

Background: A natural motif sequence was used as substrate to template hydroxyapatite (HA) formation.
Results: HA crystal formation is attributed to C-terminal amidation of a natural peptide derived from osteocalcin.
Conclusion: The chemical structure of the peptide controls HA crystal formation and growth.
Significance: HA-binding peptides are relevant for biomimetic bone synthesis, hard tissue regeneration, and improvement of osseointegration of biomedical implants.
Keywords: Biomineralization, Bone, Calcification, Electron Microscopy (EM), Peptide Chemical Synthesis, Biomineralization, C-terminal Amidation, Electron Microscopy, Hydroxyapatite, Osteocalcin-derived Peptide
Abstract
Genesis of natural biocomposite-based materials, such as bone, cartilage, and teeth, involves interactions between organic and inorganic systems. Natural biopolymers, such as peptide motif sequences, can be used as a template to direct the nucleation and crystallization of hydroxyapatite (HA). In this study, a natural motif sequence consisting of 13 amino acids present in the first helix of osteocalcin was selected based on its calcium binding ability and used as substrate for nucleation of HA crystals. The acidic (acidic osteocalcin-derived peptide (OSC)) and amidic (amidic osteocalcin-derived peptide (OSN)) forms of this sequence were synthesized to investigate the effects of different C termini on the process of biomineralization. Electron microscopy analyses show the formation of plate-like HA crystals with random size and shape in the presence of OSN. In contrast, spherical amorphous calcium phosphate is formed in the presence of OSC. Circular dichroism experiments indicate conformational changes of amidic peptide to an open and regular structure as a consequence of interaction with calcium and phosphate. There is no conformational change detectable in OSC. It is concluded that HA crystal formation, which only occurred in OSN, is attributable to C-terminal amidation of a natural peptide derived from osteocalcin. It is also proposed that natural peptides with the ability to promote biomineralization have the potential to be utilized in hard tissue regeneration.
Introduction
Most natural biomaterials composed of organic and inorganic components, such as bone, cartilage, and teeth, are difficult to synthesize. The ability to control the formation of inorganic crystals using natural peptides would be a valuable tool in tissue engineering for the restoration and regeneration of hard tissues. The formation of natural bone is thought to be regulated by templated mineralization of hydroxyapatite (HA)3 and proteins such as osteonectin, bone sialoprotein, osteocalcin, and collagen type I (1, 2). Proteins play a significant role in crystal nucleation, growth, and morphology (3–5) and enable the process of biomineralization to occur under ambient temperature and pressure conditions (6). Therefore, biological templated mineralization could be employed to synthesize bone mimetic materials. This process is thought to occur by binding the acidic groups of peptides with calcium ions and aligning them in an orientation that matches the crystal structure of HA (7). Studies have shown that a peptide motif could promote the nucleation and growth of HA crystals (8–10). Enhancing HA-binding peptides could promote biomimetic osteogenesis.
Peptide motifs are usually categorized on the basis of their origin. They could be derived from native proteins having conserved fragments in different organisms or created by artificial evolution (11). Several groups have used artificial evolutionary systems to identify HA-binding peptides (12–14). On the other hand, natural peptides are mostly fragments of bone tissue proteins that are known to be involved in the mediation of HA formation. The amino acid sequence in these proteins is capable of binding to HA and makes a template for the structural integration of organic and inorganic matrices. Noncollagenous proteins involved in this process are rich in amino acids with acidic residues that have the capacity to bind with metal ions and initiate the mineralization process (15). For example, calcium-binding peptides derived from dentin matrix protein 1 (DMP1) affect in vitro nucleation of HA (16), and the glutamic acid-rich region of bone sialoprotein is involved in HA nucleation (17). Although much research has been devoted to the application of HA-binding peptides for HA mineralization, there has been little investigation of natural peptides derived from osteocalcin.
Osteocalcin (OC) is one of the noncollagenous proteins of bone tissue secreted by mature osteoblasts and osteocytes (18, 19). The most important functions assigned to OC are regulation of glucose homeostasis and bone mineralization. It has also been postulated that uncarboxylated osteocalcin is involved in glucose metabolism (20, 21). However, most OC is found in bone tissue, and OC level in serum is considered a specific marker of bone formation (22). Gla side chains of OC bind strongly to hydroxyapatite (23) through mineral interactions (24). Determination of the crystal structure of OC has clearly shown excellent correspondence between the prism face of HA crystals and the calcium-coordinating sites of OC (25). The presence of three glutamates in the first helix of OC allows for calcium binding (26). It should be noted that OC is sensitive to several common proteases, and the proteolytic activity of these enzymes may produce fragmentation in the serum (27–29). However, the first helix of OC is located in the middle region and is resistant to fragmentation because of the structure of the molecules and protease inaccessibility (26). All fragments of OC found in vivo are acidic forms with no amidated fragments reported to date (29, 30).
C-terminal α-amide is present in the majority of peptide hormones, particularly in the nervous and endocrine systems (31). In comparison with C-terminal acids, C-terminal amides considerably enhance peptide activity. Therefore, amidation of a C terminus has been correlated to peptide bioactivity (32). Several researchers compared the molecular conformation and intermolecular interactions of C-terminal amidated peptides (C-amide) and C-terminal acidic peptides (C-acid) (33, 34). It was shown that C-terminal amidation largely influences the molecular conformation of the peptide and in turn the interaction with a receptor (35). The peptides used in such studies were, however, mostly antimicrobial and hormone peptides. Thus, the effect of C-terminal amidation on the process of biomineralization is unknown.
In our study, a natural motif sequence consisting of 13 amino acids present in the middle region and first helix of OC was selected based on its calcium binding ability. This peptide motif sequence was synthesized using the solid phase peptide synthesis method in both acidic and amidic forms to investigate the effect of C-terminal amidation (transformation of terminal COOH groups into C-terminal CONH2 groups) on HA crystal formation.
EXPERIMENTAL PROCEDURES
Peptide Synthesis, Purification, and Characterization
Comparison of OC from different organisms shows that the first helix of OC is highly conserved. The amino acid sequence of this region is LEPRREVCELNPD. We synthesized this sequence in both amidated C-terminal (Leu- … -Asn-Pro-Asp-NH2) form and free C-terminal acid (Leu- … -Asn-Pro-Asp-OH) form. From this point on, we shall refer to these forms as OSN and OSC, respectively. OSC and OSN were constructed manually by stepwise solid-phase synthesis. Peptide coupling was performed on a Fmoc-Asp-Wang resin and Fmoc amide resin according to the standard Fmoc chemistry (36, 37). The C-terminal amide was obtained using Rink amide (4-(2′,4′-dimethoxyphenyl-Fmoc-aminomethyl) phenoxy) resin. After each coupling reaction, the Fmoc group was removed by 20% piperidine in dimethylformamide for 15 min. For the final deblocking and cleavage, the dried protected peptide resin was suspended in trifluoroacetic acid (TFA), water, and triisopropylsilane (95, 2.5, and 2.5) for 2 h at room temperature with constant agitation. The peptide was filtered, and ice-cold dry diethyl ether was added to the filtrate for precipitation. The peptides were purified by reverse-phase HPLC (Pfizer Inc., Stockholm, Sweden) using C18 column with a flow rate of 3.0 ml/min and the linear gradient of solvents A (0.05% TFA in H2O) and B (0.05% TFA in CH3CN). The UV detector measured absorbance at 215 and 280 nm. OSC and OSN were checked for homogeneity by analytical reverse-phase HPLC, which indicated a purity >95%. Molecular weights were confirmed by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF; Mass Spectrometry Services, Nanyang Technological University).
In Vitro Biomineralization
Lyophilized peptides were weighed and dissolved in Tris-HCl buffer (pH = 7.4) to achieve a 2 mg/ml concentration. Calcium- and phosphate-containing solutions were prepared using reagent-grade CaCl2 and Na2HPO4 (Sigma-Aldrich, >99.0% purity) with a concentration of 10 and 5 mm, respectively. The pH of the Na2HPO4 solution was subsequently adjusted to 7.4. To investigate the effects of OSC and OSN on HA nucleation, 5 μl of each peptide solution was added to a micro test tube followed by the addition of 5 μl of CaCl2 and Na2HPO4 solutions to achieve a final concentration of 1.6 mm Na2HPO4 and 3.3 mm CaCl2, which are close to physiological concentrations (pH = 7.4) (38, 39).
Electron Microscopy Analysis
Conventional transmission electron microscopy (TEM), cryogenic-TEM (cryo-TEM), and scanning electron microscopy (SEM) were used to characterize the formation of HA crystals in the presence of OSC and OSN peptides. Cryo-TEM enables the direct imaging of self-aggregation in liquid systems as samples can be rapidly frozen, thus avoiding damage from the formation of ice crystals, which preserves ultrastructure without chemical staining or drying artifacts. This technique allows the visualization of fully hydrated samples, which is otherwise impossible in TEM. For analysis in conventional TEM, 10 μl of each solution containing OSC and OSN was immediately pipetted onto a carbon-coated Cu TEM grid. For the negative control experiments, the same concentration of CaCl2 and Na2HPO4 in Tris-HCl buffer in the absence of peptides was used. The grids were incubated for 30, 60, 90, and 120 min at room temperature. To minimize evaporation, all grids were placed in a sealed wet chamber. Three sets of each sample including OSC, OSN, and control were prepared as described and then observed in TEM and SEM analysis. TEM images were obtained using a FEI Tecnai 12 at 120 kV equipped with an AMT XR-80C charge-coupled device camera system. Samples were prepared using a similar method for SEM analysis. SEM images were taken using a Hitachi S-4700 field-emission SEM. For imaging in cryo-TEM, mineralized suspensions were applied to Quantifoil grids (Quantifoil Micro Tools GmbH, Jena, Germany) and blotted and rapidly frozen in liquid ethane using a FEI Vitrobot Mark IV (FEI Co., Eindhoven, The Netherlands). Imaging of the samples was done with a FEI Tecnai G2 F20 cryo-STEM at 200 kV equipped with a Gatan 626 single tilt cryotransfer system and Gatan UltraScan 4000 (4k × 4k) charge-coupled device camera.
Negative Staining
Negative staining was performed by pipetting 5 μl of each peptide, as well as peptides incubated with 10 mm CaCl2, onto a Formvar grid for 60 s. 10 μl of 2% uranyl acetate was then pipetted onto the grid and let stand for 1 min, and excess liquid was blotted with filter paper. Specimens were examined with a FEI Tecnai 12 TEM.
Circular Dichroism
All circular dichroism (CD) spectra were obtained using a JASCO J-810 spectropolarimeter (JASCO Corp., Tokyo, Japan) in a 1-mm quartz cell at 25 °C. The instrument was calibrated with d10-camphorsulfonic acid. Peptide stock solutions were diluted with Tris-HCl buffer (pH = 7.4) to yield a final concentration of 0.2 mg/ml. A 2,2,2-trifluroethanol (TFE) titration experiment was carried out with 10, 50, and 90% v/v of TFE. To investigate the effects of calcium and phosphate ions on the secondary structure of OSC and OSN, each peptide was incubated with 10 mm CaCl2 and 5 mm Na2HPO4 separately. The spectra were recorded with a wavelength scan of 185–250 nm with the appropriate buffer and solvent background subtraction. Each spectrum is reported as the average of five scans using 1 nm bandwidth and 0.5 nm/s scanning rate. In all CD spectra, the mean residue ellipticity [θ] is expressed in degrees cm2 dmol−1.
Particle Size and ζ Potential Measurements
The mean particle size of the assembled peptides in the absence and presence of calcium and phosphate ions was determined by dynamic light scattering using photon correlation spectroscopy. The measurements were performed using a Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, Worcestershire, UK) equipped with a helium-neon laser at 25 °C and a scattering angle of 173°. Additionally, the ζ potential of the peptides was measured with the same instrument at 25 °C by electrophoretic mobility.
Statistical Analysis
Data were analyzed using one-way analysis of variance with Tukey's multiple comparison test to a confidence level of p < 0.05.
RESULTS
Peptide Synthesis and Characterization
Fig. 1 represents the sequence of OC and the 13 amino acids selected for this study. Table 1 shows the physicochemical properties of the synthesized peptides. These data are confirmed by MALDI-TOF mass spectrometry. OSC and OSN peptides have similar molecular mass, pI, and grand average of hydropathicity (GRAVY), but different net charge at pH = 7.0, which indicates that both peptides are hydrophilic (40).
FIGURE 1.
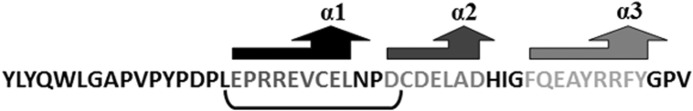
The amino acid sequence of osteocalcin. The osteocalcin sequence and the 13 amino acids in the first helix (underlined) that were selected to build the OSC and OSN are shown.
TABLE 1.
Physicochemical properties of the synthesized peptides
Physicochemical properties of OSC and OSN including pI, molecular mass, GRAVY, and net charge at pH = 7 were calculated using the Innovagen site. Both peptides have a similar molecular mass, pI, and GRAVY.
| Molecular mass | pI | Net charge at pH = 7.0 | GRAVY | |
|---|---|---|---|---|
| Da | ||||
| OSC | 1569.7 | 4.4 | −2 | −1.185 |
| OSN | 1568.8 | 4.6 | −1 | −1.185 |
Biomineralization
Electron microscopy was used to compare the effects of the synthesized peptides on the nucleation and growth of HA. HA nucleation and crystallization were investigated in conventional TEM after 30, 60, 90, and 120 min of incubation (Fig. 2). After 30 min, spherical particles were observed both in the control sample (Fig. 2a) and in the presence of OSC and OSN (Fig. 2, b and c). This morphology is usually indicative of the formation of amorphous calcium phosphate (ACP). Selected area electron diffraction (SAED) analysis confirmed the amorphous nature of these nanoparticles (Fig. 2a, inset). After 1 h, the size of the nanoparticles did not change significantly in the OSC and control groups, whereas the particles grew significantly in the presence of OSN (Fig. 2, d–f). Thin plate-like particles started to form from the surfaces of the ACP particles in the presence of both OSC and OSN after 1 h. These interweaving ridges were found to be more significant in the presence of OSN (Fig. 2f). After 90 min, the mean size of the ACP spheres increased in the control group (Fig. 2g). Thin plate-like particles formed at 60 min and increased in abundance after 90 min on the OSC sample (Fig. 2h). Crystal formation was also substantially promoted, and plate like-crystals formed and almost completely replaced the ACP spheres on the OSN sample (Fig. 2i). At 120 min, thin plate-like particles appeared in the control group from the network of amorphous calcium phosphate (Fig. 2j). There was no crystal formation on the OSC sample, whereas the OSN sample showed a complete transformation of the ACP spheres to thin plate-like crystals (Fig. 2, k and l). Fig. 3 shows high magnification TEM images of the control, OSC, and OSN samples after 120 min of incubation and clearly shows the formation of HA crystals on the OSN.
FIGURE 2.

TEM images of calcium phosphate minerals formed in the presence of OSC, OSN, and control. Calcium phosphate minerals formed in the control sample and in the presence of OSC and OSN after 30 (a–c), 60 (d–f), 90 (g–i), and 120 (j–l) min of incubation are presented. 5 μl of each peptide solution was added to a micro test tube followed by the addition of 5 μl of CaCl2 and Na2HPO4 solutions to achieve a final concentration of 1.6 mm Na2HPO4 and 3.3 mm CaCl2 (pH = 7.4). 10 μl of each solution containing OSC and OSN was immediately pipetted onto a carbon-coated Cu TEM grid. For the control experiments, the same concentration of CaCl2 and Na2HPO4 in Tris-HCl buffer in the absence of peptides was used. Amorphous calcium phosphate deposit was observed in the control and OSC samples, whereas crystalline calcium phosphate was formed in the presence of OSN. In a, the inset shows a SAED pattern that represents the ACP structure.
FIGURE 3.

TEM images of calcium phosphate minerals and related SAED patterns. High magnification TEM images of the control (a), OSC (b), and OSN (c) after 120 min of incubation are presented. The formation of HA crystals is clear in the presence of OSN. Arrows show the projection of HA crystals aligned perpendicular (white) and parallel (black) to the substrate. SAED patterns of the particles formed in the presence of OSC and control show the amorphous structure, whereas in the presence of OSN, single crystal diffraction patterns are indexed as the HA (002), (211), and (004) crystallographic planes.
The amorphous nature of the particles formed in the presence of OSC and in the control sample after 120 min was confirmed by SAED (Fig. 3, a and b). In the presence of OSN, single crystal diffraction patterns were indexed as the HA (002), (211), and (004) planes along the crystal c axis (Fig. 3c). The interplanar d-spacings of these planes relative to the diffraction planes (002), (211), and (004) were calculated as 0.28, 0.35, and 0.17 nm, respectively, which correspond to the crystallographic structure of HA. High resolution TEM images of OSC and OSN samples are presented in Fig. 4. Lattice fringes of the OSN crystals have a d-spacing of 0.36 nm, which is in agreement with theoretical data for the (211) plane of HA (41). Energy-dispersive x-ray analysis indicated that crystals formed in the presence of OSN had a Ca/P ratio of 1.67 ± 0.003, which matches the theoretical Ca/P ratio for HA, whereas a ratio of 1.27 ± 0.001 was obtained from the OSC sample.
FIGURE 4.

High resolution TEM images and energy-dispersive x-ray analysis spectra of minerals formed in the presence of OSC and OSN. High resolution TEM image of calcium phosphate mineral formed in the presence of OSC (a) and crystal lattice fringe image of OSN (b) are shown. A d-spacing of 0.36 nm was calculated for the OSN, which is in agreement with theoretical data for the (211) crystallographic plane of HA. Energy-dispersive x-ray analysis indicated the Ca/P ratio of 1.27 ± 0.001 for OSC and 1.67 ± 0.003 for OSN, which matches the theoretical Ca/P ratio for HA.
SEM was used to observe the morphology of the particles formed in the presence of OSC, OSN, and control samples after 120 min of incubation. Spherical particles were formed both in the OSC and in the control samples, having only minor differences in shape and size (Fig. 5, a and b). The OSN sample, however, had a completely different morphology characterized as noticeably large crystals with rough, spicular surfaces (Fig. 5c).
FIGURE 5.

SEM images of formed calcium phosphate minerals in the presence of OSC, OSN, and control. SEM images of control (a), OSC (b), and OSN (c) after 120 min of incubation are displayed. Spherical particles were formed both in the OSC and in the control samples, having only minor differences in shape and size. The OSN sample, however, had a completely different morphology characterized as noticeably large crystals with rough, spicular surfaces.
Fig. 6 is a high resolution cryo-TEM image of the nanocrystals formed in the presence of OSN after 120 min of incubation. Negative staining of OSC and OSN showed that both peptides underwent a similar degree of assembly and aggregation in the presence of Ca2+ (Fig. 7), which was not observed in the absence of Ca2+ (Fig. 7, a and b).
FIGURE 6.

Cryo-TEM image of HA crystals formed in the presence of OSN. For imaging in cryo-TEM, mineralized suspensions were applied to Quantifoil grids, blotted, and rapidly frozen in liquid ethane using a FEI Vitrobot Mark IV. Imaging of the samples was done with a FEI Tecnai G2 F20 cryo-STEM.
FIGURE 7.

Negatively stained TEM images of OSC and OSN. TEM images of OSC and OSN negatively stained with 2% (w/v) uranyl acetate (a and b) and after incubation with 10 mm CaCl2 (c and d), respectively, are presented. The images show a similar degree of assembly in the presence of Ca2+ for both peptides.
Circular Dichroism
CD measurements were performed to ascertain the secondary structure of the synthesized peptides. Results showed that both OSC and OSN had a random coil structure, as indicated by the π—π* transition band at 198–200 nm (Fig. 8, a and b) (42). In the absence of TFE, the negative band at 198 nm for OSN decreased to lower molar ellipticity when compared with OSC. Different percentages of TFE were added to the solution to investigate the ability of OSC and OSN to form secondary ordered structures (43). TFE is an organic solvent that provides a suitable environment for a peptide to adapt a structure in solution. It also has the ability to induce and stabilize the folded structure of a peptide (43). As seen in Fig. 8, a and b, TFE had a profound effect on the conformation of both peptides. After increasing the TFE concentration, the band at 198–200 nm disappeared, and two ellipticity bands appeared at 205–208 and 218–222 nm. These bands are relative to a π—π* transition and an n—π* transition, respectively, which are indicative of an α-helical structure (44). This indicated that the random coil structure of both peptides could be changed to a regular helical structure in the presence of TFE. CD spectra were also measured in the presence of Ca2+ and HPO42− (Fig. 8, c and d). Although Ca2+ and HPO42− did not significantly alter the random coil structure of OSC, a larger change was observed for OSN.
FIGURE 8.

Circular dichroism analysis of OSC and OSN. CD spectra of OSC (a) and OSN (b) were obtained with 10, 50, and 90% v/v of TFE. It is revealed that random coil structure of both peptides is changed to an α-helical structure in the presence of TFE. deg, degrees. OSC (c) and OSN (d) after the addition of 10 mm CaCl2 (red lines) and 5 mm Na2HPO4 (green lines), in comparison with OSC and OSN solutions without Ca2+ and HPO42− ions (blue lines), show that Ca2+ and HPO42− did not alter the random coil structure of OSC significantly, whereas a larger change was observed for OSN.
Particle Size and ζ Potential Measurements
ζ potential results showed a surface charge of −6.59 ± 1.22 mV and −14.4 ± 0.98 mV for OSN and OSC, respectively, confirming that OSN is more positive than OSC in its assembled form (Table 2). The mean particle size for OSN and OSC was 444.7 ± 19.48 and 450 ± 15.50 nm, respectively (Table 2). The addition of calcium and phosphate ions to OSN decreased both the ζ potential (−12.4 ± 0.70 and −28.5 ± 0.70 mV for calcium and phosphate, respectively) and the mean particle size (376.2 ± 14.79 and 140.3 ± 12.03 nm for calcium and phosphate, respectively) of the assembled peptide (Table 2). These reductions were much more significant with the addition of phosphate when compared with calcium ions (p < 0.006). In the case of OSC, however, the addition of calcium and phosphate ions increased the mean particle size of the peptide from 450 ± 15.50 nm to 605.0 ± 23.85 and 603.0 ± 22.55 nm for calcium and phosphate, respectively (Table 2). The ζ potential of OSC decreased after the addition of phosphate ions (from −14.4 ± 0.98 to −19.4 ± 1.20 mV) and increased after the addition of calcium ions (from 14.4 ± 0.98 to −10.1 ± 0.80 mV) (Table 2). It is therefore apparent that the addition of calcium and phosphate ions caused aggregation of OSC and disassembly of OSN.
TABLE 2.
Particle size and ζ potential analysis
The mean ± S.D. particle size and ζ potential values for OSC and OSN with and without the addition of Ca2+ and HPO42− are reported.
| Group | Sample | Particle size | ζ potential |
|---|---|---|---|
| nm | mV | ||
| 1 | OSC | 450.7 ± 15.50 | −14.4 ± 0.98 |
| 2 | OSC/Ca2+ | 605.0 ± 23.85 | −10.1 ± 0.80 |
| 3 | OSC/HPO42 − | 603.0 ± 22.55 | −19.4 ± 1.20 |
| 4 | OSN | 444.7 ± 19.48 | −6.59 ± 1.22 |
| 5 | OSN/Ca2+ | 376.2 ± 14.79 | −12.4 ± 0.70 |
| 6 | OSN/HPO42 − | 140.3 ± 12.03 | −28.5 ± 0.70 |
DISCUSSION
In this study, we show that peptide sequences composed of the 13 amino acids present in the first helix of OC in both acidic (OSC) and amidic (OSN) forms have significant effect on the mode of HA formation. Although plate-like crystals with random size and shape are formed in the presence of OSN after 120 min of incubation, only ACP spheres are produced in the presence of OSC similar to those formed in control samples without peptides.
HA crystallization is a sequential process whereby an amorphous precursor phase transforms into crystalline HA (45). Our TEM results show that ACP was similarly formed in the OSC, OSN, and control samples within the first 30 min of incubation. Gradually, thin plate-like particles start to appear from the surface of ACP particles in the presence of OSC and OSN. These particles are eventually transformed to plate-like crystals in the presence of OSN. These findings show that ACP formation is not mediated by the peptides because it is also observed in the control sample. Thus, we believe that OSN enhances the transformation of ACP to crystalline HA. To explain why OSN would have this effect, whereas OSC did not, we can consider the structure and surface charge of the two peptides. The addition of TFE to the OSC and OSN solutions used in far-ultraviolet CD experiments shows the transition of the unstructured conformation (coil) of these peptides to a folded structure (helix) in aqueous solution. The effect of calcium and phosphate ions on the conformational state of peptides measured by CD shows that although there is no significant effect on the OSC structure upon the addition of calcium and phosphate, there is a visible change in the conformation of OSN. To address the interaction of calcium and phosphate ions with OSC and OSN on the basis of their surface charges more precisely, ζ potential and particle size analyses were performed. The significant drop of ζ potential and decrease in particle size of OSN after the addition of calcium and phosphate ions suggest that both ions, but particularly phosphate, inhibit the assembly of OSN. Therefore, the number of OSN peptides in solution that could interact with phosphate and calcium increases. Conversely, the increasing size of OSC suggests that the addition of phosphate and calcium ions facilitates its assembly. Based on these results, therefore, larger changes in conformation, surface charge, and particle size are induced in OSN, in comparison with OSC, after the addition of either Ca2+ or HPO42−. This may be related to the fact that amidation brings the C-terminal group to a neutral state, which in turn decreases the repulsion of carboxyl groups in the aspartic acid. This is known to play a key role in preventing the formation of a folded conformation for the C-terminal amidated peptide (46). An open conformation for OSN would allow more side chain interaction with calcium and phosphate ions (35). Indeed, proteins involved in biomineralization, such as OC, have a disordered, open conformation that allows for enhanced intermolecular interactions at the protein-mineral interface (47, 48).
The other point that needs to be mentioned is that peptides containing acidic residues, such as Glu, Asp, and phosphoserine with a low isoelectric point, have been shown to interact efficiently with calcium ions. Because of their increased net negative charge, these peptides will promote nucleation and growth of HA (49–52). The effect of Ca+2 ions in assembly and aggregation of both OSC and OSN was evidenced by particle size analysis and negative stain results. These results, combined with the results obtained from the ζ potential and CD experiments, suggest that the increased conversion of amorphous calcium phosphate to crystalline HA observed in OSN is due to the interaction of the peptide with phosphate ions caused by the conformational changes in the OSN. According to data in the literature, the interaction of OSN with phosphate besides calcium would create a local supersaturation of ions that consequently would promote the transformation of the amorphous phase to the crystalline phase (6). Moreover, according to ζ potential results, OSN has a more positive surface charge when compared with OSC and therefore has higher affinity for negative charge. As ACP has a negative surface charge, we expect a higher degree of interaction between OSN and ACP relative to OSC (53). The decrease in activation energy of OSN-ACP binding would contribute to the specific nucleation and crystallization of HA (8). Finally, images from conventional and cryo-TEM show that HA crystals formed in the presence of OSN are plate-like (Fig. 3c, black arrows). In contrast to data presented in the literature, there is no evidence of an acicular habit of HA crystals in our study. The acicular habit often observed in TEM images is likely the projection of HA crystals aligned perpendicular to the substrate and parallel to electron beam (Figs. 3 and 6, white arrows). This is clear in the cryo-TEM image where the crystals are freely oriented in the ice.
In summary, our study demonstrates that C-terminal amidation improves the biomineralization activity of peptides. Although the exact mechanism of OSN-mediated crystal formation is not known, our study using CD and ζ potential analyses shows that OSN and OSC exhibit significant differences with respect to surface charge and conformational changes. This suggests that the transformation of COOH to CONH2 groups affects the peptide conformational state and enhances its interaction with both calcium and phosphate ions, thus effectively promoting HA crystallization. However, additional structural and conformational studies using NMR spectroscopy might provide a better understanding of the dynamics of the molecular mechanisms involved in HA crystallization.
Acknowledgments
We thank Dr. Isabelle Rouiller (McGill University) for cooperation and appreciate the cooperation and guidance of Dr. S. Kelly Sears, Line Mongeon (SEM), Dr. David Liu (TEM), and Jeannie Mui (TEM) of the Facility for Electron Microscopy Research at McGill University, Dr. Valentine Dan (McGill University), and Dr. Leila Hassani (Institute for Advanced Studies in Basic Sciences) for the help in CD.
This work was supported by the Tarbiat Modares University and Canada Research Chair Foundation (to M. C.) and the Natural Sciences and Engineering Research Council (NSERC) of Canada (to H. V.).
- HA
- hydroxyapatite
- OC
- osteocalcin
- OSC
- acidic osteocalcin-derived peptide
- OSN
- amidic osteocalcin-derived peptide
- Fmoc
- 9-fluorenyl-methoxy carbonyl
- TFE
- 2,2,2-trifluroethanol
- TEM
- transmission electron microscopy
- cryo-TEM
- cryogenic-TEM
- GRAVY
- grand average of hydropathicity
- ACP
- amorphous calcium phosphate
- SAED
- selected area electron diffraction.
REFERENCES
- 1. Young M. F., Kerr J. M., Ibaraki K., Heegaard A. M., Robey P. G. (1992) Structure, expression, and regulation of the major noncollagenous matrix proteins of bone. Clin. Orthop. Relat. Res. 281, 275–294 [PubMed] [Google Scholar]
- 2. Hunter G. K., Hauschka P. V., Poole A. R., Rosenberg L. C., Goldberg H. A. (1996) Nucleation and inhibition of hydroxyapatite formation by mineralized tissue proteins. Biochem. J. 317, 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Falini G., Albeck S., Weiner S., Addadi L. (1996) Control of aragonite or calcite polymorphism by mollusk shell macromolecules. Science 271, 67–69 [Google Scholar]
- 4. Belcher A. M., Wu X. H., Christensen R. J., Hansma P. K., Stucky G. D., Morse D. E. (1996) Control of crystal phase switching and orientation by soluble mollusc-shell proteins. Nature 381, 56–58 [Google Scholar]
- 5. Addadi L., Weiner S. (1985) Interactions between acidic proteins and crystals: Stereochemical requirements in biomineralization. Proc. Natl. Acad. Sci. 82, 4110–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beniash E. (2011) Biominerals-hierarchical nanocomposites: The example of bone. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 3, 47–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldberg H. A., Warner K. J., Li M. C., Hunter G. K. (2001) Binding of bone sialoprotein, osteopontin, and synthetic polypeptides to hydroxyapatite. Connect. Tissue Res. 42, 25–37 [DOI] [PubMed] [Google Scholar]
- 8. Chung W. J., Kwon K. Y., Song J., Lee S. W. (2011) Evolutionary screening of collagen-like peptides that nucleate hydroxyapatite crystals. Langmuir 27, 7620–7628 [DOI] [PubMed] [Google Scholar]
- 9. Fujisawa R., Kuboki Y. (1991) Preferential adsorption of dentin and bone acidic proteins on the (100) face of hydroxyapatite crystals. Biochim. Biophys. Acta 1075, 56–60 [DOI] [PubMed] [Google Scholar]
- 10. Gungormus M., Branco M., Fong H., Schneider J. P., Tamerler C., Sarikaya M. (2010) Self assembled bi-functional peptide hydrogels with biomineralization-directing peptides. Biomaterials 31, 7266–7274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shiba K. (2010) Natural and artificial peptide motifs: their origins and the application of motif-programming. Chem. Soc. Rev. 39, 117–126 [DOI] [PubMed] [Google Scholar]
- 12. Segvich S. J., Smith H. C., Kohn D. H. (2009) The adsorption of preferential binding peptides to apatite-based materials. Biomaterials 30, 1287–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roy M. D., Stanley S. K., Amis E. J., Becker M. L. (2008) Identification of a highly specific hydroxyapatite-binding peptide using phage display. Adv. Mater. 20, 1830–1836 [Google Scholar]
- 14. Gungormus M., Fong H., Kim I. W., Evans J. S., Tamerler C., Sarikaya M. (2008) Regulation of in vitro calcium phosphate mineralization by combinatorially selected hydroxyapatite-binding peptides. Biomacromolecules 9, 966–973 [DOI] [PubMed] [Google Scholar]
- 15. Gotliv B. A., Kessler N., Sumerel J. L., Morse D. E., Tuross N., Addadi L., Weiner S. (2005) Asprich: A novel aspartic acid-rich protein family from the prismatic shell matrix of the bivalve Atrina rigida. Chembiochem. 6, 304–314 [DOI] [PubMed] [Google Scholar]
- 16. He G., Dahl T., Veis A., George A. (2003) Nucleation of apatite crystals in vitro by self-assembled dentin matrix protein 1. Nat. Mater. 2, 552–558 [DOI] [PubMed] [Google Scholar]
- 17. Hunter G. K., Goldberg H. A. (1994) Modulation of crystal formation by bone phosphoproteins: Role of glutamic acid-rich sequences in the nucleation of hydroxyapatite by bone sialoprotein. Biochem. J. 302, 175–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aarden E. M., Wassenaar A. M., Alblas M. J., Nijweide P. J. (1996) Immunocytochemical demonstration of extracellular matrix proteins in isolated osteocytes. Histochem. Cell Biol. 106, 495–501 [DOI] [PubMed] [Google Scholar]
- 19. Ducy P., Desbois C., Boyce B., Pinero G., Story B., Dunstan C., Smith E., Bonadio J., Goldstein S., Gundberg C., Bradley A., Karsenty G. (1996) Increased bone formation in osteocalcin-deficient mice. Nature 382, 448–452 [DOI] [PubMed] [Google Scholar]
- 20. Lee N. K., Sowa H., Hinoi E., Ferron M., Ahn J. D., Confavreux C., Dacquin R., Mee P. J., McKee M. D., Jung D. Y., Zhang Z., Kim J. K., Mauvais-Jarvis F., Ducy P., Karsenty G. (2007) Endocrine regulation of energy metabolism by the skeleton. Cell 130, 456–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferron M., Hinoi E., Karsenty G., Ducy P. (2008) Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc. Natl. Acad. Sci. 105, 5266–5270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delmas P. D., Eastell R., Garnero P., Seibel M. J., Stepan J. (2000) The use of biochemical markers of bone turnover in osteoporosis. Committee of Scientific Advisors of the International Osteoporosis Foundation. Osteoporos. Int. 11, Suppl. 6, S2–S17 [DOI] [PubMed] [Google Scholar]
- 23. Price P. A., Poser J. W., Raman N. (1976) Primary structure of the γ-carboxyglutamic acid-containing protein from bovine bone. Proc. Natl. Acad. Sci. 73, 3374–3375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poser J. W., Price P. A. (1979) A method for decarboxylation of γ-carboxyglutamic acid in proteins: properties of the decarboxylated γ-carboxyglutamic acid protein from calf bone. J. Biol. Chem. 254, 431–436 [PubMed] [Google Scholar]
- 25. Hoang Q. Q., Sicheri F., Howard A. J., Yang D. S. C. (2003) Bone recognition mechanism of porcine osteocalcin from crystal structure. Nature 425, 977–980 [DOI] [PubMed] [Google Scholar]
- 26. Hauschka P. V., Carr S. A. (1982) Calcium-dependent α-helical structure in osteocalcin. Biochemistry 21, 2538–2547 [DOI] [PubMed] [Google Scholar]
- 27. Page A. E., Hayman A. R., Andersson L. M., Chambers T. J., Warburton M. J. (1993) Degradation of bone matrix proteins by osteoclast cathepsins. Int. J. Biochem. 25, 545–550 [DOI] [PubMed] [Google Scholar]
- 28. Novak J. F., Hayes J. D., Nishimoto S. K. (1997) Plasmin-mediated proteolysis of osteocalcin. J. Bone Miner. Res. 12, 1035–1042 [DOI] [PubMed] [Google Scholar]
- 29. Garnero P., Grimaux M., Seguin P., Delmas P. D. (1994) Characterization of immunoreactive forms of human osteocalcin generated in vivo and in vitro. J. Bone Miner. Res. 9, 255–264 [DOI] [PubMed] [Google Scholar]
- 30. Srivastava A. K., Mohan S., Singer F. R., Baylink D. J. (2002) A urine midmolecule osteocalcin assay shows higher discriminatory power than a serum midmolecule osteocalcin assay during short-term alendronate treatment of osteoporotic patients. Bone 31, 62–69 [DOI] [PubMed] [Google Scholar]
- 31. Eipper B. A., Mains R. E. (1988) Peptide α-amidation. Annu. Rev. Physiol. 50, 333–344 [DOI] [PubMed] [Google Scholar]
- 32. Strandberg E., Tiltak D., Ieronimo M., Kanithasen N., Wadhwani P., Ulrich A. S. (2007) Influence of C-terminal amidation on the antimicrobial and hemolytic activities of cationic α-helical peptides. Pure Appl. Chem. 79, 717–728 [Google Scholar]
- 33. Potetinova Z., Barbier J. R., Suen T., Dean T., Gardella T. J., Willick G. E. (2006) C-terminal analogues of parathyroid hormone: Effect of C-terminus function on helical structure, stability, and bioactivity. Biochemistry 45, 11113–11121 [DOI] [PubMed] [Google Scholar]
- 34. In Y., Fujii M., Sasada Y., Ishida T. (2001) Structural studies on C-amidated amino acids and peptides: Structures of hydrochloride salts of C-amidated Ile, Val, Thr, Ser, Met, Trp, Gln, and Arg, and comparison with their C-unamidated counterparts. Acta Crystallogr. B. 57, 72–81 [DOI] [PubMed] [Google Scholar]
- 35. In Y., Minoura K., Tomoo K., Sasaki Y., Lazarus L. H., Okada Y., Ishida T. (2005) Structural function of C-terminal amidation of endomorphin. Conformational comparison of μ-selective endomorphin-2 with its C-terminal free acid, studied by 1H-NMR spectroscopy, molecular calculation, and X-ray crystallography. FEBS J. 272, 5079–5097 [DOI] [PubMed] [Google Scholar]
- 36. Chan W. C., White P. D. (2000) Basic procedures. in Fmoc Solid Phase Peptide Synthesis: A Practical Approach (Chan W. C., White P. D., eds) pp. 41–76, Oxford University Press, New York [Google Scholar]
- 37. Howl J. (2005) Fundamentals of modern peptide synthesis. in Peptide synthesis and applications (Amblard M., Fehrentz J.-A., Martinez J., Gilles S., eds) pp. 3–24, Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 38. Deshpande A. S., Beniash E. (2008) Bio-inspired synthesis of mineralized collagen fibrils. Cryst. Growth Des. 8, 3084–3090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Linde A., Lundgren T. (1995) From serum to the mineral phase. The role of the odontoblast in calcium transport and mineral formation. Int. J. Dev. Biol. 39, 213–222 [PubMed] [Google Scholar]
- 40. Kyte J., Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 41. Vali H., McKee M. D., Ciftcioglu N., Sears S. K., Plows F. L., Chevet E., Ghiabi P., Plavsic M., Kajander E. O., Zare R. N. (2001) Nanoforms: A new type of protein-associated mineralization. Geochim. Cosmochim. Acta 65, 63–74 [Google Scholar]
- 42. Kulp J. L., 3rd, Shiba K., Evans J. S. (2005) Probing the conformational features of a phage display polypeptide sequence directed against single-walled carbon nanohorn surfaces. Langmuir 21, 11907–11914 [DOI] [PubMed] [Google Scholar]
- 43. Kulp J. L., 3rd, Minamisawa T., Shiba K., Tejani M., Evans J. S. (2007) Structural properties of an artificial protein that regulates the nucleation of inorganic and organic crystals. Langmuir 23, 3857–3863 [DOI] [PubMed] [Google Scholar]
- 44. Juban M. M., Javadpour M. M., Barkley M. D. (1997) Circular dichroism studies of secondary structure of peptides. Methods Mol. Biol. 78, 73–78 [DOI] [PubMed] [Google Scholar]
- 45. Dey A., Bomans P. H. H., Müller F. A., Will J., Frederik P. M., de With G., Sommerdijk N. A. J. M. (2010) The role of prenucleation clusters in surface-induced calcium phosphate crystallization. Nat. Mater. 9, 1010–1014 [DOI] [PubMed] [Google Scholar]
- 46. In Y., Minoura K., Ohishi H., Minakata H., Kamigauchi M., Sugiura M., Ishida T. (2001) Conformational comparison of μ-selective endomorphin-2 with its C-terminal free acid in DMSO solution, by 1H NMR spectroscopy and molecular modeling calculation. J. Pept. Res. 58, 399–412 [DOI] [PubMed] [Google Scholar]
- 47. Delak K., Collino S., Evans J. S. (2009) Polyelectrolyte domains and intrinsic disorder within the prismatic Asprich protein family. Biochemistry 48, 3669–3677 [DOI] [PubMed] [Google Scholar]
- 48. Atkinson R. A., Evans J. S., Hauschka P. V., Levine B. A., Meats R., Triffitt J. T., Virdi A. S., Williams R. J. (1995) Conformational studies of osteocalcin in solution. Eur. J. Biochem. 232, 515–521 [DOI] [PubMed] [Google Scholar]
- 49. Du C., Falini G., Fermani S., Abbott C., Moradian-Oldak J. (2005) Supramolecular assembly of amelogenin nanospheres into birefringent microribbons. Science 307, 1450–1454 [DOI] [PubMed] [Google Scholar]
- 50. Addison W. N., Miller S. J., Ramaswamy J., Mansouri A., Kohn D. H., McKee M. D. (2010) Phosphorylation-dependent mineral-type specificity for apatite-binding peptide sequences. Biomaterials 31, 9422–9430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kwak S. Y., Wiedemann-Bidlack F. B., Beniash E., Yamakoshi Y., Simmer J. P., Litman A., Margolis H. C. (2009) Role of 20-kDa amelogenin (P148) phosphorylation in calcium phosphate formation in vitro. J. Biol. Chem. 284, 18972–18979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hartgerink J. D., Beniash E., Stupp S. I. (2001) Self-assembly and mineralization of peptide-amphiphile nanofibers. Science 294, 1684–1688 [DOI] [PubMed] [Google Scholar]
- 53. Leonor I. B., Kim H.-M., Balas F., Kawashita M., Reis R. L., Kokubo T., Nakamura T. (2007) Surface potential change in bioactive polymer during the process of biomimetic apatite formation in a simulated body fluid. J. Mater. Chem. 17, 4057–4063 [Google Scholar]