedly reduced pro-ANP processing activity (26.7 ± 5.5% of WT corin, p < 0.01) (Fig. 3, E and F). When both the R539C and C540A mutations were introduced, the activity of mutant R539C/C540A was significantly higher than that of mutant C540A (55.3 ± 3.2 versus 26.7 ± 5.5%, p < 0.01) (Fig. 3, E and F), suggesting that the mutant Cys-539 residue may pair with Cys-502 in the absence of Cys-540 (Fig. 2C), thereby partially restoring the corin structure and activity.
Cleaved Corin Fragments in Conditioned Medium
Previous studies showed that corin is shed from the cell surface, producing three major soluble fragments of ∼180, ∼160, and ∼100 kDa (Fig. 4A) (32). To examine if the R539C mutation affects corin shedding, we analyzed corin fragments in the conditioned medium from transfected HEK293 cells. Three bands of ∼180, ∼160, and ∼100 kDa were detected in the conditioned medium from HEK293 cells expressing WT corin (Fig. 4B, upper panel). The sample from the inactive corin mutant R801A had only the ∼180-kDa band, consistent with previous findings that the ∼160- and ∼100-kDa bands are products of corin autocleavage (32). In the sample from the R539C mutant, a prominent extra band of ∼75 kDa was detected (Fig. 4B, upper panel). This band also was observed in the sample from the C540A mutant, but not in the samples from mutants R539A and R539C/C540A (Fig. 4B, upper panel).
FIGURE 4.
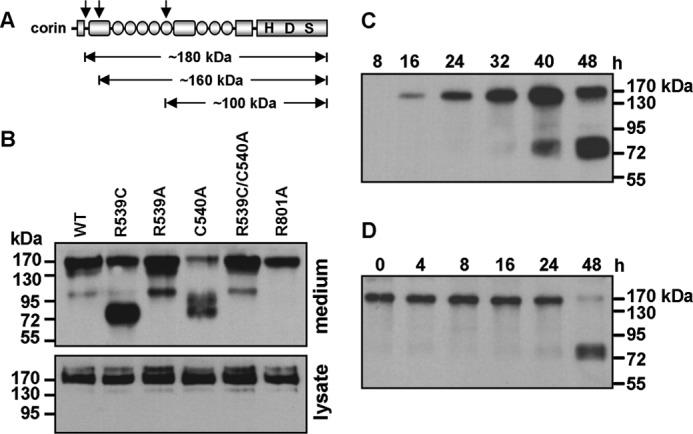
Cleaved corin fragments in conditioned medium. A, illustration of corin fragments. Cleavage sites generating three soluble fragments are indicated by arrows. B, Western analysis of corin fragments in the conditioned medium from cells expressing WT corin and mutants (upper panel). Corin proteins in cell lysate are shown as a control (lower panel). C, Western analysis of corin fragments in the conditioned medium collected over the indicated time periods from cells expressing the R539C mutant. D, Western analysis of corin fragments in the conditioned medium collected at 24 h after transfection from cells expressing the R539C mutant and then incubated at 37 °C for the indicated time periods without the cells.
In a time course experiment with the conditioned medium collected over time from HEK293 cells expressing the R539C mutant, the ∼180-kDa band appeared in samples after 16 h, whereas the ∼75-kDa band appeared only in samples after 40 h (Fig. 4C). To examine if the ∼75-kDa band was derived from the ∼180-kDa fragment, the conditioned medium was collected at 24 h after transfection and incubated at 37 °C over time without the cells. Western analysis showed the ∼75-kDa fragment in the sample incubated for 48 h (Fig. 4D). In the same sample, the level of the ∼180-kDa fragment was reduced, indicating that the ∼75-kDa fragment was derived from the ∼180-kDa fragment in this experiment.
Effect of Protease Inhibitors on Corin Cleavage
To determine the class of enzyme(s) responsible for generating the ∼75-kDa fragment, we added protease inhibitors to HEK293 cells expressing the R539C mutant and analyzed the conditioned medium by Western blotting. The level of the ∼75-kDa band was markedly reduced in the presence of benzamidine, but not metalloproteinase (GM6001 and TAPI-1) or cysteine protease (N-acetyl-leucyl-leucyl-methionine) inhibitors (Fig. 5A), indicating that the ∼75-kDa fragment was likely generated by a serine protease.
FIGURE 5.
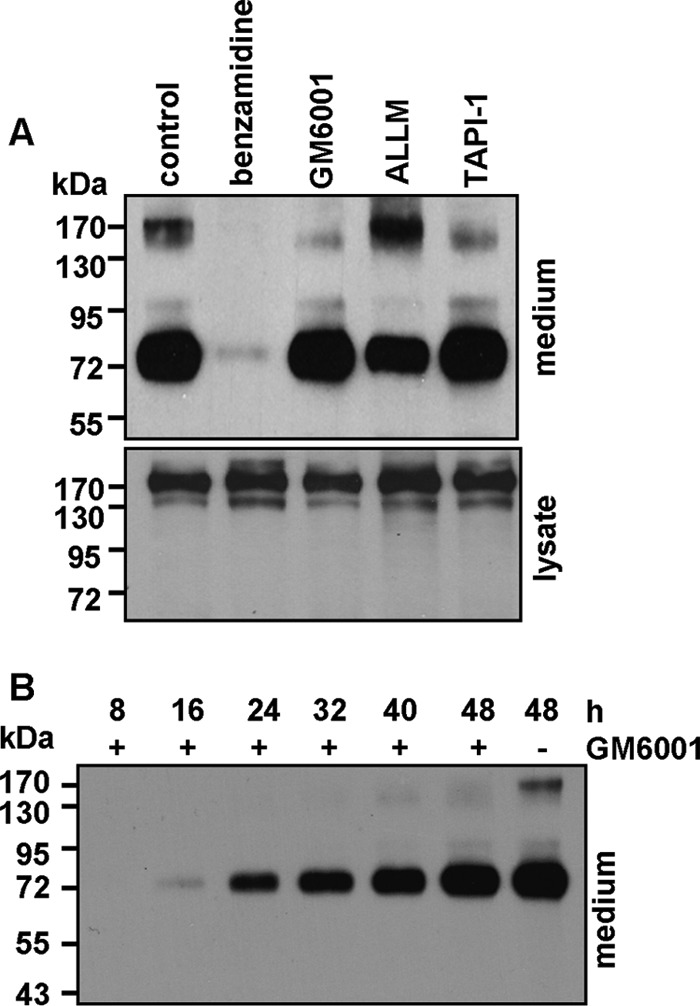
Effect of protease inhibitors. A, HEK293 cells expressing the R539C mutant were incubated with vehicle (control), benzamidine, GM6001, N-acetyl-leucyl-leucyl-methionine (ALLM), or TAPI-1. Corin fragments in the conditioned medium were analyzed by Western blotting (upper panel). Corin proteins in cell lysate are shown as a control (lower panel). Data are representative of three independent experiments. B, Western analysis of corin fragments in the conditioned medium collected over the indicated time periods from cells expressing the R539C mutant with (+) or without (−) GM6001.
As shown in Fig. 5A, GM6001 and TAPI-1 reduced the level of the ∼180-kDa fragment, consistent with a previous report that the fragment was cleaved by an ADAM protease (32). The results also indicated that the production of the ∼75-kDa fragment may not depend on the ∼180-kDa fragment. To verify this hypothesis, we added GM6001 to HEK293 cells expressing the R539C mutant and collected the conditioned medium over time. GM6001 inhibited the production of the ∼180-kDa fragment, but not the ∼75-kDa fragment (Fig. 5B), indicating that the presence of the ∼180-kDa fragment was not a prerequisite for generating the ∼75-kDa fragment.
Biological Activity of Soluble Corin Fragments
To test whether the ∼75-kDa fragment is biologically active, we incubated the conditioned medium with recombinant human pro-ANP and analyzed pro-ANP processing. Pro-ANP processing activity was detected in the conditioned medium from cells expressing WT corin (Fig. 6A). The activity was markedly lower or absent in the conditioned medium from R539C mutant-expressing cells cultured without or with GM6001, which inhibited the ∼180-kDa fragment (Fig. 6A). The results indicated that the ∼75-kDa fragment had little biological activity.
FIGURE 6.
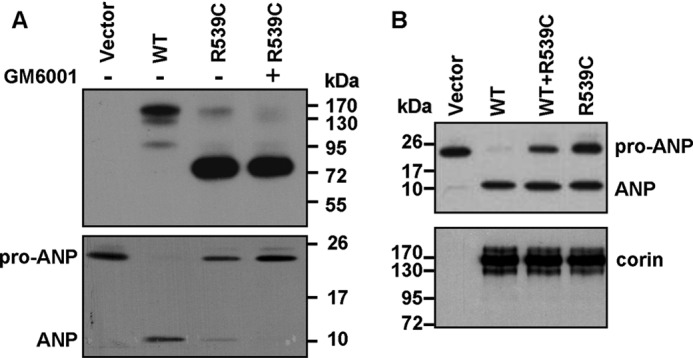
Activity of soluble corin fragments and effect of R539C on WT corin activity. A, cells expressing WT corin or mutants were cultured without (−) or with (+) GM6001. Corin fragments in the conditioned medium (upper panel) and their pro-ANP processing activities (lower panel) were analyzed by Western analysis. Data are representative of three independent experiments. B, HEK293 cells were transfected with plasmids expressing WT corin and R539C either alone or together. Corin activity was measured in a pro-ANP processing assay by Western blotting (upper panel). Corin proteins in cell lysate are shown as a control (lower panel). Data are representative of five independent experiments.
We also tested the effect of the R539C mutant on WT corin in a pro-ANP processing assay. In HEK293 cells cotransfected with plasmids expressing WT corin and the R539C mutant, pro-ANP processing activity was significantly reduced (64.4 ± 5.3% of WT corin, p < 0.01) (Fig. 6B, upper panel), indicating a dominant-negative effect of the mutant on WT corin.
Corin Autocleavage
The inhibition of the ∼75-kDa fragment by benzamidine indicated that the fragment was derived from serine protease-mediated cleavage (Fig. 5A). Corin is a serine protease, capable of generating the ∼160- and ∼100-kDa fragments by autocleavage (32). To test whether the ∼75-kDa fragment was also from autocleavage, we made a double mutant (R539C/S985A) that was catalytically inactive (Fig. 7A). Western analysis did not detect the ∼75-kDa band in the conditioned medium from cells expressing the R539C/S985A mutant (Fig. 7B), confirming that the fragment was generated by corin autocleavage.
FIGURE 7.

Corin autocleavage sites. A, illustration of Arg residues in the Fz2 and LDLR6–8 domains and the active site mutant S985A in the protease domain. H, His; D, Asp; S, Ser; S-S, disulfide bond. B, Western analysis of corin fragments in the conditioned medium from cells expressing WT corin and mutants (upper panel). Corin proteins in cell lysate are shown as a control (lower panel). Data are representative of three independent experiments. C, Western analysis of corin fragments in the conditioned medium from cells expressing corin mutants (upper panel). Corin proteins in cell lysate are shown as a control (lower panel).
Identification of Alternative Corin Autocleavage Site
Based on the size of the ∼75-kDa fragment, the cleavage site for this fragment was predicted to be in a region including the Fz2 domain and LDL receptor (LDLR) 6–8 repeats (Fig. 7A). To identify the specific cleavage site, we made a series of corin mutants by replacing single or double Arg residues with Ala within this region: R480A, R530A, R588A, R597A/R598A, R619A, and R670A (Fig. 7A). Western analysis of the conditioned medium from cells expressing these mutants showed that the R530A mutation, but not the other mutations, abolished the ∼75-kDa fragment (Fig. 7C). The results showed that the R539C mutant autocleaved at Arg-530 in the Fz2 domain to generate the alternative ∼75-kDa fragment.
DISCUSSION
In this study, we have reported a novel mutation (R539C) in the corin Fz2 domain identified in a hypertensive patient family. In functional experiments, the R539C mutation did not alter corin expression in transfected cells but impaired corin zymogen activation, as indicated by reduced amounts of activated protease domain fragment (Fig. 3, A and B). Consistently, the R539C mutant had reduced activity in processing pro-ANP (Fig. 3, C and D).
Based on a three-dimensional model of the corin Fz2 domain, Arg-539 is in an α-helix and is spatially close to Cys-502, which forms a disulfide bond with Cys-540 (Fig. 2C). The change of Arg-539 to Cys may create an alternative disulfide bond with Cys-502, thereby leaving Cys-540 unpaired and altering corin structure and function. In agreement with this hypothesis, when Cys-539 was substituted with Ala, the corin R539A mutant exhibited similar zymogen activation and activity compared with WT corin (Fig. 3, A–D), indicating that Arg at position 539 is not critical but that substitution of this Arg residue with Cys may alter the structure of corin and impair its function. In support of this hypothesis, mutant C540A, which disrupted the predicted disulfide bond between Cys-540 and Cys-520, had markedly reduced activity (Fig. 3, E and F). This defect was partially corrected when both mutations R539C and C540A were introduced (Fig. 3, E and F), suggesting that the mutant Cys-539 residue may form a disulfide bond with Cys-502, mimicking the original disulfide bond between Cys-540 and Cys-502 (Fig. 2C).
Ectodomain shedding is important in regulating protein expression and activity on the cell surface (39, 40). As a membrane protease, corin is shed by ADAM or autocleaved to generate distinct soluble fragments, which regulates corin activity in cardiac myocytes (32). In this study, we examined if the R539C mutation alters corin ectodomain shedding. Consistent with a previous report (32), three soluble fragments of ∼180, ∼160, and ∼100 kDa were detected in the conditioned medium from cells expressing WT corin. Unexpectedly, a major alternative fragment of ∼75 kDa was found in the conditioned medium from cells expressing the R539C (but not R539A) mutant (Figs. 4B and 7B), suggesting that this fragment was produced as a result of the conformational change caused by the mutant Cys-539 residue. Consistent with this hypothesis, the alternative ∼75-kDa fragment also was detected in the sample from mutant C540A (Fig. 4B), which was expected to disrupt the disulfide bond between Cys-540 and Cys-502 (Fig. 2C). When both mutations R539C and C540A were introduced, the ∼75-kDa fragment disappeared (Fig. 4B), suggesting that, in the absence of Cys-540, Cys-539 may form a disulfide bond with Cys-502, thereby partially restoring the altered conformational change in the Fz2 domain.
Using protease inhibitors and the catalytically inactive corin mutant S985A (Figs. 5A and 7B), we showed that the ∼75-kDa fragment was generated by corin autocleavage. We also found that this fragment could be generated directly from the membrane-bound corin or from the ∼180-kDa soluble fragment as an intermediate (Figs. 4D and 5B). By testing a series of mutants with replacements of single or double Arg residues in the Fz2 and LDLR6–8 domains, we showed that the ∼75-kDa fragment was from specific cleavage at Arg-530 in the Fz2 domain (Fig. 7, A and C). As indicated in the Fz2 domain model (Fig. 2C), Arg-530 is located in the N-terminal region of the α-helix that includes Arg-539 in the middle. It appears that the R539C mutation may cause a mismatched disulfide bond with Cys-502 or possibly Cys-540, resulting in a distorted conformation of the α-helix and creating an alternative autocleavage site at Arg-530.
The biological function of corin is to activate pro-ANP (6, 12). Previously, the ∼180-kDa fragment was shown to be generated by ADAM and to be biologically active, whereas the ∼160- and ∼100-kDa fragments were generated by autocleavage and were inactive (32). In this study, the ∼75-kDa fragment from alternative corin autocleavage had little pro-ANP processing activity (Fig. 6A). The results are consistent with previous findings that the Fz1 and LDLR1–5 domains, which precede the Fz2 domain (Fig. 2A), are required for pro-ANP processing (41). It is interesting that all corin fragments from autocleavage identified so far are biologically inactive, suggesting that autocleavage may serve as a mechanism to negatively regulate corin activity.
Corin is made primarily in cardiac myocytes (42). Cleaved corin fragments may enter the circulation. Corin antigen and activity have been detected in human blood (43–46). Studies indicate that the levels of plasma soluble corin and activity are significantly lower in patients with hypertension and heart failure (43, 47, 48), suggesting that the reduced corin activity may contribute to hypertensive disease.
In this study, we identified a heterozygous corin mutation in a patient family with hypertension. In cotransfected HEK293 cells, the R539C mutant inhibited WT corin in a pro-ANP processing assay (Fig. 6B). The results indicate a possible dominant-negative effect of the mutant protein, which may also occur in the patients. Within this patient family, most members with the mutant allele had higher systolic and/or diastolic blood pressure than those with two normal alleles (Fig. 1A). Individual IV-3, a 14-year-old male, appeared to be an exception; he had a mutant allele but normal blood pressure. It is known that the prevalence of hypertension increases with age (49), suggesting that age-related factors play an important role in regulating blood pressure. It would be interesting to know if this individual develops hypertension when he becomes older. Together, our results indicate that the naturally occurring R539C mutation in hypertensive patients may reduce corin activity by impairing zymogen activation and producing an inactive alternative autocleavage fragment. These findings confirm that CORIN gene mutations may be a contributing molecular mechanism in human hypertension.
This work was supported, in whole or in part, by National Institutes of Health Grant HL089298. This work was also supported by National Natural Science Foundation of China Grants 31070716, 81170247, and 31161130356; Natural Science Foundation of the Jiangsu Higher Education Institutions Grant 10KJB320017; the Priority Academic Program Development of Jiangsu Higher Education Institutions; and the Danish-Chinese Center for Proteases and Cancer.
- ANP
- atrial natriuretic peptide
- LDLR
- LDL receptor.
REFERENCES
- 1. Roger V. L., Go A. S., Lloyd-Jones D. M., Adams R. J., Berry J. D., Brown T. M., Carnethon M. R., Dai S., de Simone G., Ford E. S., Fox C. S., Fullerton H. J., Gillespie C., Greenlund K. J., Hailpern S. M., Heit J. A., Ho P. M.., Howard V. J., Kissela B. M., Kittner S. J., Lackland D. T., Lichtman J. H., Lisabeth L. D., Makuc D. M., Marcus G. M., Marelli A., Matchar D. B., McDermott M. M., Meigs J. B., Moy C. S., Mozaffarian D., Mussolino M. E., Nichol G., Paynter N. P., Rosamond W. D., Sorlie P. D., Stafford R. S., Turan T. N., Turner M. B., Wong N. D., Wylie-Rosett J. (2011) Heart disease and stroke statistics–2011 update: a report from the American Heart Association. Circulation 123, e18–e209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ambrosius W. T., Bloem L. J., Zhou L., Rebhun J. F., Snyder P. M., Wagner M. A., Guo C., Pratt J. H. (1999) Genetic variants in the epithelial sodium channel in relation to aldosterone and potassium excretion and risk for hypertension. Hypertension 34, 631–637 [DOI] [PubMed] [Google Scholar]
- 3. Choi M., Scholl U. I., Yue P., Björklund P., Zhao B., Nelson-Williams C., Ji W., Cho Y., Patel A., Men C. J., Lolis E., Wisgerhof M. V., Geller D. S., Mane S., Hellman P., Westin G., Åkerström G., Wang W., Carling T., Lifton R. P. (2011) K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331, 768–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lifton R. P., Gharavi A. G., Geller D. S. (2001) Molecular mechanisms of human hypertension. Cell 104, 545–556 [DOI] [PubMed] [Google Scholar]
- 5. Mullins L. J., Bailey M. A., Mullins J. J. (2006) Hypertension, kidney, and transgenics: a fresh perspective. Physiol. Rev. 86, 709–746 [DOI] [PubMed] [Google Scholar]
- 6. Wu Q., Xu-Cai Y. O., Chen S., Wang W. (2009) Corin: new insights into the natriuretic peptide system. Kidney Int. 75, 142–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou Y., Jiang J., Cui Y., Wu Q. (2009) Corin, atrial natriuretic peptide and hypertension. Nephrol. Dial. Transplant. 24, 1071–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hodgson-Zingman D. M., Karst M. L., Zingman L. V., Heublein D. M., Darbar D., Herron K. J., Ballew J. D., de Andrade M., Burnett J. C., Jr., Olson T. M. (2008) Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N. Engl. J. Med. 359, 158–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakayama T., Soma M., Takahashi Y., Rehemudula D., Kanmatsuse K., Furuya K. (2000) Functional deletion mutation of the 5′-flanking region of type A human natriuretic peptide receptor gene and its association with essential hypertension and left ventricular hypertrophy in the Japanese. Circ. Res. 86, 841–845 [DOI] [PubMed] [Google Scholar]
- 10. Newton-Cheh C., Larson M. G., Vasan R. S., Levy D., Bloch K. D., Surti A., Guiducci C., Kathiresan S., Benjamin E. J., Struck J., Morgenthaler N. G., Bergmann A., Blankenberg S., Kee F., Nilsson P., Yin X., Peltonen L., Vartiainen E., Salomaa V., Hirschhorn J. N., Melander O., Wang T. J. (2009) Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat. Genet. 41, 348–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rubattu S., Bigatti G., Evangelista A., Lanzani C., Stanzione R., Zagato L., Manunta P., Marchitti S., Venturelli V., Bianchi G., Volpe M., Stella P. (2006) Association of atrial natriuretic peptide and type A natriuretic peptide receptor gene polymorphisms with left ventricular mass in human essential hypertension. J. Am. Coll. Cardiol. 48, 499–505 [DOI] [PubMed] [Google Scholar]
- 12. Wu Q. (2007) The serine protease corin in cardiovascular biology and disease. Front. Biosci. 12, 4179–4190 [DOI] [PubMed] [Google Scholar]
- 13. Buckley C. L., Stokes A. J. (2011) Corin-deficient W-sh mice poorly tolerate increased cardiac afterload. Regul. Pept. 172, 44–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chan J. C., Knudson O., Wu F., Morser J., Dole W. P., Wu Q. (2005) Hypertension in mice lacking the proatrial natriuretic peptide convertase corin. Proc. Natl. Acad. Sci. U.S.A. 102, 785–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nigrovic P. A., Gray D. H., Jones T., Hallgren J., Kuo F. C., Chaletzky B., Gurish M., Mathis D., Benoist C., Lee D. M. (2008) Genetic inversion in mast cell-deficient Wsh mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am. J. Pathol. 173, 1693–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang W., Shen J., Cui Y., Jiang J., Chen S., Peng J., Wu Q. (2012) Impaired sodium excretion and salt-sensitive hypertension in corin-deficient mice. Kidney Int. 82, 26–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bugge T. H., Antalis T. M., Wu Q. (2009) Type II transmembrane serine proteases. J. Biol. Chem. 284, 23177–23181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Antalis T. M., Bugge T. H., Wu Q. (2011) Membrane-anchored serine proteases in health and disease. Prog. Mol. Biol. Transl. Sci. 99, 1–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Basel-Vanagaite L., Attia R., Ishida-Yamamoto A., Rainshtein L., Ben Amitai D., Lurie R., Pasmanik-Chor M., Indelman M., Zvulunov A., Saban S., Magal N., Sprecher E., Shohat M. (2007) Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. Am. J. Hum. Genet. 80, 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cui Y., Wu Q., Zhou Y. (2009) Iron-refractory iron deficiency anemia: new molecular mechanisms. Kidney Int. 76, 1137–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Finberg K. E., Heeney M. M., Campagna D. R., Aydinok Y., Pearson H. A., Hartman K. R., Mayo M. M., Samuel S. M., Strouse J. J., Markianos K., Andrews N. C., Fleming M. D. (2008) Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 40, 569–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guipponi M., Antonarakis S. E., Scott H. S. (2008) TMPRSS3, a type II transmembrane serine protease mutated in non-syndromic autosomal recessive deafness. Front. Biosci. 13, 1557–1567 [DOI] [PubMed] [Google Scholar]
- 23. List K., Currie B., Scharschmidt T. C., Szabo R., Shireman J., Molinolo A., Cravatt B. F., Segre J., Bugge T. H. (2007) Autosomal ichthyosis with hypotrichosis syndrome displays low matriptase proteolytic activity and is phenocopied in ST14 hypomorphic mice. J. Biol. Chem. 282, 36714–36723 [DOI] [PubMed] [Google Scholar]
- 24. Ramsay A. J., Hooper J. D., Folgueras A. R., Velasco G., López-Otín C. (2009) Matriptase-2 (TMPRSS6): a proteolytic regulator of iron homeostasis. Haematologica 94, 840–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szabo R., Bugge T. H. (2011) Membrane-anchored serine proteases in vertebrate cell and developmental biology. Annu. Rev. Cell Dev. Biol. 27, 213–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pan J., Hinzmann B., Yan W., Wu F., Morser J., Wu Q. (2002) Genomic structures of the human and murine corin genes and functional GATA elements in their promoters. J. Biol. Chem. 277, 38390–38398 [DOI] [PubMed] [Google Scholar]
- 27. Dries D. L., Victor R. G., Rame J. E., Cooper R. S., Wu X., Zhu X., Leonard D., Ho S. I., Wu Q., Post W., Drazner M. H. (2005) Corin gene minor allele defined by 2 missense mutations is common in blacks and associated with high blood pressure and hypertension. Circulation 112, 2403–2410 [DOI] [PubMed] [Google Scholar]
- 28. Rame J. E., Drazner M. H., Post W., Peshock R., Lima J., Cooper R. S., Dries D. L. (2007) Corin I555(P568) allele is associated with enhanced cardiac hypertrophic response to increased systemic afterload. Hypertension 49, 857–864 [DOI] [PubMed] [Google Scholar]
- 29. Wang W., Cui Y., Shen J., Jiang J., Chen S., Peng J., Wu Q. (2012) Salt-sensitive hypertension and cardiac hypertrophy in transgenic mice expressing a corin variant identified in blacks. Hypertension 60, 1352–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang W., Liao X., Fukuda K., Knappe S., Wu F., Dries D. L., Qin J., Wu Q. (2008) Corin variant associated with hypertension and cardiac hypertrophy exhibits impaired zymogen activation and natriuretic peptide processing activity. Circ. Res. 103, 502–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cui Y., Wang W., Dong N., Lou J., Srinivasan D. K., Cheng W., Huang X., Liu M., Fang C., Peng J., Chen S., Wu S., Liu Z., Dong L., Zhou Y., Wu Q. (2012) Role of corin in trophoblast invasion and uterine spiral artery remodelling in pregnancy. Nature 484, 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiang J., Wu S., Wang W., Chen S., Peng J., Zhang X., Wu Q. (2011) Ectodomain shedding and autocleavage of the cardiac membrane protease corin. J. Biol. Chem. 286, 10066–10072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qi X., Jiang J., Zhu M., Wu Q. (2011) Human corin isoforms with different cytoplasmic tails that alter cell surface targeting. J. Biol. Chem. 286, 20963–20969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen S., Sen S., Young D., Wang W., Moravec C. S., Wu Q. (2010) Protease corin expression and activity in failing hearts. Am. J. Physiol. Heart Circ. Physiol. 299, H1687–H1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liao X., Wang W., Chen S., Wu Q. (2007) Role of glycosylation in corin zymogen activation. J. Biol. Chem. 282, 27728–27735 [DOI] [PubMed] [Google Scholar]
- 36. Dann C. E., Hsieh J. C., Rattner A., Sharma D., Nathans J., Leahy D. J. (2001) Insights into Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains. Nature 412, 86–90 [DOI] [PubMed] [Google Scholar]
- 37. Chenna R., Sugawara H., Koike T., Lopez R., Gibson T. J., Higgins D. G., Thompson J. D. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Becherer J. D., Blobel C. P. (2003) Biochemical properties and functions of membrane-anchored metalloprotease-disintegrin proteins (ADAMs). Curr. Top. Dev. Biol. 54, 101–123 [DOI] [PubMed] [Google Scholar]
- 40. Garton K. J., Gough P. J., Raines E. W. (2006) Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J. Leukoc. Biol. 79, 1105–1116 [DOI] [PubMed] [Google Scholar]
- 41. Knappe S., Wu F., Madlansacay M. R., Wu Q. (2004) Identification of domain structures in the propeptide of corin essential for the processing of proatrial natriuretic peptide. J. Biol. Chem. 279, 34464–34471 [DOI] [PubMed] [Google Scholar]
- 42. Tran K. L., Lu X., Lei M., Feng Q., Wu Q. (2004) Up-regulation of corin gene expression in hypertrophic cardiomyocytes and failing myocardium. Am. J. Physiol. Heart Circ. Physiol. 287, H1625–H1631 [DOI] [PubMed] [Google Scholar]
- 43. Dong N., Chen S., Yang J., He L., Liu P., Zheng D., Li L., Zhou Y., Ruan C., Plow E., Wu Q. (2010) Plasma soluble corin in patients with heart failure. Circ. Heart Fail. 3, 207–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dong N., Dong J., Liu P., Xu L., Shi S., Wu Q. (2010) Effects of anticoagulants on human plasma soluble corin levels measured by ELISA. Clin. Chim. Acta 411, 1998–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ichiki T., Huntley B. K., Heublein D. M., Sandberg S. M., McKie P. M., Martin F. L., Jougasaki M., Burnett J. C., Jr. (2011) Corin is present in the normal human heart, kidney, and blood, with pro-B-type natriuretic peptide processing in the circulation. Clin. Chem. 57, 40–47 [DOI] [PubMed] [Google Scholar]
- 46. Peleg A., Jaffe A. S., Hasin Y. (2009) Enzyme-linked immunosorbent assay for detection of human serine protease corin in blood. Clin. Chim. Acta 409, 85–89 [DOI] [PubMed] [Google Scholar]
- 47. Dong N., Chen S., Wang W., Zhou Y., Wu Q. (2012) Corin in clinical laboratory diagnostics. Clin. Chim. Acta 413, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ibebuogu U. N., Gladysheva I. P., Houng A. K., Reed G. L. (2011) Decompensated heart failure is associated with reduced corin levels and decreased cleavage of pro-atrial natriuretic peptide. Circ. Heart Fail. 4, 114–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ong K. L., Cheung B. M., Man Y. B., Lau C. P., Lam K. S. (2007) Prevalence, awareness, treatment, and control of hypertension among United States adults 1999–2004. Hypertension 49, 69–75 [DOI] [PubMed] [Google Scholar]