

Background: NrdH-redoxins provide the electrons for the reduction of ribonucleotides.
Results: We characterized NrdH-redoxin from Mycobacterium tuberculosis and Corynebacterium glutamicum.
Conclusion: Both NrdH-redoxins are monomers but form non-swapped dimers at high protein concentration. They are reduced by thioredoxin reductase and not by mycothiol.
Significance: NrdH-redoxin is a potential anti-tuberculosis drug target, and new structural and functional insights help to understand its mode of action.
Keywords: Actinobacteria, Disulfide, Electron Transfer, Kinetics, Redox, X-ray Crystallography, Corynebacterium glutamicum, Mycobacterium tuberculosis, NrdH-redoxin, Mycothiol
Abstract
NrdH-redoxins are small reductases with a high amino acid sequence similarity with glutaredoxins and mycoredoxins but with a thioredoxin-like activity. They function as the electron donor for class Ib ribonucleotide reductases, which convert ribonucleotides into deoxyribonucleotides. We solved the x-ray structure of oxidized NrdH-redoxin from Corynebacterium glutamicum (Cg) at 1.5 Å resolution. Based on this monomeric structure, we built a homology model of NrdH-redoxin from Mycobacterium tuberculosis (Mt). Both NrdH-redoxins have a typical thioredoxin fold with the active site CXXC motif located at the N terminus of the first α-helix. With size exclusion chromatography and small angle x-ray scattering, we show that Mt_NrdH-redoxin is a monomer in solution that has the tendency to form a non-swapped dimer at high protein concentration. Further, Cg_NrdH-redoxin and Mt_NrdH-redoxin catalytically reduce a disulfide with a specificity constant 1.9 × 106 and 5.6 × 106 m−1 min−1, respectively. They use a thiol-disulfide exchange mechanism with an N-terminal cysteine pKa lower than 6.5 for nucleophilic attack, whereas the pKa of the C-terminal cysteine is ∼10. They exclusively receive electrons from thioredoxin reductase (TrxR) and not from mycothiol, the low molecular weight thiol of actinomycetes. This specificity is shown in the structural model of the complex between NrdH-redoxin and TrxR, where the two surface-exposed phenylalanines of TrxR perfectly fit into the conserved hydrophobic pocket of the NrdH-redoxin. Moreover, nrdh gene deletion and disruption experiments seem to indicate that NrdH-redoxin is essential in C. glutamicum.
Introduction
Ribonucleotide reductases (RNRs)3 are enzymes converting ribonucleotides into deoxyribonucleotides, thus providing the precursors for DNA synthesis and repair. RNRs are essential to all living cells except for a few parasites and obligate endosymbionts (1, 2). They are divided into three main classes, depending on the different metal cofactors modulating their catalytic activity. Class I RNRs are oxygen-dependent enzymes containing a di-iron cluster (2). Oxygen-independent class II enzymes have a cobalamin cofactor (vitamin B12), whereas the activity of oxygen-sensitive class III RNRs depends on an iron-sulfur cluster (3–5). Class I RNRs are further divided into class Ia (NrdAB) and Ib (NrdEF); class Ia is present in eukaryotes, prokaryotes, and viruses, whereas class Ib is only found in prokaryotes (6). Many prokaryotes, including Corynebacterium glutamicum (Cg) and the pathogens Bacillus anthracis, Staphylococcus aureus, and Mycobacterium tuberculosis (Mt) use class Ib enzymes as their primary RNR system under aerobic conditions, which makes them interesting drug targets (1, 7).
During the catalytic cycle of RNR, electrons are needed to reconstitute their active site radical. These electrons are provided by glutaredoxin or thioredoxin for Class Ia and II RNR or formate for Class III RNR (8). Class Ib RNR is reduced by NrdH-redoxin or thioredoxin (8–11). NrdH-redoxins are small glutaredoxin-like proteins often coded within the same operon as nrdE and nrdF, the two subunits of the class Ib RNR (12). They have a sequence identity in the range of 35–85% with glutaredoxins (Grx) but receive electrons from thioredoxin reductase (TrxR) (10).
It has been noted that NrdH-redoxins are widespread among prokaryotes lacking glutathione (GSH) (10, 11), but the absence of GSH does not imply the absence of a low molecular weight thiol buffer in these organisms. Multiple alternative low molecular weight thiols have been identified in bacteria and eukaryotes (13). Actinomycetes, like C. glutamicum and M. tuberculosis, produce millimolar concentrations of mycothiol (MSH), whereas Bacillus species, like B. anthracis, contain bacillithiol (14). Recently, Gustafsson et al. (15) have shown that NrdH-redoxin from B. anthracis is not reduced by bacillithiol but accepts electrons from TrxR. Within the actinomycetes, it has been shown that the NrdH-redoxin of Corynebacterium ammoniagenes (Ca) accepts electrons from TrxR, although the possible reduction of NrdH-redoxin by MSH has not been examined (9).
Recently, we identified mycoredoxins as a novel class of oxidoreductases in C. glutamicum and M. tuberculosis (16, 17). These enzymes use MSH to reduce disulfides in the cell and can be considered as the Grx analog of the actinomycetes. Ordóñez et al. (16) identified two potential mycoredoxins, Mrx1 and Mrx2, of which only Mrx1 was shown to be a true mycoredoxin. Mrx2, on the other hand, was specifically reduced by TrxR and not by MSH. Furthermore, it has an amino acid sequence identity of 42 and 75% with E. coli and C. ammoniagenes NrdH-redoxin, respectively. The gene coding for Mrx2 (NCg12445) is located within an operon containing nrdE and nrdF, the two subunits of the class Ib RNR. Altogether, this indicates that Mrx2 is the NrdH-redoxin of C. glutamicum.
We investigate the NrdH-redoxins of C. glutamicum and M. tuberculosis. We determined the crystal structure of Cg_NrdH-redoxin and used it as a template for homology modeling of Mt_NrdH-redoxin. With a kinetic study, we found that the NrdH-redoxins specifically receive electrons from TrxR and not from MSH. The pKa of the active site cysteines were determined, and the lack of nrdh gene deletion and disruption mutant in C. glutamicum showed that the protein might be essential for the cell.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of Cg_Mtr, Cg_TrxR, Cg_Trx, Mt_Mtr, Mt_TrxR, and Mt_TrxC
The cloning protocol of genes for Cg_Mtr, Cg_TrxR, and Cg_Trx was described by Ordóñez et al. (16). The genes of Mt_NrdH-redoxin and Mt_TrxC were cloned in a pET28a vector with a C-terminal His6 tag using the NcoI and XhoI restriction sites. The gene of Mt_TrxR was cloned in a pET28a vector with an N-terminal His6 tag using the NdeI and HindIII restriction sites. The obtained vectors for Mt_NrdH-redoxin, Mt_TrxC, and Mt_TrxR were used to transform E. coli BL21(DE3), and the cells were grown in Luria broth (LB) with 50 μg/ml kanamycin at 37 °C. At a cell density A600 nm of 0.7, 1 mm IPTG was added to the cultures. Cells were harvested after 4 h at 30 °C and resuspended in 20 mm Tris/HCl (pH 8.0), 5 mm imidazole, 1 m NaCl, 1 mm DTT, 0.1 mg/ml 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, and 1 μg/μl leupeptin and lysed using French press disruption. The cell debris was removed by centrifugation, and the supernatants was loaded on a Ni2+-IMAC affinity column and eluted with a linear gradient to 1 m imidazole in the buffer solution. The proteins were further purified on a Superdex75 PG (16/90) column (GE Healthcare) equilibrated in 20 mm Tris/HCl (pH 8.0), 150 mm NaCl, 1 mm DTT, 1 mm EDTA. Mycothiol disulfide reductase (Mtr) and TrxR were stored as an 80% ammonium sulfate precipitate at 4 °C, whereas Trx and NrdH-redoxin were flash-frozen in liquid nitrogen and stored at −20 °C. All buffers were argon-flushed, and the purification was performed at room temperature.
Cloning, Expression, and Purification of Cg_NrdH-redoxin
The cloning, expression, and purification of Cg_NrdH-redoxin, denoted as Mrx2, was described by Ordóñez et al. (16). Briefly, Cg_NrdH-redoxin was cloned into a pTYB12 vector with a C-terminal intein tag (New England Biolabs, Ipswich, MA) and transformed in E. coli BL21(DE3) cells. The cells were grown overnight at 37 °C in a LB broth with 100 μg/ml ampicillin. The culture was diluted 100-fold into LB and further grown at 37 °C. Cells were induced with 1 mm IPTG at a cell density of 0.8 and grown overnight at 15 °C. Cells were harvested at an A600 nm of 3.1 and resuspended in 20 mm Tris/HCl (pH 8.0), 0.5 m NaCl, 1 mm EDTA, 0.1 mg/ml 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, and 1 μg/μl leupeptin. After French press cell lysis, 50 μg/ml DNase I and 20 mm MgCl2 were added to the cell lysate. After a 30-min incubation at room temperature, cell debris was removed by centrifugation for 30 min at 10,000 rpm at 4 °C. The lysate was loaded on a 20-ml chitin column equilibrated with 20 mm Tris/HCl (pH 8.0), 0.5 m NaCl, 1 mm DTT, and 1 mm EDTA. After extensive washing with 1 m NaCl in the same buffer solution, the column was treated with 50 mm DTT with 3 column volumes of 50 mm DTT and kept overnight at room temperature. The protein was eluted with 20 mm Tris/HCl (pH 9.0), 1 m NaCl, 1 mm DTT, 1 mm EDTA and purified as indicated. Purity was checked on SDS-PAGE, and the sample was flash-frozen for storage. Cg_ and Mt_NrdH-redoxin were injected on a calibrated (Bio-Rad gel filtration standard) Superdex 75 HR (10/30) column and eluted in 50 mm Hepes, pH 8.0, 150 mm NaCl or 50 mm Tris/HCl (pH 8.0), 3 m NaCl.
Purification of Mycothiol from C. glutamicum
MSH was purified as described by Ordóñez et al. (16).
Coupled Enzyme Assays and Kinetic Study
To obtain an oxidized sample of Cg_NrdH-redoxin or Cg_Trx, an aliquot of the protein was incubated with a ten molar excess of diamide for 30 min at room temperature. Subsequently, the samples were injected on a Superdex75 HR column equilibrated in 50 mm Hepes/NaOH (pH 7.5), 150 mm NaCl to obtain pure oxidized protein. Cg_TrxR and Cg_Mtr, stored as 80% ammonium sulfate precipitates, were resuspended in the same buffer and treated with 20 mm DTT for 30 min at room temperature prior to dialysis in a Slide-A-Lyzer Dialysis Cassette (Thermo Scientific) against 50 mm Hepes/NaOH (pH 8.0).
The assay with TrxR was prepared by diluting all components except for oxidized Cg_NrdH-redoxin in the 50 mm Hepes/NaOH (pH 8.0) buffer to a final concentration of 250 nm Cg_TrxR and 500 μm NADPH. The Mtr assay mixture consisted of 0.5 μm Cg_Mtr, 500 μm MSH, and 500 μm NADPH. All concentrations were calculated taking into account the subsequent addition of oxidized Cg_NrdH-redoxins. The mixtures were incubated for 20 min at 37 °C in a 96-well plate prior to the addition of 50 μm oxidized Cg_NrdH-redoxin (final concentration). The oxidation of NADPH was measured by the decrease in absorption at 340 nm. A duplicate well without oxidized NrdH-redoxin was included as background control.
To determine the steady state kinetic parameters of the reduction of NrdH-redoxin or Trx by TrxR, the sample contained 250 nm Cg_TrxR and 500 μm NADPH in a 50 mm Hepes/NaOH (pH 8.0), buffer. The samples were preincubated at 37 °C for 20 min. The assay was started by the addition of different dilution series of substrate (oxidized NrdH-redoxin or Trx). The reduction of NrdH/Trx was recorded by the oxidization of NADPH (Δϵ340 = 6,220 m−1 cm−1). Initial rates were calculated with SPECTRA-maxPro (Molecular Devices) and corrected for the path length of each sample. Kinetic plots were made with Prism version 5.0. The assays of Mt_NrdH-redoxin and Mt_TrxC have an identical setup.
DTNB Reductase Activity of NrdH-redoxin
Reaction mixtures containing 500 μm NADPH, 5 μm Cg_TrxR, and 500 nm Cg_NrdH-redoxin or Cg_Trx (final concentrations taking into account the addition of DTNB) were prepared. Control samples with one of the above reagents omitted were included. The reaction was initiated by the addition of 80 μm DTNB to the mixture, and the formation of thionitrobenzene was followed at A412 nm, whereas the NADPH consumption was monitored at A340 nm. To determine the kinetic parameters, a reaction mixture of 500 μm NADPH, 1.5 μm Cg_TrxR, and 100 nm Cg_NrdH-redoxin or Cg_Trx was used. Different concentrations of DTNB were added to the mixture, and the increase in A412 nm was used as a readout to calculate the initial velocities (using the decrease in A340 nm gave similar results). The reduction of one molecule of DTNB results in the release of two molecules of TNB2−, which both yield an increase in A412 nm of 13,600 cm−1. Thus, the reduction of one disulfide by Cg_NrdH-redoxin (one molecule of DTNB) corresponds to an increase in A412 nm of 27,200 cm−1. Independent duplicates of each DTNB concentration were used to fit the Michaelis-Menten equation and determine the kinetic parameters. The assays of Mt_NrdH-redoxin and Mt_TrxC have an identical setup with minor differences; the assay conditions for Mt_NrdH-redoxin and Mt_TrxC are 500 μm NADPH, 1.5 μm Mt_TrxR, and 500 nm Mt_NrdH-redoxin or Mt_TrxC.
Determination of the pKa of the Active Site Cysteines
Reduced Cg_NrdH-redoxin was prepared by incubation with 10 mm DTT for 15 min at room temperature. Oxidized Cg_NrdH-redoxin was prepared by incubation with a 10-fold molar excess of diamide for 30 min at room temperature. The excess of DTT or diamide in each sample was removed on a Superdex75 HR column (GE Healthcare) equilibrated with the polybuffer: 10 mm sodium citrate, 10 mm sodium borate, 10 mm sodium phosphate, pH 8.5, for the pKa of the nucleophilic cysteine or 10 mm boric acid, 10 mm CHES, 10 mm CAPS, 10 mm sodium phosphate, pH 13.0, for the pKa of the C-terminal cysteine. The pH of the samples was stepwise decreased with HCl. Every 0.1 pH unit, the absorbance at 280 and 240 nm was recorded on a Cary100 Bio UV-visible spectrophotometer (Varian, Palo Alto, CA). The absorption at 240 nm was standardized with the absorption at 280 nm for each pH value (A240 nm/A280 nm) to compensate for the dilution caused by the addition of HCl. The corrected absorption (A240/A280)red/(A240/A280)ox was plotted against the pH and fitted with the Henderson-Hasselbalch equation (18). An identical protocol was used for the determination of the cysteine pKa of Mt_NrdH-redoxin.
Crystallization of Cg_NrdH-redoxin
Cg_NrdH-redoxin was concentrated to 9.35 mg/ml in a buffer solution of 20 mm Tris/HCl (pH 8.0), 100 mm NaCl, and 1 mm DTT. Crystallization conditions were screened using the Hampton Crystal Screen II by using the hanging drop technique. Small needles were found in 12% glycerol, 0.1 m Tris/HCl (pH 8.5), and 1.5 m ammonium sulfate. By changing the ammonium sulfate concentration, we obtained an optimal condition in 12% glycerol, 0.1 m Tris/HCl (pH 8.5), and 1.8 m ammonium sulfate. The crystals were flash-frozen in 40% glycerol, 0.1 m Tris/HCl (pH 8.5), and 1.1 m ammonium sulfate as cryoprotectant.
Structure Solving and Refinement
Cg_NrdH-redoxin data were collected at the ID29 beam line of the European Synchrotron Radiation Facilities (Grenoble, France) using a liquid nitrogen stream at 100 K. The data were processed and scaled to 1.5 Å resolution using the XDS package (19). The Cg_NrdH-redoxin structure was solved by molecular replacement using BALBES (20). The models designated by the PDB accession numbers 1R7H, 1H75, 3IC4, and 1ABA were used for molecular replacement (9, 21, 22). The solution of the Balbes run was submitted to the ARP/wARP server (23) for model building. The model was further refined by iterative model building and refinement with TLS using the programs COOT (24) and PHENIX (25). The TLS groups were defined using the TLS server (26, 27). Data and model statistics are presented in Table 1.
TABLE 1.
Data collection and refinement statistics
| Cg_NrdH-redoxin | |
|---|---|
| Beamline | ID29 |
| Wavelength (Å) | 0.9793 |
| Space group | P6 (5)22, 179 |
| Unit cell | |
| a (Å) | 61.77 |
| b (Å) | 61.77 |
| c (Å) | 77.19 |
| α (degrees) | 90.00 |
| β (degrees) | 90.00 |
| γ (degrees) | 120.00 |
| Resolution limits | 28.675–1.50 |
| No. of measured reflections | 277,985 (9918) |
| No. of unique reflections | 11,824 |
| Completeness | 98.4.0 (94.9) |
| Rmerge | 0.053 (0.440) |
| 〈I/σ(I)〉 | 37.0 (8.12) |
| R-factor | 0.1944 (0.269) |
| Rfree-factor | 0.2225 (0.341) |
| Ramachandran profile | |
| Core | 98.51% |
| Other allowed | 0.00% |
| Disallowed | 1.49% |
| Root mean square deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (degrees) | 1.060 |
| PDB entry | 4FIW |
Modeling and Molecular Dynamics
Sequence alignments for homology modeling were created using Kalign (28). MODELLER (29) was used to generate homology models of monomeric Mt_NrdH-redoxin and dimeric Mt_NrdH-redoxin. For the monomer Mt_NrdH-redoxin, Cg_NrdH-redoxin (this study) was used as a template. In the case of dimeric Mt_NrdH-redoxin, an initial non-swapped dimer was created by overlaying of two monomeric Mt_NrdH-redoxins on the swapped dimer solution of Ca_NrdH-redoxin. The initial model of the complex of Cg_NrdH-redoxin and Ec_TrxR was constructed by structurally overlaying Cg_NrdH-redoxin on Ec_Trx in the Ec_Trx·TrxR complex (PDB code 1F6M) (21, 30). To model the Mt_TrxR·Mt_NrdH-redoxin complex, the crystal structure of Mt_TrxR was modified from the oxidized FO conformation to the reduced FR conformation, as described by Akif et al. (31). This FR model was used to construct the initial model of the Mt_TrxR·Mt_NrdH-redoxin complex. These initial models were optimized using molecular dynamics. The molecular dynamics (MD) simulations in water were performed using the OPLS force field within GROMACS (32). All simulations were done at a constant temperature of 300 K and a constant pressure of 1 atm over a simulation time of 1 ns. For the simulation of the non-swapped Mt_NrdH-redoxin dimer, the simulation was extended to 10 ns because equilibrium was not reached. The electrostatic surface potential was calculated with the PDB2PQR server (33) using the Adaptive Poisson Boltzmann solver and the PARSE force field (34). ClustalW was used for the amino acid sequence alignment of the NrdH-redoxins and TrxR (35).
Small Angle X-ray Scattering
SAXS experiments were performed at the P12 EMBL beamline (Desy, Hamburg). The scattering intensities of Mt_NrdH-redoxin in solution were collected at concentrations of 10.1, 5.1, 2.5, and 1.3 mg/ml under reducing conditions in a buffer containing 50 mm Tris/HCl (pH 7.5), 150 mm NaCl, and 1 mm tris(2-carboxyethyl)phosphine. Data were processed using PRIMUS (36). The radius of gyration (Rg) was evaluated using the Guinier approximation (37) and also from the entire scattering curve using Porod's law (38) and the distance distribution function p(r) using GNOM (39). Because the scattering curves systematically varied among the different concentrations measured, no merging of the different concentrations was performed, and the curves were analyzed as separate monomer-dimer mixtures. OLIGOMER was used to evaluate the protein concentration-dependent percentage of monomeric and dimeric Mt_NrdH-redoxin (40). The data are therefore considered as a linear combination of the components (monomer and dimer) present in solution.
NrdH Gene Disruption and Gene Deletion Analyses in C. glutamicum
In order to investigate the role of NrdH-redoxin in vivo, we decided to construct both nrdh disruption and deletion mutants in C. glutamicum. For the construction of nrdh disruption mutants, we PCR-amplified the nrdh gene (NCgl2445) from a total DNA sample of C. glutamicum 13032 using the primer pair mrx2int-Fw and mrx2int-Rv (Table 2). The 149-base pair fragment was BamHI-HindIII-digested and subcloned into the mobilizable plasmid pK18mob and used to transform E. coli strain TOP10. The recombinant plasmid was isolated, sequenced, and named pKmrx2-int. pKmrx2-int was transferred to C. glutamicum RES167 (recipient strain) using a conjugation protocol with the pKmrx2-int containing E. coli S17-1 as donor strain (41). The plasmid pK18mob is a suicide plasmid in the recipient strain (lack of replication origin), and therefore only C. glutamicum cells where the plasmid is integrated (by single recombination) are viable under kanamycin selection. For gene deletion analysis, we used the double recombination system based on vectors containing the sacB gene (42, 43). Briefly, a 500-base pair fragment located just upstream and downstream of the nrdh gene was PCR-amplified using the primer pairs mrx2sac-upFw/mrx2sac-upRv (upstream) and mrx2sac-downFw/mrx2sac-downRv (downstream). Both PCR products are ligated via a PCR cycle to form a 1000-bp fragment using the primers mrx2sac-upFw and mrx2sac-downRv. The PCR-amplified fragment was digested with BamHI and HindIII and subcloned into the mobilizable plasmid pK18mobsacB to generate plasmid pKSDmrx2. E. coli S17–1 containing pKSDmrx2 was used as donor for plasmid conjugation to strain RES167 as described for the disruption mutants. The transconjugant clones were selected first in complex medium trypticase soy agar (Oxoid, Basingstoke, UK) containing 10% sucrose, and the resistant colonies were replated onto complex medium with and without kanamycin (12.5 μg/ml). To validate our approach, a gene deletion and disruption mutant of mrx1 (NCgl0808) was constructed (primers are provided in Table 2). From the selected clones, total DNA sample was prepared for sequence analysis to confirm the absence or disruption of the mrx1 gene.
TABLE 2.
Primers used for construction of the mrx1 (NCgl0808) and nrdh (NCgl2445) disruption and deletion mutants
| Primer name | Sequence (restriction sites are underlined) | Purpose |
|---|---|---|
| mrx1int-Fw | TAATTTAAA GGATCCACCTCATCGACGTCGACC (DraI/BamHI) | mrx1 disruption |
| mrx1int-Rv | TAATTTAAA AAGCTTATCGGAGTAGCGCAC (DraI/HindIII) | mrx1 disruption |
| mrx1sac-upFw | TAATTTAAA GAATTCCATTGATGCAGTTCAATGGGGGTAAATTG (DraI/EcoRI) | mrx1 deletion |
| mrx1sac-upRv | GTTTAAGGAGCAAACTCTAGAGCAAAAAAGAGCCTTTC (XbaI) | mrx1 deletion |
| mrx1sac-downFw | GAAAGGCTCTTTTTTGCTCTAGAGTTTGCTCCTTAAAC (XbaI) | mrx1 deletion |
| mrx1sac-downRv | TAATTTAAA AAGCTT ATTTATGACAACCACAAGGAACAGGTCG (DraI/HindIII) | mrx1 deletion |
| mrx2int-Fw | TAATTTAAA GGATCCTGCGTCCAGTGCAATGCCA (DraI/BamHI) | nrdh disruption |
| mrx2int-Rv | TAATTTAAA AAGCTTGGGAGCCATCTGCAACGACAA (DraI/HindIII) | nrdh disruption |
| mrx2sac-upFw | TAA TTTAAA GGATCC ATGCGAAAGTGAGTATTGCG (DraI/BamHI) | nrdh deletion |
| mrx2sac-upRv | CGATTAAACAGGTCGTGCCTCTAGAGACCTTCCCTTATCGGC (XbaI) | nrdh deletion |
| mrx2sac-downFw | GCCGATAAGGGAAGGTCTCTAGAGGCACGACCTGTTTAATCG (XbaI) | nrdh deletion |
| mrx2sac-downRv | TAATTTAAAAAGCTTAAACCTGATGACTTGTGG (DraI/HindIII) | nrdh deletion |
RESULTS
NrdH-redoxin Is Specifically Coupled to the TrxR Electron Transfer Pathway
We started by identifying the electron transfer pathway utilized by Cg_NrdH-redoxin (Cg2789) and Mt_NrdH-redoxins (Rv3053c). The TrxR and MSH electron transfer pathways of both species were reconstructed as described by Van Laer et al. (17) and Ordóñez et al. (16). Briefly, the TrxR pathway uses the flavoprotein TrxR to shuffle electrons from NADPH to a protein disulfide, whereas the MSH pathway utilizes MSH, the flavoprotein Mtr, and NADPH to reduce protein disulfides (44). Oxidized NrdH-redoxin, with a disulfide formed between both active site cysteines, was added to each pathway of the corresponding species, and the oxidation of NADPH, due to the reduction of NrdH-redoxin, was monitored as a decrease in absorption at 340 nm (Fig. 1A). Both Cg_NrdH-redoxin and Mt_NrdH-redoxin are reduced by the TrxR pathway. No reduction by the MSH pathway was observed (Fig. 1B).
FIGURE 1.

A schematic view of the TrxR (A) and MSH (B) electron transfer pathway is shown. Both pathways in C. glutamicum and M. tuberculosis were reconstructed in vitro. The consumption of NADPH was followed in function of time at 340 nm in the presence (▴) or absence (○) of oxidized NrdH-redoxin.
NrdH-redoxin Catalytically Reduces Disulfide Bonds
Having identified the reduction pathway of the NrdH-redoxins, we explored the capability of the proteins to reduce exposed disulfide bonds. DTNB was used as an artificial disulfide substrate for the NrdH-redoxins in a coupled assay with the TrxR pathway. Cleavage of the disulfide of DTNB results in an increased absorption at 412 nm, whereas the NADPH consumption decreases the absorption at 340 nm (Fig. 2). Only the complete NrdH-redoxin/TrxR pathway gives a catalytically relevant reduction of DTNB with a simultaneous consumption of NADPH. The Mt_TrxR pathway could slowly reduce DTNB in the absence of NrdH-redoxin although only at high TrxR (1.5 μm) and DTNB (50 μm) concentrations. The Cg_TrxR pathway did not show any reduction of DTNB even at a high Cg_TrxR concentration of 5 μm. Next, we determined the steady state kinetics of the reduction of DTNB by the NrdH-redoxins. The coupled assay with Cg_NrdH-redoxin, Cg_TrxR, and NADPH was reconstructed, and different concentrations of DTNB were added as substrate. The increase in absorption at 412 nm was used to calculate the initial velocities. The specificity constant (kcat/Km) for the reduction of DTNB by Cg_NrdH-redoxin is 5.6 × 106 m−1 min−1 (Fig. 2). For comparison, the reduction of DTNB by Cg_Trx (coupled to the TrxR pathway) has a 2-fold higher specificity constant (1.1 × 107 m−1 min−1) (data not shown). The kinetic parameters of Mt_NrdH-redoxin and Mt_TrxC, one of two active Trx in M. tuberculosis (45), were determined as described for Cg_NrdH-redoxin. A control sample lacking Mt_NrdH-redoxin or TrxC was included for each DTNB concentration. This control sample corrects for the slow reduction of DTNB by Mt_TrxR. Mt_TrxC displays a 2-fold higher specificity constant than Mt_NrdH-redoxin (3.4 × 106 and 1.9 × 106 m−1 min−1, respectively) (Fig. 2).
FIGURE 2.

Reduction of DTNB by NrdH-redoxin coupled to the TrxR electron pathway of C. glutamicum and M. tuberculosis. Top, progress curves in which each compound of the electron transfer pathway was sequentially omitted. The reduction of DTNB was recorded as an increase in absorption at 412 nm (dotted line, right y axis), whereas the consumption of NADPH was recorded at 340 nm (solid line, left y axis). Bottom, the Michaelis-Menten curves of the reduction of DTNB in the coupled enzyme assays with the NrdH-redoxin/TrxR/NADPH pathway.
Cg_TrxR Has a Similar Kinetic Turnover with Oxidized Cg_Trx and Cg_NrdH-redoxin as Substrate
The kinetics of NrdH-redoxins and Trx in the coupled enzyme assay were obtained in the presence of a 3- to 10-fold molar excess of TrxR (see “Experimental Procedures”). It allowed us to determine the kinetics of NrdH-redoxin and Trx independently of their specificity for TrxR. Here, we investigate the specificity of Cg_TrxR for the reduction of oxidized Cg_NrdH-redoxin and Cg_Trx. Varying concentrations of oxidized Cg_NrdH-redoxin or Cg_Trx were added as substrates to the TrxR pathway. The initial velocities of the reaction were calculated from the decrease in absorption at 340 nm due to NADPH consumption (Fig. 3). Cg_TrxR has similar kcat and Km values for both substrates (Cg_NrdH-redoxin or Cg_Trx), resulting in a kcat/Km value in the range of ∼5 × 105 m−1 min−1.
FIGURE 3.

The steady state kinetics of Cg_TrxR with oxidized Cg_NrdH-redoxin and Cg_Trx as substrate. The kinetic parameters were determined at pH 8.0 with 500 μm NADPH and 250 nm Cg_TrxR.
NrdH-redoxins Have a Thioredoxin Structural Fold
We solved the crystal structure of oxidized Cg_NrdH-redoxin at 1.5 Å resolution. It displays the minimal version of the Trx fold that composes a central four-stranded β-sheet surrounded by three α-helices (Fig. 4A). The active site CXXC motif is located at the N terminus of the first α-helix (Fig. 4). The N-terminal cysteine is surface-exposed, whereas the C-terminal cysteine is buried.
FIGURE 4.

A, the amino acid sequence alignment of Cg_, Mt_, Ec_, and Ca_NrdH-redoxins. The active site CVQC motif is in boldface type, and the NrdH-specific WSGFRPE motif is boxed in gray. The crystal structure of Cg_NrdH-redoxin (B) and the monomeric homology model of Mt_NrdH-redoxin (C) are shown. The CVQC active site motif is located at the N terminus of helix α1, and the WSGFRP(E/D) linker motif is denoted with an asterisk. The N and the C terminus of the protein are indicated.
Currently, only two structures of NrdH-redoxin are deposited in the Protein Data Bank. Stehr et al. (21) reported the structure of Ec_NrdH-redoxin (PDB code 1H75), providing structural insights into the specific reduction of Ec_NrdH-redoxin by TrxR. The Cg_NrdH-redoxin structure reported here strongly resembles the structure of Ec_NrdH-redoxin (root mean square deviation of 0.70 Å for 65 Cα atoms). The second NrdH-redoxin structure in the PDB is from C. ammoniagenes (PDB code 1R7H). This crystal structure differs significantly from the structures of both Ec_NrdH-redoxin and Cg_NrdH-redoxin despite the high amino acid sequence identity with both proteins (41 and 75%, respectively). The Ca_NrdH-redoxin crystal structure appears as a domain-swapped dimer (9). However, the authors noted that Ca_NrdH-redoxin is a monomer in solution. The physiological relevance of this swapped dimer is questionable because domain swapping can occur in almost any protein if subjected to certain mainly non-physiological conditions (9, 46–49). These conditions include a high local protein concentration and a destabilizing environment, which are commonly used for protein crystallization. Nevertheless, a homology model of Mt_NrdH-redoxin as a swapped dimer, based on the Ca_NrdH-redoxin structure, is available in the Swiss-Model Repository (50, 51). We used the Cg_NrdH-structure to construct a non-swapped monomeric model of Mt_NrdH-redoxin (70% sequence identity). This model was further refined by an MD simulation in water (Fig. 4B and supplemental Structure S1). The final model of Mt_NrdH-redoxin has a root mean square deviation of 0.65 Å (for 63 Cα atoms) with Ec_NrdH-redoxin (PDB code 1H75).
NrdH-redoxin of Cg and Mt Have a Protein Concentration-dependent Dimerization Profile
To determine the multimerization state of Mt_ and Cg_NrdH-redoxin (monomer or swapped dimer) in solution, we analyzed reduced NrdH-redoxin by size exclusion chromatography (SEC). The SEC analysis was initially performed in a buffer containing 150 mm NaCl and a protein concentration of ∼1 mg/ml. Under these conditions, both Cg_NrdH-redoxin and Mt_NrdH-redoxin elute as a single peak with a molecular mass of ∼12 kDa, whereas the molecular masses of the Cg and Mt monomer are 8.4 and 9.4 kDa, respectively (Fig. 5, A and B). Increasing the ionic strength of the buffer (using 3 m NaCl) shifted the retention volume to the molecular mass of 8 and 8.5 kDa for monomeric Cg_ and Mt_NrdH-redoxin, respectively (Fig. 5, A and B). Identical results are obtained when the oxidized forms of both proteins are used (data not shown), indicating that this behavior is independent of the redox state (oxidized or reduced) of the protein. We speculated that the proteins are present as a mixture of monomer and dimer in the buffer solution with a low ionic strength (150 mm NaCl). This dimer is formed by electrostatic interactions because a high ionic strength buffer (3 m NaCl) shifts the equilibrium to the monomeric state. Furthermore, the observation that the proteins elute as a single peak between the retention volume of a monomer and dimer indicates a fast exchange rate between the monomeric and dimeric state. As such, it is highly unlikely that domain swapping causes that dimerization.
FIGURE 5.

A and B, the SEC elution profile of Cg_NrdH-redoxin (A) and Mt_NrdH-redoxin (B) in a low and high ionic strength buffer. A clear shift toward the monomeric state is observed in a buffer with high ionic strength. The model of the dimer of Mt_NrdH-redoxin obtained from the SAXS analysis (C) is stabilized by several salt bridges and hydrogen bonds (D).
We confirmed our hypothesis by SAXS analysis. Mt_NrdH-redoxin and a buffer with a low ionic strength (150 mm NaCl) was used throughout the SAXS experiments. Four different protein concentrations (10.1, 5.1, 2.5, and 1.3 mg/ml) were tested. All samples indicated a monomer/dimer mixture (Fig. 6). Because the SEC analysis of Mt_NrdH-redoxin favors a non-swapped dimer over a swapped dimer, we examined the possibility that Mt_NrdH-redoxin forms a non-swapped dimer. A non-swapped dimer model of Mt_NrdH-redoxin was constructed based on the orientation of the Ca_NrdH-redoxin swapped dimer structure (PDB code 1R7H). This model has a dimer interface of 445 Å2, and the dimer has a V-shaped form. The maximal distance obtained within the dimer is 58 Å. This model was subsequently subjected to an MD simulation. The interface was not stable, and during the simulation a stable and more elongated dimer arrangement was obtained (Fig. 5C and supplemental Structure S2). In this elongated dimer, the dimer interface is 330 Å2, and the maximal distance within the dimer is 70 Å, which is in better agreement with the Dmax obtained from the SAXS data using GNOM (Dmax, SAXS = 76 Å). Although the dimer interface is formed by the same residues in both models, the different orientation of the subunits in the elongated model allows the formation of several salt bridges and hydrogen bonds (Fig. 5D). These interactions stabilize the elongated dimer.
FIGURE 6.

The small angle x-ray scattering curves (log(I) versus S) at different Mt_NrdH-redoxin concentrations. The profile shows a concentration-dependent shift at low angles.
The active site cysteines of both subunits are on opposite sides of the elongated dimer and remain surface-exposed. With the structural models of the non-swapped dimer and the monomer, we calculated the fractions of monomer and dimer present in the SAXS samples (Fig. 7). In the sample with 10.1 mg/ml protein, 54% of the protein is present as a dimer, whereas in the sample with 1.3 mg/ml protein, the dimer fraction decreases to 30%. The dimer formation is concentration-dependent, with a lower fraction of dimer at a lower protein concentration.
FIGURE 7.
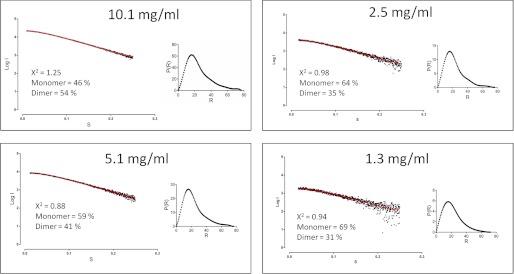
The small angle x-ray scattering curves (dots) and the corresponding fit (red line) using a linear combination of monomer and dimer for each Mt_NrdH-redoxin concentration using the OLIGOMER software. The distance distribution function p(r) is shown for each sample.
NrdH-redoxins Have a Conserved Hydrogen Bond Network
A typical difference between the structure of Trx, Mrx, NrdH-redoxins, and Grx is the orientation of helix α3 (numbering in Mrx, Grx, and NrdH-redoxin) relative to strand β4 (9, 21). In Grx, these two secondary structure elements are connected by a short linker, which orients them perpendicularly. A longer linker between the helix α3 (helix α4 in Trx) and the strand β4 (strand β5 in Trx) in Trx results in an antiparallel orientation of helix α3. This more compact orientation allows the binding of TrxR to Trx (30). NrdH-redoxins have, similar to Trx, helix α3 and strand β4 in an antiparallel orientation (Figs. 4 and 8). The linker between both secondary structure elements is formed by the NrdH-redoxin-specific WSGFRP(D/E) motif. The amino acids of this motif participate in a conserved hydrogen bond network, which positions helix α3 antiparallel to strand β4 (Fig. 8). In all structures of NrdH-redoxins (Ec, Ca, and Cg), a central water molecule controls this hydrogen bond network (9, 21). We included the Ca_NrdH-redoxin swapped dimer structure in this analysis because none of the amino acids of the hydrogen bond network are involved in domain swapping. The central water molecule interacts with Oγ1 of Thr-17 (numbering is identical in Ec, Ca, Cg, and Mt), the main-chain oxygen of Gly-63, the main-chain nitrogen of Arg-65, and Nϵ1 of Trp-61 (Fig. 8). In Ec_NrdH-redoxin, an extra interaction between Nϵ of Arg-65 and Oδ2 of Asp-67 is present. In NrdH-redoxins from Cg, Ca, and Mt, Arg-65 adopts a different orientation preventing this interaction. Instead, a salt bridge is formed between the Nϵ of Arg-65 and the side-chain oxygen of Ser-62 in Cg_NrdH-redoxin. Further, Cg_NrdH redoxin and Mt_NrdH-redoxin have a hydrogen bond between Arg-65 and Arg-68. In Ca_ and Mt_NrdH-redoxin, Arg-68 forms an extra hydrogen bond with Oϵ2 of Glu-59 or Oϵ2 of Asp-59, respectively. This hydrogen bond is absent in the NrdH-redoxins from Cg and Ec.
FIGURE 8.

The conserved hydrogen bond network in Cg (A) and Mt (B) NrdH-redoxin orientates helix α3 antiparallel to strand β4. A global view (left) and a 180° turned detailed view (right) are shown. The residues involved in hydrogen bonding are in stick representations, and the hydrogen bonds and salt bridges are indicated with black dotted lines.
NrdH-redoxin Has a TrxR Binding Site near Its CXXC Motif
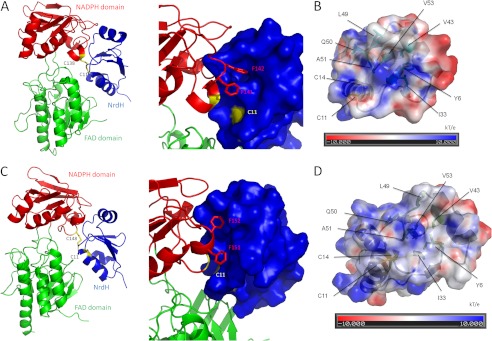
In Mt_NrdH-redoxin and Cg_NrdH-redoxins, a hydrophobic pocket is located near the active site. The hydrophobic amino acids of this pocket are identical in both NrdH-redoxins, and the hydrophobicity of these residues is conserved among all NrdH-redoxins (9, 21). In the NrdH-redoxins of Mt and Cg, this pocket consists of Tyr-6, Ile-33, Val-43, Leu-49, Gln-50, Ala-51, and Val-53 (Fig. 9, B and D). A similar hydrophobic pocket in Ec_Trx is involved in the interaction with Ec_TrxR (30). Two consecutive phenylanalines (Phe-142 and Phe-143) of Ec_TrxR occupy this hydrophobic pocket in the Trx·TrxR complex (PDB code 1F6M). These two Phe residues are conserved in both Cg_TrxR and Mt_TrxR. Stehr et al. (21) built a model of the complex between Ec_NrdH-redoxin and Ec_TrxR by structurally aligning Ec_NrdH-redoxin on Trx of the Ec_Trx·TrxR complex. In their model, the two Phe residues of TrxR occupied the hydrophobic pocket of NrdH-redoxin, although some steric hindrance was observed in the model. Using a similar approach, we modeled the complex between Cg_NrdH-redoxin and Ec_TrxR (no structure of Cg_TrxR is available). An MD simulation resolved the steric hindrance present in the model (Fig. 9A and supplemental Structure S3). Further, Cys-11 of Cg_NrdH-redoxin and Cys-139 of Ec_TrxR are disulfide-bonded (distance between both sulfur atoms is 2.04 Å). In this optimized model, the two conserved Phe residues of Ec_TrxR fit nicely into the hydrophobic pocket of Cg_NrdH-redoxin (Fig. 9A).
FIGURE 9.
Cg (A and B) and Mt (C and D) NrdH-redoxin have a TrxR binding site near their active site CXXC motif. A ribbon model of the complex between Cg_NrdH-redoxin and Ec_TrxR (A) and of the complex between Mt_NrdH-redoxin and Mt_TrxR (C) are shown. NrdH-redoxin is in blue, and the NADPH domain and FAD domain of TrxR are in red and green, respectively. The interface between both proteins has been enlarged, and the surface representation of NrdH-redoxin is shown. The two Phe residues of TrxR (red sticks) fit in the hydrophobic pocket of NrdH-redoxin. The active site cysteines of TrxR and NrdH-redoxin are in yellow. The residues forming the hydrophobic pocket in Cg (B) and Mt (D) NrdH-redoxin are conserved in both proteins. The electrostatic surface potential of the NrdH-redoxins is shown. The electrostatic potential is given from −10 (red) to 10 (blue) kT/e with k the Boltzmann constant, T the temperature, and e the elementary charge (charge of a proton). Areas with a negative electrostatic potential are colored in red, whereas areas with a positive electrostatic potential are shown in blue. The residues forming the hydrophobic pocket and the active site cysteines are annotated.
As a next step, we constructed a model of the complex between Mt_NrdH-redoxin and Mt_TrxR. A crystal structure of the oxidized conformation (FO) of Mt_TrxR is available in the Protein Data Bank (PDB code 2A87) (31). In the FO conformation, the active site cysteines of TrxR are buried at the interface between the FAD- and NADPH-binding domains. A rotation of the FAD binding domain is needed to bring TrxR into the reduced (FR) conformation (30). In this conformation, the active site cysteines are exposed, allowing the reduction of NrdH-redoxin/Trx. We started by modeling Mt_TrxR in the FR conformation as described by Akif et al. (31) and structurally aligned it with Ec_TrxR in the structure of the Ec_Trx·TrxR complex. The monomeric Mt_NrdH-redoxin was aligned with Ec_Trx. Finally, the model of the Mt_NrdH-redoxin·TrxR complex was optimized with an MD simulation (supplemental Structure S4). In this model, the active site cysteines of Mt_NrdH-redoxin and Mt_TrxR are disulfide-bonded (distance between both sulfur atoms is 2.04 Å), and the two Phe residues of TrxR fit into the hydrophobic pocket of NrdH-redoxin (Fig. 9, C and D).
The pKa of the Active Site Cysteines of Cg_ and Mt_NrdH-redoxin
The reduction of a disulfide by NrdH-redoxins proceeds via a thiol-disulfide exchange mechanism. The reaction rate of these reactions depends, among others factors, on the pKa of the thiols involved (52, 53).
Using the difference in absorption at 240 nm of the protonated thiol and the deprotonated thiolate form of cysteines, we determined the pKa of the active site cysteines of Cg_NrdH-redoxin and Mt_NrdH-redoxin. We determined a pKa of 6.3 and 6.2 for the nucleophilic cysteine of Mt_NrdH-redoxin and Cg_NrdH-redoxin, respectively (Fig. 10). Like other members of the thioredoxin fold family (Trx, Grx, Mrx1), the nucleophilic cysteines of Cg_NrdH-redoxin and Mt_NrdH-redoxin are located at the N terminus of an α-helix, (17, 53, 54). The α-helix stabilizes the thiolate form of these cysteine, lowering their pKa (55). Additionally, in Trx, Grx, and Mrx1, neighboring residues form hydrogen bonds with the N-terminal cysteine, further decreasing its pKa (17, 53, 54). We solved the crystal structure of the oxidized form of Cg_NrdH-redoxin. From this structure, we cannot conclude that hydrogen bonds lower the pKa of the N-terminal cysteine. For the C-terminal cysteine of Mt_NrdH-redoxin and Cg_NrdH-redoxin, we obtained a pKa of 10.6 and 9.9, respectively (Fig. 10). Also for Trx, Grx, and Mrx1, a high pKa value for the C-terminal cysteine was obtained (17, 53, 54). The C-terminal cysteine will only become nucleophilic in the mixed disulfide complex formed after the nucleophilic attack of the N-terminal cysteine (56).
FIGURE 10.

The pKa of the active site cysteines of Cg (A) and Mt (B) were determined. The absorption (A240/A280)red/(A240/A280)ox was plotted in function of the pH. The curves were fitted with the Henderson-Hasselbalch equation to determine the pKa of the cysteines.
NrdH Seems to Be an Essential Gene in C. glutamicum
Attempts to delete or disrupt the nrdh gene in C. glutamicum failed to yield viable colonies. In order to confirm our methodology and as a positive control, we constructed a gene deletion and disruption mutant of Mrx1 (mrx1) using the same approach. Viable colonies were obtained, and the absence or disruption of the mrx1 gene was confirmed by sequencing. As such, we reasoned that the failure to obtain a viable mutant in nrdh (deleted or disrupted) might imply that the gene is essential in C. glutamicum. Currently, no data are available on the viability of nrdh disruption or deletion mutants in M. tuberculosis.
DISCUSSION
NrdH-redoxins play an important function in the cell as the reductase of NrdEF, a class Ib RNR (57). We investigate the NrdH-redoxins of the actinomycetes C. glutamicum and M. tuberculosis. This group of organisms does not produce the low molecular weight thiol GSH but instead produce MSH at millimolar concentrations. The NrdH-redoxin of the actinomycete C. ammoniagenes has been studied previously, but possible reduction of NrdH-redoxin by MSH has not been excluded (9). Recently, we described a novel MSH-specific reductase, Mrx1, in C. glutamicum and M. tuberculosis (16, 17). These proteins show a 40–50% amino acid sequence similarity with NrdH-redoxins (in Mt and Cg, respectively) but are linked to MSH as electron donor during the reduction reaction. As such, it is not unlikely that NrdH-redoxins from actinomycetes might use MSH as an alternative electron donor. Here, we show that this is not the case. Both Cg_NrdH-redoxin and Mt_NrdH-redoxin do not accept electrons from MSH but are only reduced by TrxR (Fig. 1).
The NrdH-redoxins catalytically reduce the disulfide of DTNB when linked to the TrxR electron pathway (Fig. 2). The first step of this reduction pathway involves the transfer of the disulfide bond of DTNB to NrdH-redoxin, forming oxidized NrdH-redoxin. In this step, the specificity constant of the NrdH-redoxins is only a factor of 2 lower than for Trx. One of the factors influencing the rate of this reaction is the pKa of the active site cysteines (52, 53). The N-terminal, nucleophilic cysteine of Cg_NrdH-redoxin and Mt_NrdH-redoxin has a pKa of 6.2 and 6.3, respectively (Fig. 10). This value is in the same range as the pKa reported for the N-terminal cysteine of Trx and Mrx1, whereas the pKa of the N-terminal cysteine of Grx is typically lower and in the range of 4–5 (17, 54, 58). The C-terminal cysteines of Cg_NrdH-redoxin and Mt_NrdH-redoxin have a higher pKa value (9.9 and 10.6, respectively).
The second step in the reduction of DTNB by the NrdH-redoxin/TrxR or Trx/TrxR pathway is the reduction of the oxidized NrdH-redoxin or Trx by TrxR. Cg_TrxR has a similar specificity constant for the reduction of Cg_NrdH-redoxin and Cg_Trx (5 × 105 m−1 min−1). This relatively slow turnover rate is probably due to the huge conformational change needed during catalysis (30, 31).
During this turnover, a complex is formed between TrxR and NrdH-redoxin. We modeled the complex between Cg_NrdH-redoxin and Ec_TrxR and between Mt_NrdH-redoxin and Mt_TrxR (Fig. 9). The latter might be of medical interest because TrxR is essential in M. tuberculosis and differs from its human homolog (57, 59). Both complexes show two conserved Phe residues of TrxR occupying a conserved hydrophobic pocket on the surface of the NrdH-redoxins. This hydrophobic pocket is one of the key structural features of NrdH-redoxins that determine their specificity for TrxR (21).
A second feature is the orientation of helix α3 in NrdH-redoxins. In NrdH-redoxins and in Trx, this helix is orientated antiparallel to strand β4. This orientation prevents steric hindrance upon reduction by TrxR. Mrx1 shares this feature with Trx and NrdH-redoxin, but Mrx1 is not reduced by TrxR (17). The linker between helix α3 and strand β4 is conserved among mycoredoxins and is most probably involved in MSH binding. In NrdH-redoxins, this linker contains the conserved WSGFRP(D/E) motif (21). A hydrogen bond network formed by the amino acids of this motif orients helix α3 antiparallel to strand β4 (Fig. 8). When comparing the NrdH-redoxin structures of several species (e.g. Ec, Ca, Cg, and Mt), only minor differences are observed in this hydrogen bond network.
The homology model of Mt_NrdH-redoxin and the crystal structure of Cg_NrdH-redoxin are both monomers (Fig. 4). Currently, a homology model of Mt_NrdH-redoxin, based on the swapped dimer structure of Ca_NrdH-redoxin, is present in the Swiss-Model Repository (50, 51). Our data do not support the swapped dimer model of Mt_NrdH-redoxin. Furthermore, Ca_NrdH-redoxin is a monomer in solution. As such, it is likely that the domain swapping observed in Ca_NrdH-redoxin is a crystallization artifact (9, 47–49). SEC analysis and SAXS of Mt_NrdH-redoxin shows a concentration dependent dimerization profile. At high protein concentrations, a fraction of the proteins forms a dimer that is stabilized by several salt bridges (Fig. 5). The active sites of both subunits in the dimer remain surface-exposed, but the conserved hydrophobic pocket of one of the subunits is buried in the dimer interface. The physiological relevance of a NrdH-redoxin dimer is questionable. At a protein concentration of 1.3 mg/ml, the majority is present as monomer. The intracellular concentrations of NrdH-redoxins in M. tuberculosis are not known, but we made a rough estimation, based on data from B. anthracis. In B. anthracis, 1 μg of total protein extract contains 12 pg of NrdH-redoxin (15), and the total intracellular protein concentration is 135 mg/ml (see General Molecular Biology Data, protein data on the New England Biolabs web site). Thus, an intracellular NrdH-redoxin concentration of ∼1 μg/ml is a reasonable estimate with the assumption that B. anthracis and M. tuberculosis have similar NrdH-redoxin expression levels. This concentration is about 3 orders of magnitude lower than the 1.3 mg/ml used in our SAXS experiment. As such, it is highly unlikely that NrdH-redoxin will be present as a dimer in the cell, although molecular crowding inside the cell might favor dimer formation even at these low concentrations (60).
The physiological importance of NrdH-redoxin in the cell is not known. Currently, no data are available on deletion or disruption mutants of nrdh in M. tuberculosis, but it is noteworthy that both interaction partners of NrdH, NrdEF and TrxR, have been shown to be essential (57). We attempted to construct nrdh deletion and disruption mutants in C. glutamicum, but no viable clones mutated in nrdh were detected. A nrdh-deleted mutant in S. aureus is viable and shows no increased sensitivity to oxidative stress (11). The lack of a phenotype in S. aureus was rationalized by the observation that Trx functions as a backup reducing system of NrdEF in S. aureus. In contrast, E. coli Ec_NrdEF cannot be reduced by Trx (10). We speculate that the essentiality of NrdH-redoxin in C. glutamicum might imply that Cg_NrdH-redoxin is the sole reductase of Cg_NrdEF in vivo. Whether this is also true in M. tuberculosis remains to be investigated.
Acknowledgments
We thank Efrén Ordóñez, Toon Bosmans, and Karolien Van Belle for help. The data collection experiments were performed on the ID29 beamline at the European Synchrotron Radiation Facility (ESRF) (Grenoble, France). We are grateful to the local contact at ESRF for providing assistance in using beamline ID29. The SAXS data were recorded on the P12 beamline at the EMBL (Hamburg, Germany). We are grateful to Yann Sterckx and Abel Garcia-Pino for data collection.
This work was supported by Agentschap voor Innovatie door Wetenschap en Technologie (IWT), the Vlaams Instituut voor Biotechnologie (VIB), and the HOA-project of the Onderzoeksraad of Vrije Universiteit Brussel.

This article contains supplemental Structures S1–S4.
The atomic coordinates and structure factors (code 4FIW) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- RNR
- ribonucleotide reductase
- Trx
- thioredoxin
- TrxR
- thioredoxin reductase
- Grx
- glutaredoxin(s)
- MSH
- mycothiol
- Mtr
- mycothiol disulfide reductase
- DTNB
- 5,5′-dithiobis-(2-nitrobenzoic acid)
- Cg
- Corynebacterium glutamicum
- Mt
- Mycobacterium tuberculosis
- Ec
- Escherichia coli
- Ca
- Corynebacterium ammoniagenes
- MD
- molecular dynamics
- SAXS
- small angle x-ray scattering
- SEC
- size exclusion chromatography
- Mrx
- mycoredoxin
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- PDB
- Protein Data Bank.
REFERENCES
- 1. Lundin D., Torrents E., Poole A. M., Sjöberg B. M. (2009) RNRdb, a curated database of the universal enzyme family ribonucleotide reductase, reveals a high level of misannotation in sequences deposited to GenBank. BMC Genomics 10, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nordlund P., Reichard P. (2006) Ribonucleotide reductases. Annu. Rev. Biochem. 75, 681–706 [DOI] [PubMed] [Google Scholar]
- 3. Eklund H., Uhlin U., Färnegardh M., Logan D. T., Nordlund P. (2001) Structure and function of the radical enzyme ribonucleotide reductase. Prog. Biophys. Mol. Biol. 77, 177–268 [DOI] [PubMed] [Google Scholar]
- 4. Reichard P., Ehrenberg A. (1983) Ribonucleotide reductase. A radical enzyme. Science 221, 514–519 [DOI] [PubMed] [Google Scholar]
- 5. Nilsson O., Lundqvist T., Hahne S., Sjöberg B. M. (1988) Structure-function studies of the large subunit of ribonucleotide reductase from Escherichia coli. Biochem. Soc. Trans. 16, 91–94 [DOI] [PubMed] [Google Scholar]
- 6. Jordan A., Pontis E., Atta M., Krook M., Gibert I., Barbé J., Reichard P. (1994) A second class I ribonucleotide reductase in Enterobacteriaceae. Characterization of the Salmonella typhimurium enzyme. Proc. Natl. Acad. Sci. U.S.A. 91, 12892–12896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mowa M. B., Warner D. F., Kaplan G., Kana B. D., Mizrahi V. (2009) Function and regulation of class I ribonucleotide reductase-encoding genes in mycobacteria. J. Bacteriol. 191, 985–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kolberg M., Strand K. R., Graff P., Andersson K. K. (2004) Structure, function, and mechanism of ribonucleotide reductases. Biochim. Biophys. Acta 1699, 1–34 [DOI] [PubMed] [Google Scholar]
- 9. Stehr M., Lindqvist Y. (2004) NrdH-redoxin of Corynebacterium ammoniagenes forms a domain-swapped dimer. Proteins 55, 613–619 [DOI] [PubMed] [Google Scholar]
- 10. Jordan A., Aslund F., Pontis E., Reichard P., Holmgren A. (1997) Characterization of Escherichia coli NrdH. A glutaredoxin-like protein with a thioredoxin-like activity profile. J. Biol. Chem. 272, 18044–18050 [DOI] [PubMed] [Google Scholar]
- 11. Rabinovitch I., Yanku M., Yeheskel A., Cohen G., Borovok I., Aharonowitz Y. (2010) Staphylococcus aureus NrdH redoxin is a reductant of the class Ib ribonucleotide reductase. J. Bacteriol. 192, 4963–4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boal A. K., Cotruvo J. A., Jr., Stubbe J., Rosenzweig A. C. (2010) Structural basis for activation of class Ib ribonucleotide reductase. Science 329, 1526–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Laer K., Hamilton C. J., Messens J. (2012) Low-molecular-weight thiols in thiol-disulfide exchange. Antioxid. Redox. Signal., doi: 10.1089/ars.2012.4964 [DOI] [PubMed] [Google Scholar]
- 14. Newton G. L., Rawat M., La Clair J. J., Jothivasan V. K., Budiarto T., Hamilton C. J., Claiborne A., Helmann J. D., Fahey R. C. (2009) Bacillithiol is an antioxidant thiol produced in Bacilli. Nat. Chem. Biol. 5, 625–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gustafsson T. N., Sahlin M., Lu J., Sjöberg B. M., Holmgren A. (2012) Bacillus anthracis thioredoxin systems, characterization and role as electron donors for ribonucleotide reductase. J. Biol. Chem. 287, 39686–39697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ordóñez E., Van Belle K., Roos G., De Galan S., Letek M., Gil J. A., Wyns L., Mateos L. M., Messens J. (2009) Arsenate reductase, mycothiol, and mycoredoxin concert thiol/disulfide exchange. J. Biol. Chem. 284, 15107–15116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Laer K., Buts L., Foloppe N., Vertommen D., Van Belle K., Wahni K., Roos G., Nilsson L., Mateos L. M., Rawat M., van Nuland N. A., Messens J. (2012) Mycoredoxin-1 is one of the missing links in the oxidative stress defence mechanism of Mycobacteria. Mol. Microbiol. 86, 787–804 [DOI] [PubMed] [Google Scholar]
- 18. Roos G., Garcia-Pino A., Van Belle K., Brosens E., Wahni K., Vandenbussche G., Wyns L., Loris R., Messens J. (2007) The conserved active site proline determines the reducing power of Staphylococcus aureus thioredoxin. J. Mol. Biol. 368, 800–811 [DOI] [PubMed] [Google Scholar]
- 19. Kabsch W. (1988) Evaluation of single-crystal X-ray-diffraction data from a position-sensitive detector. J. Appl. Crystallogr. 21, 916–924 [Google Scholar]
- 20. Long F., Vagin A. A., Young P., Murshudov G. N. (2008) BALBES. A molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stehr M., Schneider G., Aslund F., Holmgren A., Lindqvist Y. (2001) Structural basis for the thioredoxin-like activity profile of the glutaredoxin-like NrdH-redoxin from Escherichia coli. J. Biol. Chem. 276, 35836–35841 [DOI] [PubMed] [Google Scholar]
- 22. Eklund H., Ingelman M., Söderberg B. O., Uhlin T., Nordlund P., Nikkola M., Sonnerstam U., Joelson T., Petratos K. (1992) Structure of oxidized bacteriophage T4 glutaredoxin (thioredoxin). Refinement of native and mutant proteins. J. Mol. Biol. 228, 596–618 [DOI] [PubMed] [Google Scholar]
- 23. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 25. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Painter J., Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 27. Painter J., Merritt E. A. (2006) TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 39, 109–111 [Google Scholar]
- 28. Lassmann T., Sonnhammer E. L. (2005) Kalign. An accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics 6, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics, Chapter 5, Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lennon B. W., Williams C. H., Jr., Ludwig M. L. (2000) Twists in catalysis. Alternating conformations of Escherichia coli thioredoxin reductase. Science 289, 1190–1194 [DOI] [PubMed] [Google Scholar]
- 31. Akif M., Suhre K., Verma C., Mande S. C. (2005) Conformational flexibility of Mycobacterium tuberculosis thioredoxin reductase. Crystal structure and normal-mode analysis. Acta Crystallogr. D Biol. Crystallogr. 61, 1603–1611 [DOI] [PubMed] [Google Scholar]
- 32. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4. Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 33. Dolinsky T. J., Nielsen J. E., McCammon J. A., Baker N. A. (2004) PDB2PQR. An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems. Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) ClustalW and ClustalX version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 36. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS. A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 37. Guinier A., Fournet G. (1955) Small-angle Scattering of X-rays, John Wiley & Sons, Inc., New York [Google Scholar]
- 38. Porod G. (1982) Small Angle X-ray Scattering, pp. 17–51, Academic Press, Inc., New York [Google Scholar]
- 39. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 40. Konarev P. V., Petoukhov M. V., Volkov V. V., Svergun D. I. (2006) ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fernández-Gonzalez C., Gil J. A., Mateos L. M., Schwarzer A., Schäfer A., Kalinowski J., Pühler A., Martín J. F. (1996) Construction of l-lysine-overproducing strains of Brevibacterium lactofermentum by targeted disruption of the hom and thrB genes. Appl. Microbiol. Biotechnol. 46, 554–558 [DOI] [PubMed] [Google Scholar]
- 42. Jäger W., Schäfer A., Pühler A., Labes G., Wohlleben W. (1992) Expression of the Bacillus subtilis sacB gene leads to sucrose sensitivity in the gram-positive bacterium Corynebacterium glutamicum but not in Streptomyces lividans. J. Bacteriol. 174, 5462–5465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ordóñez E., Thiyagarajan S., Cook J. D., Stemmler T. L., Gil J. A., Mateos L. M., Rosen B. P. (2008) Evolution of metal(loid) binding sites in transcriptional regulators. J. Biol. Chem. 283, 25706–25714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Patel M. P., Blanchard J. S. (1999) Expression, purification, and characterization of Mycobacterium tuberculosis mycothione reductase. Biochemistry 38, 11827–11833 [DOI] [PubMed] [Google Scholar]
- 45. Akif M., Khare G., Tyagi A. K., Mande S. C., Sardesai A. A. (2008) Functional studies of multiple thioredoxins from Mycobacterium tuberculosis. J. Bacteriol. 190, 7087–7095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Garcia-Pino A., Martinez-Rodriguez S., Wahni K., Wyns L., Loris R., Messens J. (2009) Coupling of domain swapping to kinetic stability in a thioredoxin mutant. J. Mol. Biol. 385, 1590–1599 [DOI] [PubMed] [Google Scholar]
- 47. Rousseau F., Schymkowitz J. W., Itzhaki L. S. (2003) The unfolding story of three-dimensional domain swapping. Structure 11, 243–251 [DOI] [PubMed] [Google Scholar]
- 48. Liu Y., Eisenberg D. (2002) 3D domain swapping. As domains continue to swap. Protein Sci. 11, 1285–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Newcomer M. E. (2001) Trading places. Nat. Struct. Biol. 8, 282–284 [DOI] [PubMed] [Google Scholar]
- 50. Kiefer F., Arnold K., Künzli M., Bordoli L., Schwede T. (2009) The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 37, D387–D392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leiting W. U., Jianping X. I. (2010) Comparative genomics analysis of Mycobacterium NrdH-redoxins. Microb. Pathog. 48, 97–102 [DOI] [PubMed] [Google Scholar]
- 52. Jensen K. S., Hansen R. E., Winther J. R. (2009) Kinetic and thermodynamic aspects of cellular thiol-disulfide redox regulation. Antioxid. Redox. Signal. 11, 1047–1058 [DOI] [PubMed] [Google Scholar]
- 53. Roos G., Foloppe N., Messens J. (2013) Understanding the pKa of redox cysteines. The key role of hydrogen bonding. Antioxid. Redox. Signal. 18, 94–127 [DOI] [PubMed] [Google Scholar]
- 54. Collet J. F., Messens J. (2010) Structure, function, and mechanism of thioredoxin proteins. Antioxid. Redox. Signal. 13, 1205–1216 [DOI] [PubMed] [Google Scholar]
- 55. Roos G., Loverix S., Geerlings P. (2006) Origin of the pKa perturbation of N-terminal cysteine in α- and 310-helices. A computational DFT study. J. Phys. Chem. B 110, 557–562 [DOI] [PubMed] [Google Scholar]
- 56. Roos G., Foloppe N., Van Laer K., Wyns L., Nilsson L., Geerlings P., Messens J. (2009) How thioredoxin dissociates its mixed disulfide. PLoS Comput. Biol. 5, e1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sassetti C. M., Boyd D. H., Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 [DOI] [PubMed] [Google Scholar]
- 58. Harris T. K., Turner G. J. (2002) Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life 53, 85–98 [DOI] [PubMed] [Google Scholar]
- 59. Williams C. H., Arscott L. D., Müller S., Lennon B. W., Ludwig M. L., Wang P. F., Veine D. M., Becker K., Schirmer R. H. (2000) Thioredoxin reductase two modes of catalysis have evolved. Eur. J. Biochem. 267, 6110–6117 [DOI] [PubMed] [Google Scholar]
- 60. Chebotareva N. A., Kurganov B. I., Livanova N. B. (2004) Biochemical effects of molecular crowding. Biochemistry 69, 1239–1251 [DOI] [PubMed] [Google Scholar]