

Abstract
Background
One of the major issues in the fight against infectious diseases is the notable increase in multiple drug resistance in pathogenic species. For that reason, newly acquired high-throughput data on virulent microbial agents attract the attention of many researchers seeking potential new drug targets. Many approaches have been used to evaluate proteins from infectious pathogens, including, but not limited to, similarity analysis, reverse docking, statistical 3D structure analysis, machine learning, topological properties of interaction networks or a combination of the aforementioned methods. From a biological perspective, most essential proteins (knockout lethal for bacteria) or highly conserved proteins (broad spectrum activity) are potential drug targets. Ribosomal proteins comprise such an example. Many of them are well-known drug targets in bacteria. It is intuitive that we should learn from nature how to design good drugs. Firstly, known antibiotics are mainly originating from natural products of microorganisms targeting other microorganisms. Secondly, paleontological data suggests that antibiotics have been used by microorganisms for million years. Thus, we have hypothesized that good drug targets are evolutionary constrained and are subject of evolutionary selection. This means that mutations in such proteins are deleterious and removed by selection, which makes them less susceptible to random development of resistance. Analysis of the speed of evolution seems to be good approach to test this hypothesis.
Results
In this study we show that pN/pS ratio of genes coding for known drug targets is significantly lower than the genome average and also lower than that for essential genes identified by experimental methods. Similar results are observed in the case of dN/dS analysis. Both analyzes suggest that drug targets tend to evolve slowly and that the rate of evolution is a better predictor of drugability than essentiality.
Conclusions
Evolutionary rate can be used to score and find potential drug targets. The results presented here may become a useful addition to a repertoire of drug target prediction methods. As a proof of concept, we analyzed GO enrichment among the slowest evolving genes. These may become the starting point in the search for antibiotics with a novel mechanism.
Background
Endless modifications of existing antibiotics might lead to the appearance of cross-resistance; therefore there is a need for parallel efforts of developing new types of antimicrobials. The preceding step is often finding a new drug target for these drugs. However, given the wealth of information provided by genome sequencing, mass spectrometry and microarray experiments, the selection of a potential drug target for pathogenic species and their relatives is not a trivial task. The repertoire of approaches starts with simple similarity searches, during which sequences of bacterial proteins are compared to known drug targets and human proteins [1]. Other methods are more extensive and implement analyses of metabolic and interaction networks [2] - approaches that, to our knowledge, were first tested with human protein drug targets [3]. There have been attempts to use machine learning methods to mine the substantial amount of data that can be found and derived for bacterial proteins. These approaches may focus on proteins as targets, providing lists of features (sequence length, mostly beta or alpha secondary structure, cytoplasmic/membrane bound, enzyme/non-enzyme, etc.) for a typical drug target [4]. Alternatively, they can focus on protein-protein interactions [5]. Finally, given a small molecule, one can dock it to the set of protein structures described in [6], which aids in the identification of new, potential drug targets for known compounds.
In general, all essential proteins of a given organism constitute potential drug targets [7,8]. The most prominent, essential proteins, such as the ribosomal proteins, are already approved drug targets in bacteria [9]. Their importance for the cell results in their evolutionary conservation [10]. For over 30 years, it has been commonly thought that essential genes are likely subject to stronger negative (purifying) selection (with a less frequent occurrence of mildly deleterious substitutions) [11] compared to nonessential genes. However, the prokaryotic and eukaryotic kingdoms seem to differ in this respect. No statistical difference in the relative rate of evolution between essential and nonessential genes was evident for the mouse, if immune genes were excluded [12]. Analysis of the yeast genome sheds light on eukaryotic species, by explaining why it was difficult to see a statistically significant difference in that case [13] (they observed a significantly higher rate of evolution of nonessential proteins when they compared essential proteins with the “most dispensable” half of nonessential proteins). In bacteria, stronger negative selection on essential genes was shown by Jordan [14] in the case of Escherichia coli K12. By applying an orthology-based essentiality transfer from Escherichia coli, the authors also predicted that this should be true for pathogenic species in the Neisseria and Helicobacter genera. This finding has been used for prediction of essential genes on its own or in conjunction with other methods.
No doubt we can learn a lot about choosing good drug targets from nature. Antibiotics are mainly originating from natural fungal and bacterial products [15]. Microorganisms have been using them for millions of years to combat (successfully) competing organisms. This impressive finding has been confirmed recently using paleontological data [16]. From the evolutionary point of view it may suggest that good drug targets are evolutionary constrained and are subject to purifying selection, which makes them less susceptible to random development of resistance. The efforts to validate this hypothesis and to find its application in drug design workflows comprise the aim of this study.
Analysis of evolutionary rates to identify putative drug targets has been already suggested by Searls [17], but no comprehensive study has been published so far. Moreover, two methods were proposed to identify evolutionary constrained residues in drug targets. Durand and co-workers [18] assessed purifying selection on individual sites in Plasmodium falciparum drug targets using the dN/dS ratio. The method is called “evolutionary patterning” (EP). A second method called “evolutionary tracing” (ET) [19], was proposed by Lichtarge in 1996. The Lichtarge method is based solely on evolutionary conservation. In both methods the key assumption is that a good drug should bind to the slowly evolving protein pocket. This expectation is based on intuition that at such sites development of drug resistance will be less probable. Both methods do not provide the overall picture of the evolutionary rates of genes of pathogenic species as they focus on individual sites, not the whole genes.
Generally speaking the key question we wanted to answer in this study was whether proteins which are targeted by antibiotics tend to evolve slowly. For this purpose we analyzed relative rate of evolution of genes from seven bacterial pathogens and from E. coli. We used polymorphism analysis, i.e. pN/pS ratio (which represents an appropriate measure of purifying selection in the case of comparison of inter-species diversity) and reproduced the analysis with dN/dS ratio (which is better for comparing sequences derived from different species) [20]. In both cases the rate of evolution of known drug targets, was not only significantly lower than the genome average but was also significantly lower than that for the essential genes, suggesting a higher selective force acting on a wide spectrum of drug targets. This finding suggests that calculation of evolutionary rate can aid in scoring during the process of drug target selection and can provide additional insights into whether a particular protein might or might not be an attractive drug target. As such, it complements EP/ET approaches.
Methods
Data preparation
The input set for our analysis consisted of bacterial genomes for which experimental data for the identification of essential genes existed (Table 1). Data on essential genes were obtained from the DEG database [21]. We obtained alignments of clusters of coding sequences (CDS) from whole-genome alignments of the reference genome (strain with experimental list of essential genes) and genomes of other strains from the ATGC (Alignable Tight Genomic Clusters) database [22].
Table 1.
Summary of the data used at pN/pS analysis
| Reference genome (strain with experimental data on essential genes; NCBI Taxonomy ID in brackets) | Other strains with complete pairwise alignments with reference genome (NCBI Taxonomy IDs) | The number of all genes* | The number of essential genes* | The number of drug targets* |
|---|---|---|---|---|
|
Escherichia coli K12 (83333) |
155864, 199310, 316407, 331111, 331112, 362663, 364106, 386585, 405955 |
4294 |
771 |
41 |
| (3104) |
(616) |
(31) |
||
|
Francisella novicida U112 (401614) |
119857, 177416, 393011, 393115, 418136, 458234 |
1719 |
391 |
34 |
| (1065) |
(320) |
(32) |
||
|
Haemophilus influenzae Rd KW20 (71421) |
262727, 262728, 281310, 374927, 374928, 374930, 374931, 374932, 374933, 375063, 375177, 375432 |
1581 |
477 |
39 |
| (1024) |
(399) |
(34) |
||
|
Helicobacter pylori 26695 (85962) |
357544, 85963 |
1576 |
336 |
30 |
| (992) |
(292) |
(28) |
||
|
Pseudomonas aeruginosa UCBPP-PA14 (208963) |
208963, 381754 |
5892 |
335 |
40 |
| (4530) |
(305) |
(36) |
||
|
Salmonella typhimurium LT2 (99287) |
209261, 220341, 295319, 321314 |
4425 |
481 |
41 |
| (3140) |
(403) |
(31) |
||
|
Staphylococcus aureus N315 (93061) |
158878, 158879, 196620, 273036, 282458, 282459, 359786, 359787, 367830, 418127, 426430, 93062 |
2892 |
351 |
35 |
| (1918) |
(277) |
(28) |
||
|
Streptococcus pneumoniae TIGR4 (170187) |
171101, 373153, 406556, 406557, 406558, 406559, 406560, 406561, 406562, 406563 |
1965 |
195 |
37 |
| (1532) | (175) | (33) |
*All genes, essential genes and drug targets comprised the three groups of genes compared in our study. The final analysis was restricted to genes for which no recombination events were detected, numbers of which are presented in brackets.
For further comparison, we chose only reference genomes with at least two alignments with strains (subspecies) with complete genomes available in the ATGC database. The majority of cluster alignments we have obtained consisted of two sequences (one-to-one orthology assignments). However, in a small number of cases (~5%) we had more than one orthologous sequence representing a particular cluster in the compared genomes. Such duplications were resolved using reciprocal BLAST [23] on the corresponding protein sequences.
All genes were divided into three sets (see Table 1). The first group contained all genes from a particular organism, and the second group contained all of its essential genes (from the DEG database). The third group, referred to later as “potential wide-spectrum drug targets”, consisted of genes belonging to one of the orthology groups (as defined by KEGG KO [24]) covering bacterial drug targets with a known broad-spectrum activity acting as antagonist, inhibitor or in an adduct. In the case of duplications, (more than one gene in a single KO), the bidirectional best hit was selected using the KEGG SSDB database. The most comprehensive database containing FDA-approved existing drug targets is DrugBank [25]. We used provided data and then manually verified them. The verification included removal of beta-lactamases, which are drug targets and drug resistance enzymes at the same time which leads to a completely different evolutionary pattern than a typical drug target. However, we have included the enoyl-acyl carrier protein reductase (fabI gene), as this protein is a known drug target of the antibacterial agent Triclosan [26]. Data for Triclosan are not in DrugBank yet, however, this compound was used in an antibiotic profiling study in Escherichia coli[27], and it seemed reasonable to use it for further analysis. The final list of drug targets for each species is shown in additional table file (see Additional file 1).
All three groups were mutually exclusive. We removed the genes corresponding to known drug targets from the group of essential genes. Similarly, in the group of all genes, those known as lethal genes or known as drug targets were excluded. The rationale for such approach was to avoid biasing the p-value tests.
Estimation of evolutionary rate
For each MSA of orthologous sequences, we evaluated polymorphism (the pN/pS ratio) using polyDnDs software [28]. We chose simple statistics based on a number of nonsynonymous and synonymous mutations (not taking into account number of possible places where mutations can occur).
Assessment of pN/pS differences
For each species, we assessed the statistical difference of relative speed of evolution between the three aforementioned groups of genes (all, essential and potential wide-spectrum drug targets). We used the Mann–Whitney U test [29]. The p-values for a difference between sets were calculated using R implementation of the test (wilcox.test function; two sided).In our statistical approach we tested 24 hypotheses (three sets compared in one combination for eight species). We corrected our p-values using FDR approach (Benjamini-Yokutieli correction for multiple testing approach) [30].
Gene ontology analysis
We used ontologies from Gene Ontology [31] (file gene_ontology_edit.obo; 10.07.2011), while annotations were obtained from EBI (Uniprot-GOA [32]). More than 60% of genes for all the species had at least one GO term assigned. For each species, 10% of the slowest evolving genes were selected as study set, while all genes in the species comprised the population set. Analysis was performed using command line version of Ontologizer [33].
Plasmodium falciparum pN/pS analysis
We estimated evolutionary rate of all Plasmodium falciparum genes and ranked them on this parameter. It enabled the assessment of evolutionary rate of the two genes used in the evolutionary patterning (EP) study, i.e. dihydrofolate synthase (DHFR-TS) and glycerol kinase (GK).
The pN/pS ratio was estimated using the approach proposed by Krzyczmonik et al. [34]. Thus we calculated pN/pS using nonsynonymous and synonymous SNPs from PlasmoDB [35]. We used SNP observable for the Plasmodium falciparum 3d7 strain and other strains of this species. As it was shown by Krzyczmonik et al. [34] in many cases P. falciparum genes have only nonsynonymous genes and under such conditions it is impossible to calculate pN/pS ratio (as pS equals zero). We applied the correction suggested by those authors, i.e. in such cases pS were approximated by 1.
Additional tests
In addition to the above analyses, we have conducted the tests using omega (dN/dS) instead of pN/pS. While such an approach is obviously biased, we were interested if the overall results would be different. Detailed methods, incorporating correction on recombinant genes, are provided in the supplementary materials.
Results
The evolutionary rate of potential wide-spectrum drug targets
We have analyzed genomes of seven pathogenic species. The results are summarized in Figure 1 and Figure 2 (see Additional file 2 for more statistical details). Potential drug targets had significantly lowered values of pN/pS compared to all genes from a given genome, as assessed by the average pN/pS ratio per orthologous group (although in the case of S. aureus and H. influenzae the differences were not statistically significant). Also, we observed lower pN/pS values for potential drug targets in comparison to essential genes (for all except F. novicida and S. aureus). In most genomes (all except H. influenzae and H. pylori), essential genes showed higher negative selection than the genome average, confirming the results of Jordan [14].
Figure 1.
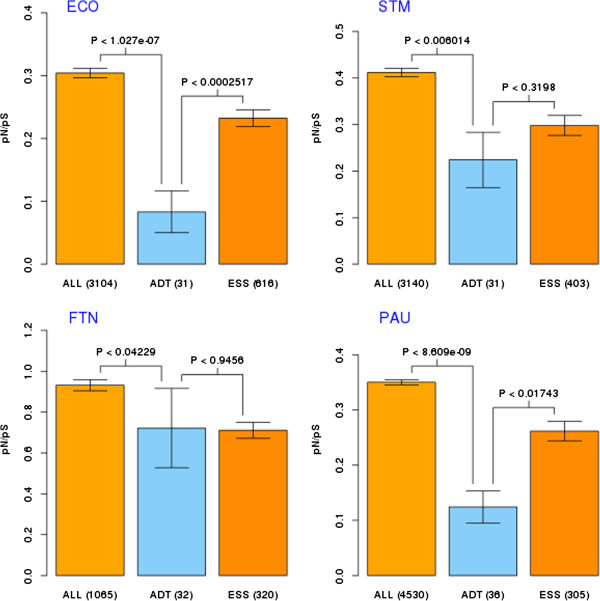
Evolutionary rate differences of four Enterobacteriaceae species. Evaluation of evolutionary rate differences between three sets of genes of interest: ALL -all genes, ESS - essential genes and ADT - approved drug targets). Evolutionary rate was estimated using (pN/pS ratio). In this case pN/pS values were compared using Mann–Whitney U test (wilcox.test in R language, two sided hypothesis tested). Box plots of means of pN/pS with 95% confidence intervals are presented (number of genes in given set are shown in brackets). Result for four species from Enterobacteriaceae. Abbreviations: ECO: Escherichia coli, STM – Salmonella typhimurium, PAU – Pseudomonas aeruginosa, FTN – Francisella novicida.
Figure 2.
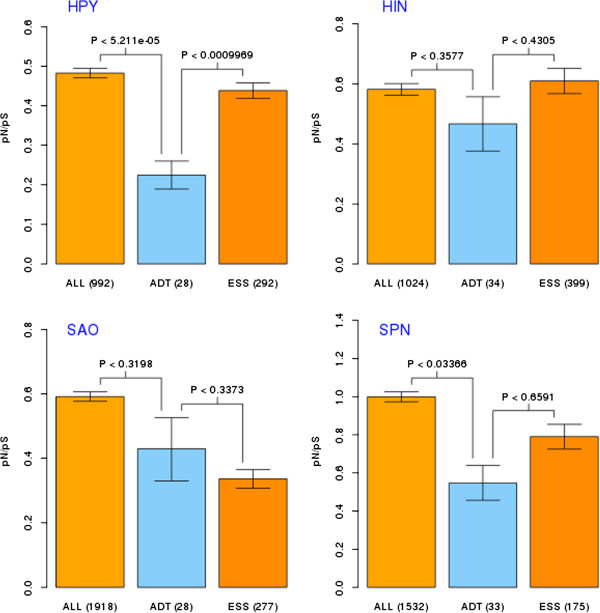
Evolutionary rate differences of four non-Enterobacteriaceae species. Evaluation of evolutionary rate differences between three sets of genes of interest: ALL -all genes, ESS - essential genes and ADT - approved drug targets). Evolutionary rate was estimated using (pN/pS ratio). In this case pN/pS values were compared using Mann–Whitney U test (wilcox.test in R language, two sided hypothesis tested). Box plots of means of pN/pS with 95% confidence intervals are presented (number of genes in given set are shown in brackets). Result for four species not from Enterobacteriaceae taxon. Abbreviations: HPY: Helicobacter pylori, HIN – Haemophilus influenzae, SAO – Staphylococcus aureus, SPN – Streptococcus pneumoniae.
We also performed the same analysis on the genome of the non-pathogenic species, Escherichia coli strain K12. In this case, essential genes had lower pN/pS values than the genome average, and potential drug targets had lower pN/pS values than essential genes and the genome average. All those differences were highly significant (p-value < 0.01).
It should be noted, that the results of the analysis above are similar when using omega (dN/dS) instead of pN/pS. Details can be found in Supplementary Materials (see Additional file 3 and Additional file 4).
Characterization of the orthology groups of drug targets
The assignment of known drug targets to KEGG KO orthology groups (see Methods) resulted in 44 representative groups (see Table 2). The three largest groups were various ribosomal proteins (~30%), proteins related to DNA processing and penicillin binding proteins. We ranked all of the orthology groups according to the average dN/dS of genes belonging to a given group across the eight genomes (seven pathogenic species and Escherichia coli K12) (see Additional file 1 for more details). Ribosomal and DNA processing enzymes ranked at the top and were the most attractive drug targets from an evolutionary perspective. The same could be said about penicillin binding protein (PBP) - 2 and PBP3 but not PBP4, PBP5/6 and PBP7. These results agree well with experimental data on these proteins [36]. PBP2 and PBP3 are bound by beta-lactam antibiotics with high affinity and are known to be the main/lethal target of these drugs. Affinity is lower in the cases of PBP4, PBP5/6 and PBP7. PBP4 to 7 contribute to penicillin resistance but are considered auxiliary drug targets.
Table 2.
Ranking of known drug targets (from the evolutionary perspective)
| Drugs | KEGG KO | Mean pN/pS | SD | Gene name; description |
|---|---|---|---|---|
| Cycloserine |
ko:K01921 |
0.5813 |
0.136342 |
ddl; D-alanyl-alanine synthetase A |
| |
ko:K01775 |
0.4017 |
0.260427 |
alr; alanine racemase |
| Triclosan |
ko:K00208 |
0.2425 |
0.084719 |
fabI; enoyl-(acyl carrier protein) reductase |
| Trimethoprim |
ko:K00287 |
0.6356 |
0.265319 |
folA; dihydrofolate reductase |
| Sulphonamides |
ko:K00796 |
0.5708 |
0.213370 |
folP; dihydropteroate synthase |
| Fusidic acid |
ko:K02355 |
0.1284 |
0.104805 |
fusA; elongation factor G |
| Mupirocin |
ko:K01870 |
0.3031 |
0.139443 |
ileS; isoleucyl-tRNA synthetase |
| Quinolones |
ko:K02469 |
0.3063 |
0.167897 |
gyrA; DNA gyrase subunit A |
| |
ko:K02470 |
0.2195 |
0.192711 |
gyrB; DNA gyrase subunit B |
| |
ko:K02621 |
0.3364 |
0.158633 |
parC; DNA topoisomerase IV subunit A |
| |
ko:K02622 |
0.3016 |
0.231233 |
parE; DNA topoisomerase IV subunit B |
| Rifampin |
ko:K03046 |
0.1263 |
0.047384 |
rpoC; DNA-directed RNA polymerase subunit beta' |
| |
ko:K03043 |
0.1423 |
0.092346 |
rpoB; DNA-directed RNA polymerase subunit beta |
| |
ko:K03040 |
0.4072 |
0.388638 |
rpoA; DNA-directed RNA polymerase subunit alpha |
| Macrolides |
ko:K02926 |
0.2090 |
0.242629 |
rplD; 50S ribosomal protein L4 |
| |
ko:K02890 |
0.2212 |
0.408734 |
rplV; 50S ribosomal protein L22 |
| |
ko:K02864 |
0.4581 |
0.364803 |
rplJ; 50S ribosomal protein L10 |
| |
ko:K02911 |
0.5297 |
0.438268 |
rpmF; 50S ribosomal protein L32 |
| Tetracyclines |
ko:K02986 |
0.4107 |
0.343409 |
rpsD; 30S ribosomal protein S4 |
| |
ko:K02982 |
0.1927 |
0.187825 |
rpsC; 30S ribosomal protein S3 |
| |
ko:K02965 |
0.1139 |
0.171661 |
rpsS; 30S ribosomal protein S19 |
| |
ko:K02992 |
0.3173 |
0.327775 |
rpsG; 30S ribosomal protein S7 |
| |
ko:K02954 |
0.4495 |
0.467237 |
rpsN; 30S ribosomal protein S14 |
| |
ko:K02994 |
0.1578 |
0.203180 |
rpsH; 30S ribosomal protein S8 |
| |
ko:K02996 |
0.1692 |
0.216565 |
rpsI; 30S ribosomal protein S9 |
| Glacycyline |
ko:K02952 |
0.6584 |
0.422977 |
rpsM; 30S ribosomal protein S13 |
| Retapamulin |
ko:K02906 |
0.2112 |
0.307444 |
rplC; 50S ribosomal protein L3 |
| Aminoglycosides |
ko:K02946 |
0.1711 |
0.293128 |
rpsJ, nusE; 30S ribosomal protein S10 |
| |
ko:K02878 |
0.1300 |
0.151462 |
rplP; 50S ribosomal protein L16 |
| |
ko:K02950 |
0.4372 |
0.425007 |
rpsL; 30S ribosomal protein S12 |
| beta-lactam antibiotics |
ko:K07258 |
0.4060 |
0.280471 |
dacA; D-alanyl-D-alanine carboxypeptidase fraction A |
| |
ko:K05515 |
0.2293 |
0.118958 |
mrdA; penicillin-binding protein 2 |
| |
ko:K03693 |
0.4693 |
0.000000 |
penicillin-binding protein 1B |
| |
ko:K03587 |
0.2658 |
0.077185 |
ftsI; division specific transpeptidase (PBP3) |
| |
ko:K00687 |
0.3279 |
0.000000 |
penicillin-binding protein 2B |
| |
ko:K12553 |
0.4829 |
0.000000 |
penicillin-binding protein 3 |
| |
ko:K05365 |
0.3425 |
0.334610 |
mrcB, ponB; penicillin-binding protein 1B |
| |
ko:K05366 |
0.3644 |
0.086939 |
mrcA; penicillin-binding protein 1A |
| |
ko:K07262 |
0.3863 |
0.118514 |
pbpG; D-alanyl-D-alanine endopeptidase |
| |
ko:K12556 |
0.3431 |
0.000000 |
penicillin-binding protein 2X |
| |
ko:K12555 |
0.5859 |
0.000000 |
penicillin-binding protein 2A |
| |
ko:K05367 |
0.7132 |
0.001414 |
pbpC; penicillin-binding protein 1C |
| |
ko:K07259 |
0.3973 |
0.161482 |
dacB; D-alanyl-D-alanine carboxypeptidase/endopeptidase |
| Fosfomycin | ko:K00790 | 0.2551 | 0.183465 | murA; UDP-N-acetylglucosamine 1-carboxyvinyltransferase |
Drug targets are ordered by drug class. Mean pN/pS as well as standard deviation (SD) were calculated using data from eight species analyzed in the study.
Among protein groups with relatively high rate of evolution are alanine racemase and d-alanine ligase, which are targeted by sulphonamides [37]. Both of these proteins seem to be only moderately attractive drug targets from an evolutionary perspective. The D-alanine ligase gene has paralogs in Escherichia coli and Salmonella typhimurium, and such genes generally do not constitute good drug targets [38]. The same can be said about folate reductase and dihydropteroate synthase, two genes from the folate pathway targeted by two distinct classes of drugs. In the case of these genes, the fast appearance of resistance is commonly known [39]. Thus, drugs for these targets are often applied in combination. It is worth to note that all these proteins (alanine racemase, alanine ligase, folate reductase, dihydropteroate synthase) are drug targets of human designed antibiotics (i.e. synthetic as opposite to semisynthetic antibiotics being derivatives of bacterial natural products). Thus we see clearly how difficult it is to find a good novel drug target without referring to evolutionary history of pathogenic species. This is probably one of the key reasons why there has been no new class of antibiotic introduced into the market for the past twenty years [40].
Functional classes among slowly evolving genes
Additionally, we analyzed slowly evolving genes by means of GO enrichment. Results partially overlap with common functional classes characterizing known drug targets (see Table 3). Statistically significant terms were “rRNA binding” and “structural molecule activity” corresponding to ribosomal proteins or “nucleic acid binding” corresponding to topoisomerases, RNA polymerases and gyrases. However we also identified some novel classes, usually not associated with known drug targets, such as succinate dehydrogenase or metal binding proteins. These may become interesting starting points in finding new drug targets with a unique mechanism of action.
Table 3.
GO analysis for slowly evolving genes
| GO | P-value | Study-fraction | Population-fraction | Description |
|---|---|---|---|---|
| GO:0005198 |
2.7e–37 |
0.00523 |
0.0359 |
structural molecule activity |
| GO:0005515 |
1.8e–12 |
0.0257 |
0.0685 |
protein binding |
| GO:0005488 |
2.9e–12 |
0.274 |
0.373 |
binding |
| GO:0019843 |
1.4e–06 |
0.0028 |
0.0194 |
rRNA binding |
| GO:0015453 |
0.00043 |
0.000997 |
0.00627 |
oxidoreduction-driven active transmembrane transporter activity |
| GO:0000104 |
0.0076 |
0.000436 |
0.00285 |
succinate dehydrogenase activity |
| GO:0016667 |
0.011 |
0.00567 |
0.012 |
oxidoreductase activity, acting on a sulfur group of donors |
| GO:0003735 |
0.014 |
0.00392 |
0.0308 |
structural constituent of ribosome |
| GO:0015078 |
0.027 |
0.00361 |
0.00856 |
hydrogen ion transmembrane transporter activity |
| GO:0046872 | 0.045 | 0.0756 | 0.105 | metal ion binding |
Enrichment of GO terms (molecular function domain): data for slowly evolving genes. Ten percent of genes (of eight studied species) with the lowest pN/pS rank comprised the study set. Genes with reliable pN/pS (of eight studied species) comprised the population set. Analysis was performed using Ontologizer (with Benjamini-Hochberg correction for multiple testing and Parent–child-Union Settings).
Complementation of other evolutionary approaches for drug target discovery
In our opinion the approach presented here could be a good complementation to other drug target discovery methods based on evolutionary data: evolutionary tracing (ET) and evolutionary patterning (EP). As a proof of concept we analyzed the data of both approaches and put them into the context of our approach.
In the case of EP, Durand et al. analyzed the position specific evolutionary rate for two Plasmodium falciparum genes: known drug target, dihydrofolate reductase (DHFR-TS) and drug target candidate, glycerol kinase (GK). We estimated pN/pS genome wide, for almost all genes of Plasmodium falciparum (3d7 strain) and ranked genes using this parameter. Then we were able to evaluate those genes in the context of observed genome-wide distribution of pN/pS. We observed high purifying selection (slow evolutionary rate) for glycerol kinase (ranked in 32-nd percentile), which allows this gene to be considered as an attractive drug target from our perspective. We also observed rather weak purifying selection in the case of dihydrofolate reductase (ranked in 76-th percentile). It agrees well with the fact that antifolate resistance in the malaria parasite is well recognized [41]. Moreover, orthologous dihydrofolate reductases being known antibacterial drug targets are also under relatively weak selection pressure (as we pointed out earlier; see also Table 2).
In the case of evolutionary tracing (ET) Adikesavan et al. [42] presented in 2011 a first application of their approach to prokaryotes. They identified evolutionarily important surface amino acids involved in Escherichia coli RecA functions. RecA is already known as a drug target or co-drug target (in species for which gene knockout results in higher effectiveness of antibiotics). We compared evolutionary rate of the recA gene in the eight bacterial species analyzed in our study. In all cases (except for the Streptococcus pneumoniae) the recA gene was under strong evolutionary pressure (ranked in the lower quartile for these species and in the upper quartile in the case of S. pneumoniae) which makes it a good drug target from the perspective of our approach.
Discussion
Antibiotics are mainly natural products used by micro-organisms against other micro-organisms. They seem to be relatively evolution proof, i.e. resistance is sufficiently rare and it is still beneficial for microorganism to use antibiotics against competing microorganisms.
We have shown that in most of the analyzed pathogenic genomes potential drug targets have statistically significant higher negative selection than essential genes or the genome average. Our explanation for this phenomenon is that such proteins are evolutionary constrained, i.e. they are overall highly sensitive to perturbations, which could correspond to relatively infrequent point mutations (including those leading to resistance).
One may ask the question whether our observation is not the result of the fact that bacteria have already been subject to considerable “drug” pressure - either by medical usage of compounds [43] or by more ancient and long term exposures to the natural products on which the antibiotics are based [44]. The observed purifying selection may, in this case, be the result of selective sweeps brought about by the drugs. If it would be the case more sensitive variants would be removed due to selection caused by the drug. The dN/dS analysis suggests that this possibility should be excluded. This test compares relative rate of evolution observed in the comparison of two closely related species. In contrast to the pN/pS test which takes in account existing allelic diversity, dN/dS takes into account only fixed mutations (with frequency = 1 in the population). Therefore, it is much more robust to the observed selective sweeps caused by antibiotics.
In some cases in our study we observed exceptions to the described general pattern. For example for Staphylococcus aureus and Haemophilus influenzae we did not observe a statistical difference in average pN/pS values between essential genes and potential drug targets (although drug targets evolve much slower than other genes). It is likely that for many drug targets, directed positive selection has led to intrinsic resistance; many Staphylococcus aureus strains are known to be resistant to vancomycin (VRSA) as well as methicillin (MRSA). Similarly, many Haemophilus influenzae strains have intrinsic resistance to beta-lactam drugs. It makes the observable differences in evolutionary rate between drug targets and other analyzed groups (essential genes and all genes) being lower than they in fact are.
Drug targets also have a higher negative selection when assessed by dN/dS ratio (omega). We consider these results supplementary as omega analysis has certain drawbacks (e.g. sensitiveness to recombination, limited range of dS for which dN/dS ratio is considered to be reliably estimated or non-linear dependency on time). Nevertheless, dN/dS analysis confirms the results obtained by the pN/pS approach.
When thinking about developing drugs against a certain target, one must also consider issues such as resistance mechanism (efflux pumps, other resistance proteins), drug target accessibility or host-related factors. Considering whether a gene is essential (even in a broader context than is commonly considered [45]) or conserved is often not sufficient. The assessment of evolutionary rate (e.g. by pN/pS values) helps substantially in the evaluation of potential drug targets. The resulting targets have an evolutionary history suggesting that they are less likely to randomly develop resistance via point mutations. And while it seems that the only cases one could find are the “obvious” ones, we show that this approach identified metal ion binding genes and succinate dehydrogenases - neither of which corresponds to well-studied wide-spectrum drug targets. Our results show that pN/pS analyses are an attractive addition to drug target prediction pipelines.
On the other hand one should be aware of the biases in our method. First, we used only whole genomes to limit the cases where low quality alignments will substantially affect pN/pS estimation. Because of that the eight chosen species are among the most common infective bacteria (they were among the first sequenced species). This, of course, is advantageous to address concerns like MRSA and VRSA and other antibiotic resistant pathogens, but then there is no evidence that the conclusions of this study apply to the less common human pathogens, to veterinary pathogens, or to other bacteria which could be antibiotic targeted. Second, we needed to limit the analysis to only eight species as the approach undertaken requires data on essential genes. This raises a concern whether Gram-negative species dominating in this study led to biased results. And finally, predicting evolutionary rate with pN/pS is limited to alignments of closely related sub-species. The last issue can be avoided by performing dN/dS analysis, but as we mentioned above dN/dS methodology has its own drawbacks.
Conclusions
In this study we showed that good drug targets evolve slowly and that the rate of evolution is a better predictor of drugability than essentiality. This to some extent explains why known antibiotics (usually being of microbial origin) have been efficiently targeting other microorganisms for millions of years of evolution [16].
Our study also shows that evolutionary rate can be used to score and find potential drug targets. Generally our approach can be considered a useful complementation to EP (Evolutionary Patterning) and ET (Evolutionary Tracing) approaches. Both those methods can be useful in designing a drug that targets a specific site and has a known mechanism of operation. Our approach can be considered an attractive solution in the preceding step, i.e. finding the targets which could be analyzed in detail by ET or EP.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AG performed the analysis. AG, SK, PS and PZ participated in the design of the study. AG and PS drafted the manuscript. SK and PZ revised the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Essentiality and ranking of known drug targets. The essentiality and ranking (from an evolutionary perspective) of known drug targets. Drug targets are ordered by drug class. Data for eight species of interest are presented in separate columns. Red: essential genes, blue: nonessential genes, grey: no data addressing essentiality, white: no ortholog with reliable omega.
Statistical data - pN/pS analysis. Mean pN/pS ratio for three sets of genes (ADT - drug targets, ESS - essential genes, ALL - other genes) presented. Data for eight species analyzed in the study. P-values where corrected for multiple testing with FDR (Benjamini-Yekutieli algorithm). Abbreviations: ciu - confidence interval upper limit; cil - confidence interval lower limit, eco – Escherichia coli, ftn – Francisella novicida, hin – Haemophilus influenzae, hpy – Helicobacter pylori, pau – Pseudomonas aeruginosa, sao – Staphylococcus aureus, spn – Streptococcus pneumoniae, stm – Salmonella typhimurium.
Statistical data - dN/dS analysis. Mean dN/dS ratio for three sets of genes (ADT - drug targets, ESS - essential genes, ALL - other genes) presented. Data for eight species analyzed in the study. P-values where corrected for multiple testing with FDR (Benjamini-Yekutieli algorithm). Abbreviations: ciu - confidence interval upper limit; cil - confidence interval lower limit, eco – Escherichia coli, ftn – Francisella novicida, hin – Haemophilus influenzae, hpy – Helicobacter pylori, pau – Pseudomonas aeruginosa, sao – Staphylococcus aureus, spn – Streptococcus pneumoniae, stm – Salmonella typhimurium.
Dn/Ds analysis - materials, methods and results. Analysis of evolutionary rate (dN/dS ratio) of known drug targets. Materials and methods as well as Results are presented in the file. Details about recombination (detection and removal procedures) can be also found.
Contributor Information
Arkadiusz Gladki, Email: arek@ibb.waw.pl.
Szymon Kaczanowski, Email: szymon@ibb.waw.pl.
Pawel Szczesny, Email: szczesny@ibb.waw.pl.
Piotr Zielenkiewicz, Email: piotr@ibb.waw.pl.
Acknowledgements
We acknowledge grant 772/N-COST/2010 from the Polish Ministry of Science.
References
- Toomey D, Hoppe HC, Brennan MP, Nolan KB, Chubb AJ. Genomes2Drugs: identifies target proteins and lead drugs from proteome data. PLoS One. 2009;4(7):e6195. doi: 10.1371/journal.pone.0006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha SK, Shakya M. Protein interaction network analysis--approach for potential drug target identification in Mycobacterium tuberculosis. J Theor Biol. 2010;262(2):284–294. doi: 10.1016/j.jtbi.2009.09.029. [DOI] [PubMed] [Google Scholar]
- Zhu M, Gao L, Li X, Liu Z. Identifying drug-target proteins based on network features. Sci China C Life Sci. 2009;52(4):398–404. doi: 10.1007/s11427-009-0055-y. [DOI] [PubMed] [Google Scholar]
- Bakheet TM, Doig AJ. Properties and identification of human protein drug targets. Bioinformatics. 2009;25(4):451–457. doi: 10.1093/bioinformatics/btp002. [DOI] [PubMed] [Google Scholar]
- Sugaya N, Ikeda K. Assessing the druggability of protein-protein interactions by a supervised machine-learning method. BMC Bioinformatics. 2009;10:263. doi: 10.1186/1471-2105-10-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Gao Z, Kang L, Zhang H, Yang K, Yu K, Luo X, Zhu W, Chen K, Shen J. et al. TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006;34(Web Server issue):W219–W224. doi: 10.1093/nar/gkl114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker AF, Lunsford RD. Rational identification of new antibacterial drug targets that are essential for viability using a genomics-based approach. Pharmacol Ther. 2002;95(1):1–20. doi: 10.1016/S0163-7258(02)00222-X. [DOI] [PubMed] [Google Scholar]
- Cole ST. Comparative mycobacterial genomics as a tool for drug target and antigen discovery. Eur Respir J Suppl. 2002;36:78s–86s. doi: 10.1183/09031936.02.00400202. [DOI] [PubMed] [Google Scholar]
- Poehlsgaard J, Douthwaite S. The bacterial ribosome as a target for antibiotics. Nat Rev Microbiol. 2005;3(11):870–881. doi: 10.1038/nrmicro1265. [DOI] [PubMed] [Google Scholar]
- Ridley M. Evolution. 2. Cambridge, Mass. u.a: Blackwell Science; 1996. [Google Scholar]
- Wilson AC, Carlson SS, White TJ. Biochemical evolution. Annu Rev Biochem. 1977;46:573–639. doi: 10.1146/annurev.bi.46.070177.003041. [DOI] [PubMed] [Google Scholar]
- Hurst LD, Smith NG. Do essential genes evolve slowly? Curr Biol. 1999;9(14):747–750. doi: 10.1016/S0960-9822(99)80334-0. [DOI] [PubMed] [Google Scholar]
- Hirsh AE, Fraser HB. Protein dispensability and rate of evolution. Nature. 2001;411(6841):1046–1049. doi: 10.1038/35082561. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Rogozin IB, Wolf YI, Koonin EV. Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res. 2002;12(6):962–968. doi: 10.1101/gr.87702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livermore DM. Discovery research: the scientific challenge of finding new antibiotics. J Antimicrob Chemother. 2011;66(9):1941–1944. doi: 10.1093/jac/dkr262. [DOI] [PubMed] [Google Scholar]
- D’Costa VM, King CE, Kalan L, Morar M, Sung WW, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R. et al. Antibiotic resistance is ancient. Nature. 2011;477(7365):457–461. doi: 10.1038/nature10388. [DOI] [PubMed] [Google Scholar]
- Searls DB. Pharmacophylogenomics: genes, evolution and drug targets. Nat Rev Drug Discov. 2003;2(8):613–623. doi: 10.1038/nrd1152. [DOI] [PubMed] [Google Scholar]
- Durand PM, Naidoo K, Coetzer TL. Evolutionary patterning: a novel approach to the identification of potential drug target sites in Plasmodium falciparum. PLoS One. 2008;3(11):e3685. doi: 10.1371/journal.pone.0003685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257(2):342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15(12):496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Lin Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 2009;37(Database issue):D455–D458. doi: 10.1093/nar/gkn858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novichkov PS, Ratnere I, Wolf YI, Koonin EV, Dubchak I. ATGC: a database of orthologous genes from closely related prokaryotic genomes and a research platform for microevolution of prokaryotes. Nucleic Acids Res. 2009;37(Database issue):D448–D454. doi: 10.1093/nar/gkn684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38(Database issue):D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V. et al. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011;39(Database issue):D1035–D1041. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater-Radosti C, Van Aller G, Greenwood R, Nicholas R, Keller PM, DeWolf WE Jr, Fan F, Payne DJ, Jaworski DD. Biochemical and genetic characterization of the action of triclosan on Staphylococcus aureus. J Antimicrob Chemother. 2001;48(1):1–6. doi: 10.1093/jac/48.1.1. [DOI] [PubMed] [Google Scholar]
- Liu A, Tran L, Becket E, Lee K, Chinn L, Park E, Tran K, Miller JH. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrob Agents Chemother. 2010;54(4):1393–1403. doi: 10.1128/AAC.00906-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton K. Libsequence: a C++ class library for evolutionary genetic analysis. Bioinformatics. 2003;19(17):2325–2327. doi: 10.1093/bioinformatics/btg316. [DOI] [PubMed] [Google Scholar]
- Wittkowski KM, Song T. Nonparametric methods for molecular biology. Methods Mol Biol. 2010;620:105–153. doi: 10.1007/978-1-60761-580-4_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 2001;29(4):1165–1188. doi: 10.1214/aos/1013699998. [DOI] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrell D, Dimmer E, Huntley RP, Binns D, O’Donovan C, Apweiler R. The GOA database in 2009--an integrated Gene Ontology Annotation resource. Nucleic Acids Res. 2009;37(Database issue):D396–D403. doi: 10.1093/nar/gkn803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Grossmann S, Vingron M, Robinson PN. Ontologizer 2.0--a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics. 2008;24(14):1650–1651. doi: 10.1093/bioinformatics/btn250. [DOI] [PubMed] [Google Scholar]
- Krzyczmonik K, Switnicki M, Kaczanowski S. Analysis of immunogenicity of different protein groups from malaria parasite Plasmodium falciparum. Infect Genet Evol. 2012;12(8):1911–1916. doi: 10.1016/j.meegid.2012.07.023. [DOI] [PubMed] [Google Scholar]
- Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS. et al. PlasmoDB: a functional genomicdatabase for malaria parasites. Nucleic Acids Res. 2009;37(Databaseissue):D539–D543. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgopapadakou NH. Penicillin-binding proteins and bacterial resistance to beta-lactams. Antimicrob Agents Chemother. 1993;37(10):2045–2053. doi: 10.1128/AAC.37.10.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock L, Miller GH, Schaper KJ, Seydel JK. Sulfonamide structure-activityrelationships in a cell-free system. 2. Proof for the formation of asulfonamide-containing folate analog. J Med Chem. 1974;17(1):23–28. doi: 10.1021/jm00247a006. [DOI] [PubMed] [Google Scholar]
- Doyle MA, Gasser RB, Woodcroft BJ, Hall RS, Ralph SA. Drug target prediction and prioritization: using orthology to predict essentiality in parasite genomes. BMC Genomics. 2010;11:222. doi: 10.1186/1471-2164-11-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skold O. Resistance to trimethoprim and sulfonamides. Vet Res. 2001;32(3–4):261–273. doi: 10.1051/vetres:2001123. [DOI] [PubMed] [Google Scholar]
- Walsh C. Antibiotics: actions, origins, resistance. Washington, D.C: ASM Press; [Oxford: Enfield, distributor]; 2003. [Google Scholar]
- Gregson A, Plowe CV. Mechanisms of resistance of malaria parasites to antifolates. Pharmacol Rev. 2005;57(1):117–145. doi: 10.1124/pr.57.1.4. [DOI] [PubMed] [Google Scholar]
- Adikesavan AK, Katsonis P, Marciano DC, Lua R, Herman C, Lichtarge O. Separation of recombination and SOS response in Escherichia coli RecA suggests LexA interaction sites. PLoS Genet. 2011;7(9):e1002244. doi: 10.1371/journal.pgen.1002244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez JL. Antibiotics and antibiotic resistance genes in natural environments. Science. 2008;321(5887):365–367. doi: 10.1126/science.1159483. [DOI] [PubMed] [Google Scholar]
- Forsberg KJ, Reyes A, Wang B, Selleck EM, Sommer MO, Dantas G. The shared antibiotic resistome of soil bacteria and human pathogens. Science. 2012;337(6098):1107–1111. doi: 10.1126/science.1220761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Elia MA, Pereira MP, Brown ED. Are essential genes really essential? Trends Microbiol. 2009;17(10):433–438. doi: 10.1016/j.tim.2009.08.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Essentiality and ranking of known drug targets. The essentiality and ranking (from an evolutionary perspective) of known drug targets. Drug targets are ordered by drug class. Data for eight species of interest are presented in separate columns. Red: essential genes, blue: nonessential genes, grey: no data addressing essentiality, white: no ortholog with reliable omega.
Statistical data - pN/pS analysis. Mean pN/pS ratio for three sets of genes (ADT - drug targets, ESS - essential genes, ALL - other genes) presented. Data for eight species analyzed in the study. P-values where corrected for multiple testing with FDR (Benjamini-Yekutieli algorithm). Abbreviations: ciu - confidence interval upper limit; cil - confidence interval lower limit, eco – Escherichia coli, ftn – Francisella novicida, hin – Haemophilus influenzae, hpy – Helicobacter pylori, pau – Pseudomonas aeruginosa, sao – Staphylococcus aureus, spn – Streptococcus pneumoniae, stm – Salmonella typhimurium.
Statistical data - dN/dS analysis. Mean dN/dS ratio for three sets of genes (ADT - drug targets, ESS - essential genes, ALL - other genes) presented. Data for eight species analyzed in the study. P-values where corrected for multiple testing with FDR (Benjamini-Yekutieli algorithm). Abbreviations: ciu - confidence interval upper limit; cil - confidence interval lower limit, eco – Escherichia coli, ftn – Francisella novicida, hin – Haemophilus influenzae, hpy – Helicobacter pylori, pau – Pseudomonas aeruginosa, sao – Staphylococcus aureus, spn – Streptococcus pneumoniae, stm – Salmonella typhimurium.
Dn/Ds analysis - materials, methods and results. Analysis of evolutionary rate (dN/dS ratio) of known drug targets. Materials and methods as well as Results are presented in the file. Details about recombination (detection and removal procedures) can be also found.