

Abstract
Background:
Mutations in DNA methyltransferase 1 (DNMT1) have been identified in 2 autosomal dominant syndromes: 1) hereditary sensory autonomic neuropathy with dementia and hearing loss (HSAN1E); and 2) cerebellar ataxia, deafness, and narcolepsy. Both syndromes have mutations in targeting sequence (TS) domain (exons 20–21), which is important in mediating DNA substrate binding to the DNMT1 catalytic domain. Frontal lobe hypometabolism has been documented in an HSAN1E family, but memory loss has been the primary cognitive complaint. The chromosomal location of the DNMT1 gene at 19p13.2 has been linked to familial late-onset Alzheimer disease.
Methods:
We sequenced 41 exons of DNMT1 and their flanking regions in 1) 2 kindreds with HSAN1E; 2) 48 patients with HSAN1 alone without dementia and hearing loss; and 3) 5 probands of familial frontotemporal dementia (FTD) kindreds. We also sequenced exon 20 and 21 in 364 autopsy-confirmed late-onset Alzheimer disease cases.
Results:
Mutations in DNMT1 were specific to 2 HSAN1E kindreds with dementia and hearing loss (no narcolepsy). One family carried previously identified mutation Tyr495Cys; the other carried a novel Tyr495His, both in the TS domain. The symptoms of these patients include prominent personality, psychiatric manifestations, and seizures in one and the onset time is later than the previously reported cases.
Conclusion:
Clinicians should consider DNMT1 mutations in patients presenting with FTD or primary memory decline who also have sensory neuropathy and hearing loss. Amino acid Tyr495 is a hot spot for HSAN1E, distinct from exon 21 mutations associated with narcolepsy.
DNA methyltransferase 1 (DNMT1) is the fundamental methylation enzyme in maintaining methylation during DNA replication and DNA repair. Proper epigenetic regulation including DNA methylation is important for development, survival, and connectivity of neural cells.1 We showed that mutations in DNMT1 cause hereditary sensory and autonomic neuropathy with dementia and hearing loss (HSAN1E).2,3 Autosomal dominant hereditary sensory and autonomic neuropathy (HSAN1) includes multiple subtypes (HSAN1A–HSAN1E) based on the specific phenotypes and genetic causes. The affected persons in our previously reported HSAN1E families had normal development and childhood, then developed stereotypic progressive-onset (teens–mid-30s) hearing loss and sensory neuropathy, followed by memory decline in their 30s and 40s.3 Autopsies in HSAN1E showed diffuse neurodegeneration of brain, spinal cord, and nerve without specific interstitial changes, i.e., no β-amyloid, TDP-43, or α-synuclein immunostaining. Nursing home placement and total care requirements were typical by the fifth decade, mainly due to memory loss and sensory ataxia, and foot ulcers or extremity amputations in about half of affected persons. The potential frontal lobe involvement and abnormal behavioral presentations were suggested by SPECT, PET images, and autopsy studies in one Japanese kinship.4,5 Recently the phenotype spectrum of DNMT1 mutations has been expanded to include narcolepsy, i.e., autosomal dominant cerebellar ataxia deafness and narcolepsy (ADCA-DN).6 Patients with ADCA-DN also have neuropathy and frequently unspecified psychosis and dementia with characteristic brain imaging studies.7 It was speculated that the rare polymorphisms located within the DNMT1 locus may account for sporadic narcolepsy,6 since a recent genome-wide association study linked sporadic narcolepsy patients to this chromosomal region.8
Two mutations causing HSAN1E occurred in exon 20 of DNMT1 (p.Tyr495Cys, p.Asp490Glu-Pro491Tyr) and 3 mutations causing ADCA-DN occurred in exon 21 (p.Ala570Val, p.Gly605Ala, p.Val606Phe) (figure 1). In both disorders, all causal mutations were found within the targeting sequence (TS) domain of DNMT1. The TS domain has been demonstrated to be indispensable in mediating the association of DNMT1 to heterochromatin during the G2 and M phase of the cell cycle. The interaction between the TS and catalytic domains is crucial for proper methylation maintenance of the genome.9 Our studied mutations in exon 20 cause premature degradation of the mutant protein, reduced methyltransferase activity of DNMT1, and impaired heterochromatin binding during the G2 cell cycle phase. Importantly, we also observed global DNA hypomethylation throughout the genome and site-specific hypermethylation, a common phenomenon in cancer cells, emphasizing the direct link between DNMT1 and neurodegeneration.3
Figure 1. DNMT1 protein structure and mutations.
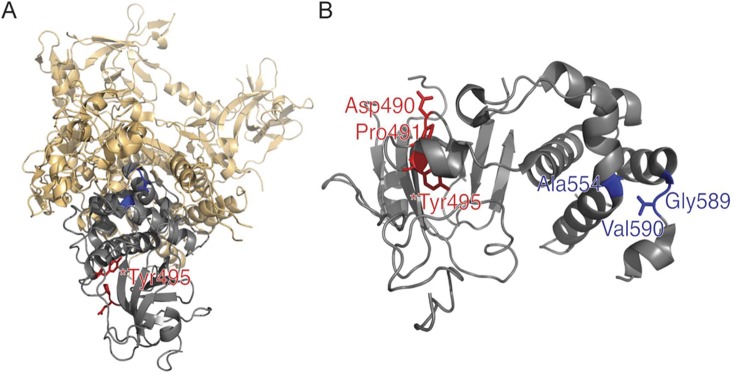
(A) DNMT1 entire protein structure with Tyr495 hot spot for mutation shown. (B) Targeting sequence (TS) domain with mutations. The TS domain (gray) is the site of known mutations for hereditary sensory and autonomic neuropathy with dementia and hearing loss (HSAN1E) (red) in exon 20 for HSAN1E. In contrast, all cases of autosomal dominant cerebellar ataxia deafness and narcolepsy arise from mutations in exon 21 (blue), also within the TS domain. Images were created using PyMol (www.pymol.org) and crystal structures of mouse DNMT1 (amino acids 357–1,608)17 and human DNA DNMT1 TS domain.18
A previous study has linked the chromosomal location of DNMT1 at 19p13.2 with familial late-onset Alzheimer disease (FLOAD).10 Herein we investigated whether DNMT1 mutations may occur in isolated Alzheimer dementia, familial frontotemporal dementia (FTD), or isolated HSAN1 without hearing loss or dementia, and whether DNMT1 mutations may be linked to a wider spectrum of clinical presentations in HSAN1E.
METHODS
Patients.
With institutional review board approval and informed patient consent, we searched the Mayo DNA Peripheral Neuropathy database and identified 48 dominantly inherited sensory neuropathy (HSAN1) patients without dementia or hearing loss and without genetic discovery. These patients had been screened for mutations in SPTLC1 and RAB7 and found negative.11 Additionally, by utilizing the autopsy-based case series at Mayo Clinic Florida, we identified 364 subjects with Alzheimer disease (AD) who had definite, neuropathologic diagnosis according to National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association criteria12 and had Braak scores of ≥4.0. These subjects had mean age at death of 75.7 ± 6.1 (SD) years and 52.2% were female (n = 190). Finally, through collaboration we identified 5 kindreds with familial FTD and 2 kindreds with a triad of sensory predominant neuropathy, hearing loss, and some cognitive concern, i.e., possible HSAN1E kindreds.
DNA analysis.
We sequenced all 41 coding exons of DNMT1 including 30 bps of flanking intron sequence in 48 identified HSAN1 patients without dementia and hearing loss, 5 indexed cases with familial FTD, and 2 possible HSAN1E kindreds. Focused sequencing of DNMT1 exon 20 and 21 was performed for AD cases. Identified variants were compared to various Web-based sequence databases, and we further confirmed the novel variants are absent in a set of 1,200 normal controls. Genomic DNA variants were identified using either Sequencher (Gene Codes) or Mutation Surveyor (Soft Genetics) software.
RESULTS
Only the 2 kindreds affected by HSAN1E (with dementia and hearing loss but without any narcolepsy phenotype) had DNMT1 mutations at Tyr495 located in exon 20 within TS domain. Specifically, a novel mutation c.1483TAT>CAT (p.Tyr495His) was found in one kindred (figure 2A), and the same mutation found in 3 previously described kindreds c.1484TAT>TGT (p.Tyr495Cys) was found in the other kindred (figure 2B). The identified mutations tracked with clinically affected status of the kinships. No mutations were found in the 48 HSAN1 patients who did not have dementia or hearing loss phenotype, nor were they found in 5 FTD probands. Among late-onset AD patients, known polymorphisms (rs75443147, rs2228613, rs721186, rs2114724, and rs74671219) were found with expected minor allele frequencies based on European HapMap data. However, no mutation of exon 20 or exon 21 was found among these 364 AD cases. Thus Tyr495 appears to be a hot spot for mutation specific to HSAN1E phenotype.
Figure 2. The 2 described kinships with DNMT1 amino acid 495 mutations tracking with hereditary sensory and autonomic neuropathy with dementia and hearing loss phenotype including both with prominent behavioral cognitive components consistent with frontotemporal dementia.

(A) Tyr495His mutation pedigree, Norwegian origin (samples II-2, III-1, and III-2 were sequenced); (B) Tyr495Cys mutation pedigree, Scottish origin (samples II-1, II-2, and II-3 were sequenced).
HSAN1 dementia hearing loss complex kindred 1 (Tyr495His).
The family is of Norwegian descent (figure 2A). The index case in this family had the onset of noticeable hearing loss in his early 40s. He had a generalized seizure at age 45 years without discovered cause at that time. Cognitive difficulties were first noted at age 50 years. MRI brain showed diffuse cerebral and cerebellar atrophy. Mini-Mental State Examination was abnormal, 23 of 30 points (normal >25, consistent with mild cognitive impairment). It was noted on bedside testing that memory was better than judgment and executive functions. He experienced odd behaviors (such as mowing the lawn in the middle of the night) and eventually was given the diagnosis of probable FTD. Peripheral neuropathy was diagnosed at age 55 years. He had tremulousness of his hands, gait ataxia, and vibratory sensory loss. Additional seizures occurred at age 57, thought to be of partial complex type. EEG was diffusely slowed especially in the frontal regions without epileptiform activity. Clinical Dementia Rating score was 1.0 at age 57, 2.0 at age 58, and 3.0 at age 59. He died at age 61 years. Neuropathology was consistent with FTD with selective frontal atrophy without distinct histopathology but including microvascular changes in frontal and temporal lobes, also with cerebellar Purkinje cell loss with Bergmann gliosis. No plaques, tangles, or Lewy bodies were appreciated with negative immunostains for tau, α-synuclein, and TDP-43. His 47-year-old daughter had developed moderate sensorineural hearing loss and mild peripheral neuropathy.
The brother of the index case is 61 years of age. In his 40s, he had onset of slowly progressive hearing loss, gait ataxia, sensorimotor neuropathy, and mild to moderate cognitive problems. His balance was much worse in the dark. He is presently unable to walk because of his ataxia and neuropathy. He also has profound hearing loss. MRI shows mild diffuse cerebral atrophy. Their father had the onset of dementia and abnormal gait at age 49 and died at age 65; he also had hearing loss. The father's mother and maternal grandmother lived in Norway and by family history were said to have been “confused and disoriented.”
HSAN1 dementia hearing loss complex kindred 2 (Tyr495Cys).
The kindred originates from Scotland, United Kingdom (figure 2B). The mother had deafness and an unspecified dementia and died at age 53 years without neuropathy evaluated. The propositus presented at age 47 years with sensory neuropathy, foot ulcers, mild deafness, and cognitive problems. The painless injuries to her foot took months to heal. She had a sensory ataxia and sensory neuropathy to all modalities in the legs extending to proximal calves. Her husband was concerned about her forgetfulness, but she denied memory problems and still works (age 50 years). Neuropsychiatric evaluation on Montreal Cognitive Assessment scored 24 (normal >27, consistent with mild cognitive impairment). MRI was reported as normal. She has 2 sisters who also have deafness and cognitive problems; both have the mutation. One is a hoarder and suffered obsessive-compulsive disorder and was unable to live without support related to her behavioral issues but denied memory problems; she also had sensory neuropathy with Charcot joints of her feet. MRI showed global cortical and cerebellar atrophy consistent with the phenotypes previously found in patients with the same DNMT1 mutation Tyr495Cyc.3,13
DISCUSSION
This study identified that amino acid Tyr495 in DNMT1 is a hot spot for mutations among patients with autosomal dominant sensory and autonomic neuropathy, dementia, and hearing loss (HSAN1E). The 2 kindreds in this report originate from Norway and Scotland, and the previously reported 4 HSAN1E kindreds with DNMT1 mutations are also from different geographical regions including Japan, England, Australia, and the United States.2 Two mutations are found in this study: one has been previously been reported (Tyr495Cys), the other (Tyr495His) is novel. To date, all mutations found in HSAN1E kinships are within exon 20 of TS domain. From our previous and this current study, a total of 6 HSAN1E families have been identified. Five of them have mutations at Tyr495 (4 kindreds with Tyr495Cys, 1 kindred with Tyr495His), and one has mutations 4 amino acids away of Tyr495 (Asp490Glu-Pro491Tyr).3
Also emphasized in this report is that DNMT1 mutations are associated with a wider phenotypic spectrum of HSAN1E, including behavioral psychiatric manifestations prior to prominent memory involvements consistent with FTD. The proband of HSAN1E kindred 1 in this study also had seizures, a recognized associative phenomenon of CNS neurodegeneration.14 The later and milder clinical onsets compared to the previous reported cases are also noted among the current cases. Both HSAN1E kindreds of this study had individuals presenting after 50 years of age, while the previously reported families all had symptom onsets no later than in their 30s. In the previously reported Japanese family with Tyr495Cys mutation, 2 family members had frontal brain atrophy or functional frontal hypometabolism with behavioral components including paranoia, and also with prominent memory decline.4 In contrast, the previously reported large American family with Tyr495Cys mutation had 11 clinically examined, affected members with memory decline but no behavioral or frontal executive concern. In this large family, brain autopsy of 3 affected patients, imaging studies, and multiple neuropsychometric data suggested a global process without selective frontal or subcortical involvements.3,13 It is worth noting that although the cognitive features are highlighted here, all HSAN1E patients have hearing loss and sensory predominant neuropathy when examined. Neither the currently reported 2 kinships nor our previously reported 4 HSAN1E kindreds had narcolepsy or cataplexy as described in ADCA-DN from mutations in exon 21 of DNMT1.6
The importance of methylation in maintaining the health of the nervous system has been emphasized by the progressive neurodegenerative diseases such as Rett syndrome caused by MeCP2 mutations15 and immunodeficiency, centromere instability, and facial anomaly syndrome caused by DNMT3b mutations.16 The defective genes in these disorders likely have diverse detrimental effects on epigenetic pathways and cellular processes. Similar to DNMT3b and MeCP2, DNMT1 is highly expressed in the brain. DNMT1 expression patterns and its intricate interactions with a series of regulatory molecules are highly complex at the different phases of cell cycle.8 The phenotypic variability emphasized in this study by the varied cognitive presentations associated with Tyr495 mutations further highlights the complexity of the disease pathogenic mechanisms. Genetic background and environmental influence can also contribute to the clinical variability of HSAN1E phenotypes. Based on our current knowledge, DNMT1 Tyr495 mutations should be considered for patients with dementia, including those with frontotemporal appearance when hearing loss and sensory predominant neuropathy are present.
GLOSSARY
- AD
Alzheimer disease
- ADCA-DN
autosomal dominant cerebellar ataxia deafness and narcolepsy
- DNMT1
DNA methyltransferase 1
- FLOAD
familial late-onset Alzheimer disease
- FTD
frontotemporal dementia
- HSAN1
autosomal dominant hereditary sensory and autonomic neuropathy
- HSAN1E
hereditary sensory and autonomic neuropathy with dementia and hearing loss
- TS
targeting sequence
AUTHOR CONTRIBUTIONS
C.J.K.: study concept and design, acquisition of the data, analysis and interpretation of data, drafting and revising the manuscript, and obtaining funding. T.B., S.J.L., R.H., Y.W., J.K., P.J.D., G.A.N.: concept and design, acquisition of the data, revising manuscript, analysis and interpretation of data. N.E.-T.: acquisition of the data, revising manuscript, analysis and interpretation of data, and obtaining funding.
STUDY FUNDING
Supported by NIH grants (K08 NS065007-01A1, NS36797, R01 AG032990, P50 AG016574, and KL2 RR024151) and a National Institute of Aging grant (9 R01 AG034676-45).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Feng J, Fan G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int Rev Neurobiol 2009;89:67–84 [DOI] [PubMed] [Google Scholar]
- 2.Klein CJ. DNMT1-related dementia, deafness, and sensory neuropathy. In: GeneReviews™. Seattle: University of Washington; 2012 [Google Scholar]
- 3.Klein CJ, Botuyan M-V, Wu Y, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 2011;43:595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hojo K, Imamura T, Takanashi M, et al. Hereditary sensory neuropathy with deafness and dementia: a clinical and neuroimaging study. Eur J Neurol 1999;6:357–361 [DOI] [PubMed] [Google Scholar]
- 5.Hojo K, Kawamata T, Tanaka C, Maeda K. Inflammatory glial activation in the brain of a patient with hereditary sensory neuropathy type 1 with deafness and dementia. Neurosci Lett 2004;367:340–343 [DOI] [PubMed] [Google Scholar]
- 6.Winkelmann J, Lin L, Schormair B, et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum Mol Genet 2012;21:2205–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melberg A, Dahl N, Hetta J, et al. Neuroimaging study in autosomal dominant cerebellar ataxia, deafness, and narcolepsy. Neurology 1999;53:2190–2192 [DOI] [PubMed] [Google Scholar]
- 8.Kornum BR, Kawashima M, Faraco J, et al. Common variants in P2RY11 are associated with narcolepsy. Nat Genet 2011;43:66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin W, Leonhardt H, Pichler G. Regulation of DNA methyltransferase 1 by interactions and modifications. Nucleus 2011;2:392–402 [DOI] [PubMed] [Google Scholar]
- 10.Wijsman EM, Daw EW, Yu CE, et al. Evidence for a novel late-onset Alzheimer disease locus on chromosome 19p13.2. Am J Hum Genet 2004;75:398–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein CJ, Wu Y, Kruckeberg KE, et al. SPTLC1 and RAB7 mutation analysis in dominantly inherited and idiopathic sensory neuropathies. J Neurol Neurosurg Psychiatry 2005;76:1022–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944 [DOI] [PubMed] [Google Scholar]
- 13.Wright A, Dyck PJ. Hereditary sensory neuropathy with sensorineural deafness and early-onset dementia. Neurology 1995;45:560–562 [DOI] [PubMed] [Google Scholar]
- 14.Mendez M, Lim G. Seizures in elderly patients with dementia: epidemiology and management. Drugs Aging 2003;20:791–803 [DOI] [PubMed] [Google Scholar]
- 15.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999;23:185–188 [DOI] [PubMed] [Google Scholar]
- 16.Xu GL, Bestor TH, Bourc'his D, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999;402:187–191 [DOI] [PubMed] [Google Scholar]
- 17.Takeshita K, Suetake I, Yamashita E, et al. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc Natl Acad Sci USA 2011;108:9055–9059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Syeda F, Fagan RL, Wean M, et al. The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J Biol Chem 2011;286:15344–15351 [DOI] [PMC free article] [PubMed] [Google Scholar]