

Abstract
Alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV) is a rare developmental lung disorder that is uniformly lethal. Affected infants die within the first few weeks of their life despite aggressive treatment, although a few cases of late manifestation and longer survival have been reported. We have shown previously that mutations and deletions in FOXF1 are a cause of this disorder. Although most of the cases of ACD/MPV are sporadic, there have been infrequent reports of familial cases. We present a family with five out of six children affected with ACD/MPV. DNA analysis identified a missense mutation (c.416G>T; p.Arg139Leu) in the FOXF1 gene that segregated in the three affected siblings tested. The same variant is also present as a de novo mutation in the mother and arose on her paternally derived chromosome 16. The two tested affected siblings share the same chromosome 16 haplotype inherited from their maternal grandfather. Their single healthy sibling has a different chromosome 16 haplotype inherited from the maternal grandmother. The results are consistent with paternal imprinting of FOXF1 in human.
Keywords: ACD/MPV, FOXF1, imprinting, angiogenesis, lung development
Introduction
Alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV; OMIM no. 265380) is a rare lung disorder that presents in the early neonatal period. It affects both parenchymal and pulmonary vasculature development.1, 2 The characteristic histologic features include malpositioned (misaligned) pulmonary veins that run alongside small pulmonary arteries both in their pre-acinar and intra-acinar course and, at times with the arteries within a common adventitial sheath, rather than in their normal location within interlobular septa or at the lobular periphery;3, 4 increased smooth muscles in small pulmonary arteries; lobular underdevelopment and a severe deficiency of normally positioned and normal sized capillaries in alveolar walls.3, 5 A third of the cases also have lymphangiectasis.3 Most cases of ACD/MPV are sporadic with only a few reported familial cases (∼10%).4, 6, 7, 8, 9 Affected infants typically present with pulmonary hypertension within a few hours of birth and despite treatment with positive pressure oxygen, nitric oxide, sildenafil and/or extracorporeal membrane oxygenation, clinical improvement is limited and survival is brief.10, 11, 12 A few cases of late presentation and longer survival have been reported.10, 13, 14 The disorder has more often been diagnosed histopathologically at autopsy of affected infants; however, increasing reports of biopsy diagnosis are being made. A very high percentage (∼80%) of ACD/MPV patients has a variety of associated anomalies particularly of the gastrointestinal, cardiovascular and genitourinary systems.3
We have been involved in research related to ACD/MPV since 2000 and have accumulated a large collection (>80) of tissue and DNA samples from patients with ACD/MPV and their immediate family members. The parents' organization ‘ACD Association (ACDA)' has been of great help in recruiting new patients and spreading the information about our research (http://www.acd-association.com). ‘The Breath of Life Project' (http://www.breathoflifeproject.com) has recently begun educating health professionals about ACD/MPV as a part of a Centers for Disease Control and Prevention (CDC)-funded awareness project.
In 2009, we showed that a segment of ACD/MPV patients in our cohort had either inactivating point mutations in the FOXF1 gene or deletions at 16q24.1, which were both genic and upstream to FOXF1.15
Materials and methods
The affected infants, their parents, healthy sibling and grandparents were enrolled in our ongoing study of the genetic basis of ACD/MPV, approved by the Baylor Institutional Review Board (H-8712) after their signed consent. Lung histology was reviewed by CL to confirm ACD/MPV (Figure 1).
Figure 1.
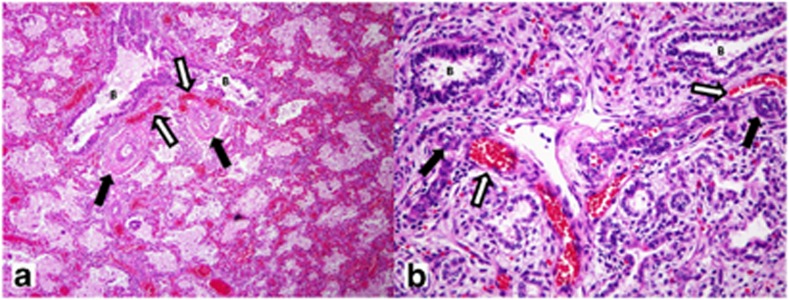
Histopathological findings in term female (a, 39 weeks gestation; 17 days life span) (Table 1: Patient 1) and 21-weeks-old male fetus (b) (Table 1: Patient 2). The lungs show characteristic histological findings of ACD/MPV with pulmonary vein misalignment, arterial medial thickening and lobular underdevelopment even in the lung of the immature male fetus (b). For both, white arrows indicate misaligned pulmonary veins and black arrows identify small pulmonary arteries with smooth muscle hyperplasia. Nearby bronchioles are identified as B. The lobular parenchyma for the term infant shows enlarged and poorly subdivided air spaces, diminished numbers of normally positioned capillaries and marked venous congestion. For the fetus (b), the lobular parenchyma appears immature for the gestational age with simple rounded air spaces without evidence of early secondary crest formation, and with persistent subnuclear glycogen evident in the uniform cuboidal epithelium.
The pedigree of this Caucasian family of Central European origin is shown in Figure 2. Among six children in the family only one is healthy (Figure 2; II 05). Three infants (Figure 2; II 01, II 03 and II 04) died in the neonatal period and two fetuses (Figure 2; II 02 and II 06) were terminated at 21 and 22 weeks of gestation, respectively, for severe urinary tract anomalies (Table 1). The parents are non-consanguineous.
Figure 2.

Pedigree of the described family. Five of the six children were affected with ACD/MPV. The arrow shows the proband. DNA from II 02, II 04 and II 06 identified the c.416G>T;p.Arg139Leu mutation. Smaller squares indicate terminations of pregnancies. The proband is shown with an arrow.
Table 1. Clinical information about the ACD/MPV infants in the family reported.
| Patient no. (pedigree no.) | Gender | Birth weight (kg) | Mother's age and others | Gestation (weeks) | Apgar 1 and 5 min | Delivery | Life span (days) | Diagnosis | Medications | ECMO | Associated anomalies |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 (II 01) | F | 3.4 | 25, G2P1 | 39 | 9, 9 | Vaginal | 17 | Autopsy | NA | NA | NA |
| 2 (II 02) | M | NA | 25 | Terminated at 21 weeks | NA | NA | NA | Autopsy | NA | NA | Uretral stenosis, megavesica, severe hydronephrosis |
| 3 (II 03) | M | 3.2 | 27, G4P2 | 37 and 1/7 | 9, 10 | Vaginal | 0.16 | Autopsy | Surfactants and catecholamines | No | Pulmonary hypoplasia, hydrothorax, severe polyhydramnion |
| 4 (II 04) | F | 3.2 | 31, G5P3 | 39 | 9, 9 | Vaginal | 1 | Autopsy | Nitric oxide, catecholamines | No | Isolated arhinencephaly |
| 5 (II 06) | M | NA | 36 | Terminated at 22 weeks | NA | NA | NA | Autopsy | NA | NA | Severe obstructive uropathy |
Abbreviations: ACD/MPV, alveolar capillary dysplasia with misalignment of pulmonary veins; ECMO, extracorporeal membrane oxygenation; NA, not available.
DNA from patient tissue samples were isolated using Qiagen's (Germantown, MD, USA) DNeasy Blood and Tissue kit using manufacturer's instructions. DNA from blood of the parents, healthy sibling and grandparents were isolated using Gentra Puregene isolation kit following the manufacturer's instruction. The PCR primers (The primers were synthesized in the Laboratory of Dr Partha Sen, Baylor College of Medicine, TX, USA, using ABI 3900 DNA Synthesizer, Foster City, CA, USA.) and cycling protocol are shown below.

Cycling conditions: 94 °C 3 min, 40 cycles of 94 °C for 30 sec, 54 °C for 30 sec, 72 °C for 1 min; final extension at 72 °C for 5 min.
Genotyping of 733,202 SNPs was done using Illumina (San Diego, CA, USA) OmniExpress beadarrays, following manufacturer's instructions, to investigate the haplotype encompassing FOXF1 on chromosome 16.
Results
Diagnosis of familial ACD/MPV was based on the identification of characteristic histopathological changes in the lungs in three of the five affected subjects (Figure 2; II 01, II 02, II 06) present even in the more immature fetal lungs. All showed lobular underdevelopment, capillary deficiency and misalignment of pulmonary veins (Figure 1). The clinical details of the patients are shown in Table 1. A missense heterozygous mutation of G>T at the 416 position that changes an arginine to leucine at position 139 of the predicted protein sequence (c416G>T; p.Arg139Leu) was identified within the DNA-binding domain of FOXF116, 17 (Figure 3). The mutation was identified in DNA samples from three affected infants (Figure 2; II 02, II 04 and II 06, and 2, 4, 5 Table 1). No DNA samples were available from the two other affected infants (Figure 2; II 01; II 03, and 1, 3 Table 1). The same mutation was also identified in the DNA sample from the mother (Figure 3) who had heart scratch in her childhood without any recognized heart defect or cardiac function that faded away later in her life. She also had a tracheal stenosis at 3 years of age, which was resolved by dilatation. She is without any respiratory problems now. The unaffected child (Figure 2; II 05) and the father of the children do not have the mutation (Figure 3) and are clinically well. The mutation in the mother arose de novo as it was not found in her parents or siblings. This mutation is not one of the SNPs reported in the dbSNP and is not cited in the Exome Variant Server, NHLBI Exome Sequencing Project (ESP; Seattle, WA, USA) (http://evs.gs.washington.edu/EVS/), which covers more than 10 000 alleles. The mutation was predicted to be deleterious by two programs viz. PolyPhen-2 and Aligned GVGD.18, 19
Figure 3.

DNA sequencing showing the heterozygous mutation (c416G>T). (a) DNA from the father; (b) DNA from the mother; (c) DNA from one of the affected children (Figure 2; II 06); (d) DNA from the unaffected child (Figure 2. II 05). The arrows indicate the mutated base. A and D are homozygous for the wild-type allele, whereas B and C show double peak for G and T indicating a heterozygous mutation.
Haplotype analysis was carried out using Illumina OmniExpress arrays that genotype 733,202 SNPs. Using an interval of approximately 1 Mb flanking the FOXF1 locus, we selected 214 SNPs with low inter-marker linkage disequilibrium (r2<0.2) using the HapMap CEU population as the reference. We then removed markers that were monomorphic among the three unrelated individuals in the pedigree (maternal grandfather, maternal grandmother and father), leaving 145 markers for haplotype analysis. Haplotypes were inferred using the EM algorithm and logic rules implemented in HAPLORE software (http://bioinformatics.med.yale.edu/ group/software.html). The results show that the affected children inherited the same haplotype from their mother. The unaffected sibling inherited the alternate maternal haplotype. By reference to the genotypes of the maternal grandparents, the haplotype bearing the FOXF1 mutation was inherited from their maternal grandfather (Figure 4).
Figure 4.

(a) Result of the haplotype analysis using Illumina Omni express platform. The analysis is centered on FOXF1 (Chr16:85,000,000–85,200,000, hg18). The colored boxes identify chromosomes in different individuals. The affected children in generation III have the same maternal chromosome from their mother that she inherited from her father. The unaffected child has the other chromosome from their mother which she inherited from her mother. (b) A detail of the informative SNPs. F: father; M: mother, AF1 and AF2: affected child 1 and 2, UN: unaffected child, MGF and MGM: maternal grandfather and grandmother, respectively. The SNP numbers are shown on the right. Identity of the inherited chromosomes are indicated by * and †.
Discussion
FOXF1 is a transcription factor expressed in the lung mesenchyme during development.16 It is essential for cell migration during embryonic and fetal development.20 Our previous work has shown that abnormalities of FOXF1 are associated with ACD/MPV.15 The mouse Foxf1 and the human FOXF1 proteins act in a haploinsufficient manner.15, 21, 22, 23 FOXF1 has been predicted bioinformatically to be paternally imprinted in the humans;24 however, a paternal uniparental disomy of chromosome 16 (UPD16pat) resulted in an otherwise normal child with only some fetal and postnatal growth retardation.25 Interestingly, all the genic and upstream deletions in our cohort of patients reported previously15 occurred on the maternal chromosome 16, which suggested that FOXF1 might be paternally imprinted. We have since identified eight additional deletions upstream of FOXF1 and two deletions involving FOXF1 in ACD/MPV patients. Seven of these deletions, for which parental origin could be determined, arose de novo on the maternal chromosome (P Szafranski, personal communication).
In this article, we present a unique familial case of ACD/MPV with five affected subjects among who were two female and one male infants that died of ACD/MPV; additionally, the mother had two pregnancy terminations because of severe obstructive uropathy (Table 1; Figure 2). The family has one unaffected child. Pathological examination of the lungs of the affected infants and the fetal terminations show typical features of ACD/MPV (Figure 1). The mutation in the infants is inherited from their healthy mother who is heterozygous for the identical mutation (Figure 3). The history of tracheal stenosis in the mother fits well with the mild end of the spectrum of FOXF1 deficiency26 and can be explained by a higher local dosage sensitiveness of the FOXF1 protein required for trachea development when compared with the lungs. The absence of the mutation in DNA sequenced from the maternal grandparents and uncle and aunt of the children confirmed that the mutation in the mother is de novo. The change of an arginine to leucine most likely affects the function of the protein as arginine is a basic amino acid and leucine a nonpolar one.27 Further, the arginine at this position is highly conserved in the FOX genes.17
It is evident that the affected children died of ACD/MPV due to the mutation in the FOXF1 gene. They inherited the mutation from their mother and therefore, the mutation is present on their maternal chromosome 16. The healthy child inherited the wild-type copy of chromosome 16 from the mother. Further, the mother has the mutation and has no features of ACD/MPV, suggesting that the mutation in her is present on the paternal chromosome 16. This clearly supports the possibility that FOXF1 is paternally imprinted with only the maternal copy being expressed. We reported a paternally inherited no-stop mutation in the second exon (last codon) in FOXF1 in a patient with ACD/MPV.15 We believe that the aberrant and extended transcript might have escaped nonsense-mediated decay and negatively acted on the maternal wild-type copy of the transcript, resulting in ACD/MPV.
The imprinted nature of FOXF1 has been predicted previously.24, 25 To confirm the lineage of the inheritance of the maternal chromosome in the affected infants and the unaffected child, we performed a haplotype analysis using Illumina OmniExpress. The result (Figure 4) implies a model in which the mother, who carries a de novo mutation in FOXF1, is healthy because the mutation is on her paternal chromosome; however, the same mutation when transmitted to her offspring results in ACD/MPV. The data strongly suggest that FOXF1 is predominantly expressed from the maternal copy. Further evidence that deletions in the 16q24.1 region in ACD/MPV patients are all on the maternal chromosome strongly supports this model. Szafranski et al (P Szafranski, personal communication) have shown that the 75-kb regulatory region mapping at 250 kb upstream of FOXF1 is differentially methylated, which indicates that the gene may be differentially expressed. This result along with the UPD16pat data suggests that the expression of FOXF1 might be complex and imprinting of this gene can be incomplete with the maternal copy being predominantly expressed. It is possible that the expression of the gene from two paternal copies might be just adequate to fulfill its functional requirements as evidenced by a normal child with UPD16pat.
Thus, our data further confirm that FOXF1 is implicated in ACD/MPV and that the gene is maternally expressed and paternally imprinted in human. We plan to investigate the role of methylation in regulating the expression of this gene. Tissue-specific methylation has been shown to regulate Foxf1 expression in mice28 and methylation patterns have been conserved in eukaryotic evolution.29, 30 It will be interesting to investigate tissue-specific pattern of methylation of FOXF1 in humans as a majority of the ACD/MPV patients also have anomalies of other organ systems.3, 4 Eventually, we would like to understand the molecular role of FOXF1 in lung development, thereby providing counseling and possibly treatment for patients with ACD/MPV and their families in future.
Acknowledgments
Part of this work was supported by a NORD grant and Pilot Project Award from the Texas Children's Hospital, Houston TX to P Sen, and NIH grant 1RO1HL101975-01 to P Stankiewicz. We would like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies that produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
The authors declare no conflict of interest.
References
- Janney CG, Askin FB, Kuhn C. Congenital alveolar capillary dysplasia – an unusual cause of respiratory distress in the newborn. Am J Clin Pathol. 1981;76:722–727. doi: 10.1093/ajcp/76.5.722. [DOI] [PubMed] [Google Scholar]
- Langston C. Misalignment of pulmonary veins and alveolar capillary dysplasia. Pediatr Pathol. 1991;11:163–170. doi: 10.3109/15513819109064753. [DOI] [PubMed] [Google Scholar]
- Sen P, Thakur N, Stockton DW, Langston C, Bejjani BA. Expanding the phenotype of alveolar capillary dysplasia (ACD) J Pediatr. 2004;145:646–651. doi: 10.1016/j.jpeds.2004.06.081. [DOI] [PubMed] [Google Scholar]
- Boggs S, Harris MC, Hoffman DJ, et al. Misalignment of pulmonary veins with alveolar capillary dysplasia: affected siblings and variable phenotypic expression. J Pediatr. 1994;124:125–128. doi: 10.1016/s0022-3476(94)70267-5. [DOI] [PubMed] [Google Scholar]
- Wagenvoort CA. Misalignment of lung vessels: a syndrome causing persistent neonatal pulmonary hypertension. Hum Pathol. 1986;17:727–730. doi: 10.1016/s0046-8177(86)80182-4. [DOI] [PubMed] [Google Scholar]
- Gutierrez C, Rodriguez A, Palenzuela S, Forteza C, Rossello JL. Congenital misalignment of pulmonary veins with alveolar capillary dysplasia causing persistent neonatal pulmonary hypertension: report of two affected siblings. Pediatr Dev Pathol. 2000;3:271–276. doi: 10.1007/s100249910035. [DOI] [PubMed] [Google Scholar]
- Shohet I, Reichman B, Schibi G, Brish M. Familial persistent pulmonary hypertension. Arch Dis Child. 1984;59:783–785. doi: 10.1136/adc.59.8.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manouvrier-Hanu S, Devisme L, Farre I, et al. Pulmonary hypertension of the newborn and urogenital anomalies in two male siblings: a new family with misalignment of pulmonary vessels. Genet Couns. 1996;7:249–255. [PubMed] [Google Scholar]
- Vassal HB, Malone M, Petros AJ, Winter RM. Familial persistent pulmonary hypertension of the newborn resulting from misalignment of the pulmonary vessels (congenital alveolar capillary dysplasia) J Med Genet. 1998;35:58–60. doi: 10.1136/jmg.35.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hathlol K, Phillips S, Seshia MK, Casiro O, Alvaro RE, Rigatto H. Alveolar capillary dysplasia. Report of a case of prolonged life without extracorporeal membrane oxygenation (ECMO) and review of the literature. Early Hum Dev. 2000;57:85–94. doi: 10.1016/s0378-3782(99)00065-1. [DOI] [PubMed] [Google Scholar]
- Plat G, Rouquette I, Marcoux MO, Bloom MC, Acar P, Dulac Y. [Alveolar capillary dysplasia and persistent pulmonary hypertension of the newborn] Arch Mal Coeur Vaiss. 2007;100:458–461. [PubMed] [Google Scholar]
- Kitayama Y, Kamata S, Okuyama H, et al. Nitric oxide inhalation therapy for an infant with persistent pulmonary hypertension caused by misalignment of pulmonary veins with alveolar capillary dysplasia. J Pediatr Surg. 1997;32:99–100. doi: 10.1016/s0022-3468(97)90105-6. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Ackerman V, Faught P, Langston C. Profound hypoxemia and pulmonary hypertension in a 7-month-old infant: late presentation of alveolar capillary dysplasia. Pediatr Crit Care Med. 2008;9:e43–e46. doi: 10.1097/PCC.0b013e31818e383e. [DOI] [PubMed] [Google Scholar]
- Eulmesekian P, Cutz E, Parvez B, Bohn D, Adatia I. Alveolar capillary dysplasia: a six-year single center experience. J Perinat Med. 2005;33:347–352. doi: 10.1515/JPM.2005.067. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Sen P, Bhatt SS, et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. 2009;84:780–791. doi: 10.1016/j.ajhg.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlapuu M, Pelto-Huikko M, Aitola M, Enerback S, Carlsson P. FREAC-1 contains a cell-type-specific transcriptional activation domain and is expressed in epithelial-mesenchymal interfaces. Dev Biol. 1998;202:183–195. doi: 10.1006/dbio.1998.9010. [DOI] [PubMed] [Google Scholar]
- Pierrou S, Hellqvist M, Samuelsson L, Enerback S, Carlsson P. Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J. 1994;13:5002–5012. doi: 10.1002/j.1460-2075.1994.tb06827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34:1317–1325. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin D, Kim IM, Boetticher E, et al. Forkhead box F1 is essential for migration of mesenchymal cells and directly induces integrin-beta3 expression. Mol Cell Biol. 2007;27:2486–2498. doi: 10.1128/MCB.01736-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinichenko VV, Gusarova GA, Kim IM, et al. Foxf1 haploinsufficiency reduces Notch-2 signaling during mouse lung development. Am J Physiol Lung Cell Mol Physiol. 2004;286:L521–L530. doi: 10.1152/ajplung.00212.2003. [DOI] [PubMed] [Google Scholar]
- Kalinichenko VV, Zhou Y, Bhattacharyya D, et al. Haploinsufficiency of the mouse Forkhead Box f1 gene causes defects in gall bladder development. J Biol Chem. 2002;277:12369–12374. doi: 10.1074/jbc.M112162200. [DOI] [PubMed] [Google Scholar]
- Yu S, Shao L, Kilbride H, Zwick DL. Haploinsufficiencies of FOXF1 and FOXC2 genes associated with lethal alveolar capillary dysplasia and congenital heart disease. Am J Med Genet A. 2010;152A:1257–1262. doi: 10.1002/ajmg.a.33378. [DOI] [PubMed] [Google Scholar]
- Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL, Hartemink AJ. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007;17:1723–1730. doi: 10.1101/gr.6584707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhase J, Janssen B, Weidenauer K, Harms K, Bartels I. First confirmed case with paternal uniparental disomy of chromosome 16. Am J Med Genet. 2000;91:190–191. doi: 10.1002/(sici)1096-8628(20000320)91:3<190::aid-ajmg6>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Shaw-Smith C. Genetic factors in esophageal atresia, tracheo-esophageal fistula and the VACTERL association: roles for FOXF1 and the 16q24.1 FOX transcription factor gene cluster, and review of the literature. Eur J Med Genet. 2010;53:6–13. doi: 10.1016/j.ejmg.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borders CL, Broadwater JA, Bekeny PA, et al. A structural role for arginine in proteins: multiple hydrogen bonds to backbone carbonyl oxygens. Protein Sci. 1994;3:541–548. doi: 10.1002/pro.5560030402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang VW, Ho Y. Structural characterization of the mouse Foxf1a gene. Gene. 2001;267:201–211. doi: 10.1016/s0378-1119(01)00400-0. [DOI] [PubMed] [Google Scholar]
- Lee TF, Zhai J, Meyers BC. Conservation and divergence in eukaryotic DNA methylation. Proc Natl Acad Sci USA. 2010;107:9027–9028. doi: 10.1073/pnas.1005440107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Han L, Zhao Z. Conservation and divergence of DNA methylation in eukaryotes: new insights from single base-resolution DNA methylomes. Epigenetics. 2011;6:134–140. doi: 10.4161/epi.6.2.13875. [DOI] [PMC free article] [PubMed] [Google Scholar]