

Abstract
Viral infection is commonly associated with virus-driven hijacking of host proteins. Here we describe a novel mechanism by which influenza virus affects host cells through the interaction of influenza non-structural protein 1 (NS1) with the infected cell epigenome. We show that the NS1 protein of influenza A H3N2 subtype possesses a histone-like sequence (histone mimic) that is used by the virus to target the human PAF1 transcription elongation complex (hPAF1C). We demonstrate that binding of NS1 to hPAF1C depends on the NS1 histone mimic and results in suppression of hPAF1C-mediated transcriptional elongation. Furthermore, human PAF1 has a crucial role in the antiviral response. Loss of hPAF1C binding by NS1 attenuates influenza infection, whereas hPAF1C deficiency reduces antiviral gene expression and renders cells more susceptible to viruses. We propose that the histone mimic in NS1 enables the influenza virus to affect inducible gene expression selectively, thus contributing to suppression of the antiviral response.
Histones are essential regulators of genome function in eukaryotic cells1,2. The amino-terminal domain of histones (the histone tail) provides a scaffold for the assembly of protein complexes controlling gene activity3. This role of a histone tail is achieved largely by post-translational histone modifications, catalysed by distinct modifying enzymes4. The combination of diversely modified histone tails establishes gene-region-specific patterns that, upon recognition by histone-binding proteins, contribute to the regulation of gene replication5, repair6 and transcription7.
The ability of histone tails to guide gene function indicates the possibility of targeted control of gene expression by artificial or naturally occurring molecules that can structurally and/or functionally mimic the histone tail. The former possibility has been demonstrated by studies that show the ability of synthetic compounds to interfere with inducible gene expression by abolishing the interaction between the acetylated histone H4 and the BET family of transcriptional regulators8,9. Histone binding to transcriptional regulators could also be interrupted by exogenous cell-permeable histone peptides10,11. This approach underscores the possibility of a competition between endogenous and exogenous histone tails for the common binding partners. Indeed, the histone H3 tail-like sequence (histone mimic) within histone methyltransferase G9a can compete, in a modification-dependent fashion, for binding to the histone-bound heterochromatin protein 1 (HP1)12. Overall, the 3–5-amino-acid-long sequences that match various parts of the histone tail can be found in numerous eukaryotic and prokaryotic proteins; however, the role of these presumptive histone mimics in the regulation of gene activity remains unknown.
Influenza virus NS1 carries a histone H3-like sequence
Pathogens have a known ability tointerfere withvitalprocesses in thehost cells by mimicking regulatory components of host protein networks13. In searching for naturally occurring histone mimics with a gene regulatory capability, we screened in silico for pathogen-derived proteins that have a known or predicted capacity to accumulate within the nuclei of infected cells, and bear a histone-tail-like unstructured domain at the amino or carboxy termini. One of these proteins was found to be the influenza A virus NS1 protein that suppresses host response to influenza virus14.
Depending on the viral subtype, NS1 is a 219–237-amino-acid-long protein14. NS1 is not essential for formation of the viral particle but is critical for counteracting the antiviral cell response14,15. In the absence of NS1, influenza is greatly attenuated15. Suppression of antiviral host response by NS1 relies partly on the ability of NS1 to interfere with cytosolic signalling processes that regulate the expression of type I interferon genes16–19. Additionally, the NS1 protein can affect host gene expression by interfering with RNA splicing and messenger RNA export20–23.
We found that NS1 of influenza A (H3N2) virus carries a sequence that resembles the histone H3 tail. Similar to the histone tail, the C terminus of NS1 comprises a non-structured and potentially highly interactive domain24,25 (Fig. 1a). Although this unstructured C-terminal domain is present in most of the human influenza A variants (Fig. 1a), only NS1 from H3N2 subtype (hereafter defined as NS1) possesses the ARSK sequence (amino acids 226–229) that is chemically analogous to the 1ARTK4 sequence that comprises the lysine 4 site (H3K4) of histone H3 (Fig. 1a). The similarity between histone H3 and NS1 tails is further strengthened by the ability of the NS1 tail to serve as a substrate for histone-modifying enzymes in vitro. Incubation of the NS1 tail peptide with lysine methyltransferase Set1 complex or Set7/9 in the presence of S-adenosylmethionine resulted in incorporation of the methyl mark into the peptide (Fig. 1b, upper panel). Accordingly, substitution of lysine 229 to arginine (K229R) prevented methylation. These results, as well as the ability of histone acetyltransferase TIP60—which acetylates histone H3 lysine 4 in yeast26—to acetylate the NS1 peptide at K229 (Fig. 1b, upper panel), support the histone mimicry by NS1. The ability of the H3K4-like sequence within the NS1 tail to serve as a substrate for chosen histone-modifying enzymes has been further confirmed by using recombinant GST fusion NS1 protein (Fig. 1b, middle panel). Finally, the H3K4-like site within NS1 could be methylated or acetylated in virally infected cells. Using a custom-made antibody against dimethylated lysine 229 in NS1 (NS1me2; Supplementary Fig. 1), or acetyl-specific antibody27, we found the presence of methylated and acetylated NS1 in infected human cells (Fig. 1b, lower panel). Point mutation of the NS1 gene that causes K229R substitution prevented modification (Fig. 1b, lower panel). It is likely that members of the H3K4 methyltransferase and acetyltransferase enzymes, that can modify NS1 in vitro, are responsible for NS1 modification in vivo. However, in view of the known redundancy between multiple H3K4 modifying complexes4,28–30, identification of a sole NS1 modifying enzyme seems unlikely.
Figure 1. Influenza NS1 contains a histone mimic.

a, The homologous carboxyterminal NS1 and the aminoterminal histone H3 sequences are shown (red letters). The table displays C-terminal NS1 sequences of the influenza A subtypes. b, Methylation or acetylation of the NS1 peptide (top panel), the GST–NS1 protein (middle panel) or of the viral NS1 in A549 infected cells (bottom panel) are shown. KR, NS1 substrates where K229 is replaced by arginine. c, Association of the NS1 histone mimic with the hPAF1C subunits and CHD1 in nuclear extracts. In, input material. d, NS1 histone mimic binds to CHD1. Unmodified or methylated NS1 (K229) or methylated H3 (K4) peptides were incubated with the recombinant CHD1 or the CHD1 double-chromodomain. Binding to NS1 was revealed by silver or Coomassie staining (top and bottom panel, respectively). e, Binding of Flag-tagged hPAF1C subunits to NS1 or histone H3 peptides was assessed by western blotting (left and right panels, respectively). IP, immunoprecipitation.
NS1 interacts with transcript-elongating proteins
Our experiments show the presence of a histone mimic within the NS1 tail. In histones, the unmodified or modified histone tail contributes to the assembly of chromatin protein complexes31–33. The nature of the histone-tail-binding proteins is commonly revealed by the identification of nuclear proteins that bind to immobilized histone peptides during affinity purification34. Using this approach (Supplementary Fig. 2), we searched for human nuclear proteins that bind to the NS1 tail peptide (amino acids 220–230). To control for the specificity of binding we used a scrambled peptide with an amino acid composition identical to NS1. Mass-spectrometry analysis of NS1-tail-bound proteins revealed the association of the NS1 tail with polypeptides that belong to the human PAF1 transcription elongation (hPAF1C) and CHD1 chromatin remodelling complexes (Supplementary Fig. 2). Binding of the NS1 tail to human PAF1, parafibromin (CDC73), SNF2l and CHD1 were validated by western blotting (Fig. 1c). In support of the histone mimicry by the NS1 tail, we observed the histone-H3-like pattern of the NS1 tail binding to CHD1 in vitro. The chromodomain of human CHD1 binds specifically to di- or trimethylated lysine 4 of histone H3 (ref. 35). Similarly, the full-length CHD1 or the purified double-chromodomain binds selectively to di- or trimethylated NS1 peptide, but not to unmodified or acetylated NS1 tail (Fig. 1d).
To start assessing the role of the NS1 histone mimic in the regulation of chromatin complexes, we focused our attention on hPAF1C. We chose this on the basis of several factors. First, hPAF1C is a potent regulator of RNA elongation36, a co-transcriptional process that contributes significantly to pathogen-induced gene expression11,37. Second, PAF1C has been implicated in the regulation of stress-induced genes in yeast38 that could be functionally equated to pathogen-induced genes in mammalian cells. Third, opposite to CHD1 that binds to the NS1 tail in a methyl-dependent fashion, purified hPAF1C binds to unmodified as well as modified NS1 tail (Supplementary Fig. 3a, left panel), thus making this interaction potentially more versatile. The interaction of hPAF1C with the NS1 tail is sequence-dependent. Neither NS1 tail of the H5N1 influenza virus, the scrambled NS1 peptide, nor the truncated NS1 tail that lacks the H3K4-like sequence were able to bind to hPAF1C (Supplementary Fig. 3b).
hPAF1C consists of six distinct subunits (PAF1, LEO1, CDC73 (also called parafibromin), SKI8 (also called WDR61), CTR9, RTF1) that have different roles in the interaction between hPAF1C and its binding partners, including RNA polymerase II (RNA Pol II)36,39,40. To identify the primary NS1-tail-interacting hPAF1C subunit(s), we tested binding of the NS1 tail to purified recombinant individual hPAF1C subunits. We found that human PAF1 is the primary binder of the unmethylated or methylated NS1 tail, but not the control peptide that lacks the histone mimic (Fig. 1e, left panel). PAF1 does not bind to acetylated NS1 tail, thus pointing to a possible in vivo regulation of NS1 binding to PAF1 by a methyl/acetyl-switch mechanism. Finally, NS1 interacts with PAF1 in vivo. Immunoprecipitation of NS1 or PAF1 from infected cell extract revealed the existence of an NS1–PAF1 complex (Supplementary Fig. 3c). Our findings show that direct binding of the NS1 histone mimic to PAF1 is responsible for the association of NS1 with hPAF1C.
Interaction of hPAF1C with the NS1 histone mimic raised the possibility of hPAF1C binding to the histone H3 tail. Although the interaction of hPAF1C with RNA Pol II and other transcriptional regulators is well established36,39,40, the direct association between hPAF1C and histone H3 has not been documented. We found that unmodified and methylated histone H3 tail peptides, but not the scrambled control peptide, bind to hPAF1C (Supplementary Fig. 3a, right panel) or to purified PAF1 protein (Fig. 1e, right panel). Similar to the NS1 histone mimic, the acetylated histone H3 tail did not bind to either hPAF1C components.
The ability of homologous tails of NS1 and histone H3 to interact directly with hPAF1C indicated a possibility of virus-mediated hijacking of the transcription elongation machinery from the host genes. We found that NS1 accumulates in the nuclei of the infected human cells (Supplementary Fig. 4a), where it reaches approximately 5 ×105 molecules per nucleus within 12 h after infection (Supplementary Fig. 4b). The salt-extraction profile of NS1 from nuclei of infected cells showed association of NS1 with chromatin (Supplementary Fig. 4c). To assess the position of NS1 on chromatin during viral infection, we generated a recombinant virus (Flag–NS1) that expresses Flag-tagged NS1 protein (Supplementary Fig. 5a, b). Genome-wide analysis of the Flag–NS1 binding at 12 h after infection revealed the presence of NS1 on induced antiviral gene promoters (Supplementary Tables 1 and 2). In infected cells, NS1 is enriched at promoters characterized by the presence of H3K4me3 and RNA Pol II (Fig. 2a). The pattern of NS1 distribution on its targets parallels the distribution of hPAF1C (Fig. 2a). Collectively, these data revealed a strategic position of NS1 at sites of active antiviral gene transcription.
Figure 2. Functional interaction between NS1 and PAF1 in infected cells.

a, The ChIP-seq profiles show the distribution of indicated proteins at inducible genes before (black line) and after (red line) infection. The induced genes were revealed by RNA-seq and ChIP-seq analysis of infected A549 cells (Supplementary Tables 1 and 2). TSS and TES, the transcriptional start and end sites, respectively. b, The NS1 levels at gene promoters in PAF1- or CHD1-deficient cells (blue and green lines, respectively). The scrambled siRNA-treated cells (red line) were used as control. The insert shows knockdown of PAF1 or CHD1 in A549 cells. c, PAF1, RNA Pol II and H3K4me3 levels at the TSS and TES of the induced genes in uninfected (ui) cells, cells infected with the wild-type (WT) or PAF1-binding mutant virus (ΔPAF). Data are representative of three independent experiments; error bars show the s.e.m.
NS1 histone mimic controls antiviral gene expression
The interaction between the NS1 tail and hPAF1C has two complementary functions that may support viral infection. The virus probably uses the NS1–hPAF1C interaction to target sites of active transcription. The short interfering RNA (siRNA)-mediated PAF1 deficiency resulted in 30% reduction of NS1 levels at the transcriptional start sites of the NS1-bound genes in infected cells (Fig. 2b). The specific role of PAF1 in recruitment of NS1 was supported by unaltered NS1 binding to its gene targets in cells treated with CHD1-specific siRNA (Fig. 2b). These findings revealed a previously unknown ability of the influenza virus to use host PAF1 to position viral NS1 protein at sites of active gene transcription.
Once targeted to transcriptionally active loci, as defined by RNA Pol II levels cross-referenced to the infected cell transcriptome (Supplementary Tables 1 and 2), NS1 interferes with PAF1 and RNA Pol II abundance at target genes. As exemplified by virus-induced IFIT1 and IFI6 genes, infection with the wild-type influenza virus resulted in decreased PAF1 and RNA Pol II levels at the transcriptional end sites (TES) and to a lesser extent at the transcriptional start sites (TSS) of the genes, as compared with PAF1-binding mutant virus (ΔPAF) (Fig. 2c). This pattern of PAF1, RNA Pol II and H3K4me3 distribution is indicative of impaired transcriptional elongation.
To determine the impact of the NS1 tail on the dynamics of antiviral gene transcription, we conducted a genome-wide nuclear ‘run-on’ analysis (global run-on sequencing, GRO-seq)41 of cells infected with the wild-type or mutant viruses that cannot bind to human PAF1 (ΔPAF). In the absence of infection, we observed accumulation of RNA species that map to the TSS of antiviral genes and low levels of RNAs that map to the gene body (Fig. 3a). Accumulation of TSS-associated short RNAs is a common feature of rapidly inducible genes that maintain a paused/poised state in the absence of an inducer41. Infection with the ΔPAF virus resulted in a marked increase in levels of RNA transcripts that map to the TSS as well as to the downstream gene region. The high abundance of transcripts associated within the gene body is indicative of effective RNA elongation, which has an essential role in regulation of the pathogen-driven host response11,37. Compared to cells infected with ΔPAF virus, the cells infected with wild-type virus displayed slightly reduced levels of TSS-associated RNAs but greatly diminished levels of transcripts associated with the gene body and 3′ end region (Fig. 3a). This pattern is indicative of impaired transcriptional elongation and is consistent with defective elongation in other experimental settings42,43. The differences between the impact of ΔPAF and wild-type viruses on the global dynamics of antiviral gene transcription are exemplified by differences in transcription of selected antiviral genes (Fig. 3a, right panel). Infection with either wild-type or ΔPAF virus has no effect on transcriptional dynamics of housekeeping genes and genes not affected by viral infection (Fig. 3b).
Figure 3. NS1 suppresses antiviral gene transcription in infected cells.

a, Left: the GRO-seq profile of inducible RNA transcripts in uninfected (ui) A549 cells (black line) or cells infected with wild-type or ΔPAF virus (green and red lines, respectively). Right: GRO-seq profile of IFIT1 and IFI6 genes in uninfected and infected cells. b, GRO-seq profile of A549-expressed genes that are not affected by virus infection (left panel) or of the HPRT1 gene (right panel). Reads from either DNA strands are indicated as +/−. The y axes display reads per million mapped reads per 25 bp.
The ability of NS1 to control directly hPAF1C-dependent transcriptional elongation was further supported by the analysis of the impact of the NS1 tail on hPAF1C-driven RNA elongation in vitro. In this assay, a chromatinized DNA template, supplemented with selected transcription factors and histone-modifying enzymes, supports hPAF1C-mediated transcription in a fashion that faithfully reproduces transcriptional elongation in vivo36 (Fig. 4a). We found that NS1 has a negative impact on hPAF1C function (Fig. 4b). The comparison between the amount of in vitro synthesized RNA in the absence or presence of hPAF1C shows the ability of hPAF1C to boost RNA synthesis (Fig. 4b). Addition of purified NS1 (Supplementary Fig. 5c) to the transcription reaction reduced the amount of synthesized RNA to background levels (Fig. 4b). Addition of purified NS1 that lacks the PAF1-binding sequence had no effect on RNA synthesis (Fig. 4b). Both wild-type NS1 and NS1 that lacks the PAF1 binding sequence have no impact on transcription of non-chromatinized template in vitro (Supplementary Fig. 6). This selective and direct impact of NS1 on virus-induced gene expression has been further supported by a lack of NS1 requirement for the association between the viral and the host RNA polymerases in infected cells44,45 (Supplementary Fig. 7).
Figure 4. NS1 inhibits transcriptional elongation in vitro.
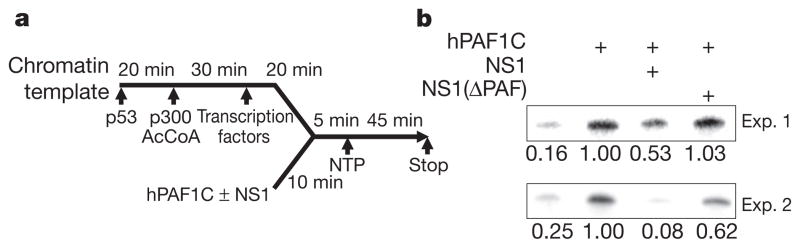
a, The full-length NS1 protein (NS1) or NS1 lacking the PAF1-binding sequence (NS1(ΔPAF)) (Supplementary Fig. 5c) was added to the RNA elongation reaction as indicated. b, The amount of the 390-nt RNA elongation product was quantified by ImageJ. The results of two independent experiments are shown.
Our findings provide unambiguous proof for the ability of the NS1 tail to interfere directly with hPAF1C-driven transcription of antiviral genes.
hPAF1C controls antiviral gene expression and infection
The significance of NS1–hPAF1C interaction during infection was evaluated by two independent experimental approaches. Deletion of the hPAF1C binding sequence in NS1 resulted in reduced viral titres of the ΔPAF virus as compared to those of the virus carrying full-length NS1 (Supplementary Fig. 8). This finding is in agreement with the previously reported positive role of the NS1 carboxy terminus in influenza A virulence46.
We also revealed the central role of hPAF1C in transcriptional response induced by various viruses, as well as by the viral RNA analogue poly(I:C) and type I interferon. To study the impact of PAF1 on influenza-induced antiviral gene expression we used the NS1-deficient influenza virus (PR8/ΔNS1) that elicits an extremely potent antiviral response and is therefore particularly useful to study host factors that regulate the antiviral response14,15. Infection with PR8/ ΔNS1 of control siRNA-treated cells resulted in a strong upregulation of numerous antiviral genes (Fig. 5a). This response was reduced in PAF1-deficient cells (Fig. 5a, Supplementary Fig. 9, left panel, and Supplementary Table 3). The negative effect of PAF1 deficiency on antiviral response is not a consequence of impaired virus life-cycle caused by a reduction in viral gene expression (Supplementary Fig. 10).
Figure 5. PAF1 controls antiviral response.

a, b, The expression levels of mRNAs in influenza infected (a) or IFN-β1-treated (b) control (siCtrl) or PAF1-deficient (siPAF) A549 cells. The tables show the siPAF-affected gene categories. Ut, untreated with siRNA. c, Dynamics of virus replication in control or PAF1-deficient A549 cells. p.f.u. plaque-forming units. Data are representative of three independent experiments. Error bars show the s.e.m.
PAF1 deficiency also attenuated the transcriptional response to wild-type influenza A H1N1 (Supplementary Fig. 11, left panel). The general positive role of PAF1 in antiviral gene expression was underscored by reduced expression of antiviral genes in PAF1-deficient cells infected with vesicular stomatitis virus (VSV) as well as cells treated with poly(I:C) (Supplementary Fig. 11 and Supplementary Tables 4–6). PAF1 deficiency also downregulates the transcriptional response to type I interferon that governs the antiviral immunity (Fig. 5b, Supplementary Fig. 9, right panel and Supplementary Table 7). The impaired antiviral response by PAF1-deficient cells contributes to a nearly tenfold increase of NS1-deficient PR8/ΔNS1 virus replication, as compared to infected control cells (Fig. 5c). The impact of PAF1 deficiency on antiviral gene expression is selective. PAF1 deficiency does not alter the expression of housekeeping genes (Supplementary Fig. 12 and Supplementary Table 8).
Concluding remarks
We have shown that H3N2 influenza A virus interferes with host gene expression by exploiting the very basic principles of the epigenetic control of gene regulation. By mimicking the histone H3K4 sequence, which has a key role in positive regulation of gene transcription, the influenza virus gains access to histone-interacting transcriptional regulators that govern inducible antiviral gene expression. The histone-like unstructured NS1 tails are present in the majority of human influenza A isolates, but their primary sequence differs between viruses. The presence of potentially reactive amino acid residues within the unstructured NS1 tails and the nuclear localization of NS1 predict a multitude of nuclear proteins interacting with influenza NS1 in a sequence- and/or modification-specific fashion. In extension of the histone code that has provided a conceptual framework for understanding chromatin-based gene regulation47–49, we propose the existence of the ‘NS1 code’ that guides influenza virus interference with host chromatin. The interference with chromatin function may complement other immunosuppressive functions of NS1 and provide the influenza virus with an opportunity to affect host gene expression in a highly selective fashion by recognizing and using epigenetic patterns of the host cells. This mechanism may contribute to the influenza-variant-specific pathogenesis and disease progression that differ between viruses carrying the histone-like NS1 tail and those viruses, such as human H1N1 (including pandemic 2009 virus), that lack the NS1 tail50. It is possible that by targeting the actively transcribed antiviral genes, the NS1 tail helps influenza to balance its virulence against the preservation of the infected cell function. Such a mechanism could help the virus to maintain its long-term presence within the human population. Our findings could also be applied to the development of novel anti-influenza therapies that will interfere in a highly selective fashion with the NS1 tail binding to its chromatin partners. Finally, identification of hPAF1C as a key mediator of antiviral and pro-inflammatory gene response may pave the way for the development of anti-inflammatory therapies that—similar to the recently described ‘epigenetic’ therapies8,9—will interfere selectively with hPAF1C binding to its partners.
METHODS SUMMARY
The methylation and acetylation in vitro has been performed as described previously12,26. The analysis of NS1 methylation or acetylation in vivo has been done by using custom-made methyl-specific or commercial acetyl-specific antibodies27. Isolation of the NS1 binding proteins, their analysis by mass-spectrometry and western blotting were performed according to a protocol used to identify histone-bound proteins34. The recombinant hPAF1C and its individual components were prepared as described before36. Interaction between hPAF1C and its individual components with peptides were performed as reported32,33. The transcriptional assays have been performed as described36. Genome-wide distribution of NS1 and other chromatin-bound proteins were accessed by ChIP-sequencing as previously described9, and analysed using standard bioinformatic approaches. Recombinant influenza viruses were generated using standard reverse genetics approaches15,17. Infection of cells with viruses was performed according to the standard methods17,46 and viral titres were quantified by plaque assay. Gene expression analysis has been done by microarray using Illumina Human HT-12 v4 Expression BeadChips or by RNA-sequencing and verified by quantitative real-time PCR9. GRO-sequencing was performed as described41. All methodological details are additionally outlined in Supplementary Information.
Supplementary Material
Supplementary Table 1. Genes affected by Influenza Infection
FPKM values for genes that are upregulated (A) or downregulated (B) by greater than 2 fold (shown as natural log of fold change; ln(Fold Change)) between 0h and 12h post influenza infection are shown. RNA from virally infected cells was collected at 0h and 12h post infection and used to prepare libraries for RNA-sequencing.
Supplementary Table 2. List of genes used for the integrated profiles
(A) Virus-induced genes were identified by cross-referencing RNA expression and RNA Pol II and H3K4me3 ChIP-sequencing profiles. Expression of antiviral RNA levels were determined by RNA-sequencing (Suppl. Table 1)
(B) Number of aligned reads in ChIP-Seq and RNA-Seq experiments shown in Fig. 2c
(C) Number of aligned reads in ChIP-Seq experiments shown in Fig. 2d
(D) Number of aligned reads in GRO-Seq experiments shown in Fig. 3
Supplementary Table 3. siPAF dependent genes in PR8/ΔNS1 infected cells
siPAF regulated genes in PR8/ΔNS1 infected cells were identified by microarray analysis of infected A549 cells. The impact of siPAF on virally regulated genes was determined by identifying siPAF regulated genes (A) and cross referencing them with PR8/ΔNS1 regulated genes (B). siCtrl regulated genes are listed in (C). Functional analyses of siPAF dependent genes were also carried out with IPA software (D).
(A) siPAF regulated genes in PR8/ΔNS1 infected A549 cells. siPAF regulated genes are defined as any gene that show ≥2.0 fold difference (p <0.001) in response magnitude between siPAF treated cells and siCtrl upon stimulation.
(B) PR8/ΔNS1 regulated genes in infected A549 cells. PR8/ΔNS1 regulated genes are defined as any gene that show ≥2.0 fold difference (p < 0.001) between stimulated and unstimulated cells, regardless of siRNA treatment.
(C) siCtrl dependent genes in infected A549 cells. siCtrl dependent genes are defined as any gene that show ≥1.5 fold difference (p <0.001) in response magnitude between siCtrl treated cells and mock transfected cells (ut) upon stimulation.
(D) Functional Classification of siPAF regulated genes. All siPAF dependent genes identified in (A) were used as the input for functional analyses in IPA software. The full defined list of higher order functions used for the analysis is shown.
Supplementary Table 4. siPAF dependent genes in Influenza (H1N1) infected cells
siPAF regulated genes in Influenza (H1N1) infected cells were identified by microarray analysis of infected A549 cells. The impact of siPAF on virally regulated genes was determined by identifying siPAF regulated genes (A) and cross referencing them with Influenza (H1N1) regulated genes (B). siCtrl regulated genes are listed in (C). Functional analyses of siPAF dependent genes were also carried out with IPA software (D).
(A) siPAF regulated genes in Influenza (H1N1) infected A549 cells. siPAF regulated genes are defined as any gene that show ≥2.0 fold difference (p <0.001) in response magnitude between siPAF treated cells and siCtrl upon stimulation. Genes that show ≥2.5 fold difference, as displayed in Supplementary Fig. 12 due to space constraints, are highlighted.
(B) Influenza (H1N1) regulated genes in infected A549 cells. Influenza (H1N1) regulated genes are defined as any gene that show ≥2.0 fold difference (p < 0.001) between stimulated and unstimulated cells, regardless of siRNA treatment.
(C) siCtrl dependent genes in infected A549 cells. siCtrl dependent genes are defined as any gene that show ≥1.5 fold difference (p <0.001) in response magnitude between siCtrl treated cells and mock transfected cells (ut) upon stimulation.
(D) Functional Classification of siPAF regulated genes. All siPAF dependent genes identified in (A) were used as the input for functional analyses in IPA software. The full defined list of higher order functions used for the analysis is shown.
Supplementary Table 5. siPAF dependent genes in VSV infected cells
siPAF regulated genes in VSV infected cells were identified by microarray analysis of infected A549 cells. The impact of siPAF on virally regulated genes was determined by identifying siPAF regulated genes (A) and cross referencing them with VSV regulated genes (B). siCtrl regulated genes are listed in (C). Functional analyses of siPAF dependent genes were also carried out with IPA software (D).
(A) siPAF regulated genes in VSV infected A549 cells. siPAF regulated genes are defined as any gene that show ≥2.0 fold difference (p <0.001) in response magnitude between siPAF treated cells and siCtrl upon stimulation.
(B) VSV regulated genes in infected A549 cells. VSV regulated genes are defined as any gene that show ≥2.0 fold difference (p < 0.001) between stimulated and unstimulated cells, regardless of siRNA treatment.
(C) siCtrl dependent genes in infected A549 cells. siCtrl dependent genes are defined as any gene that show ≥1.5 fold difference (p <0.001) in response magnitude between siCtrl treated cells and mock transfected cells (ut) upon stimulation.
(D) Functional Classification of siPAF regulated genes. All siPAF dependent genes identified in (A) were used as the input for functional analyses in IPA software. The full defined list of higher order functions used for the analysis is shown.
Supplementary Table 6. siPAF dependent genes in Poly(I:C) transfected cells
siPAF regulated genes in Poly(I:C) transfected cells were identified by microarray analysis of transfected A549 cells. The impact of siPAF on virally regulated genes was determined by identifying siPAF regulated genes (A) and cross referencing them with Poly(I:C) regulated genes (B). siCtrl regulated genes are listed in (C). Functional analyses of siPAF dependent genes were also carried out with IPA software (D).
(A) siPAF regulated genes in Poly(I:C) transfected A549 cells. siPAF regulated genes are defined as any gene that show ≥2.0 fold difference (p <0.001) in response magnitude between siPAF treated cells and siCtrl upon stimulation.
(B) Poly(I:C) regulated genes in transfected A549 cells. Poly(I:C) regulated genes are defined as any gene that show ≥2.0 fold difference (p < 0.001) between stimulated and unstimulated cells, regardless of siRNA treatment.
(C) siCtrl dependent genes in transfected A549 cells. siCtrl dependent genes are defined as any gene that show ≥1.5 fold difference (p <0.001) in response magnitude between siCtrl treated cells and mock transfected cells (ut) upon stimulation.
(D) Functional Classification of siPAF regulated genes. All siPAF dependent genes identified in (A) were used as the input for functional analyses in IPA software. The full defined list of higher order functions used for the analysis is shown.
Supplementary Table 7. siPAF dependent genes in IFNβ1 treated cells
iPAF regulated genes in IFNβ1 treated cells were identified by microarray analysis of treated A549 cells. The impact of siPAF on IFNβ1 regulated genes was determined by identifying siPAF regulated genes (A) and cross referencing them with IFNβ1 regulated genes (B). No significantly (≥1.5 fold, p<0.001) siCtrl regulated genes were identified. Functional analyses of siPAF dependent genes were carried out with IPA software (D).
(A) siPAF regulated genes in IFNβ1 treated A549 cells. siPAF regulated genes are defined as any gene that show ≥1.5 fold difference (p <0.001) in response magnitude between siPAF treated cells and siCtrl upon stimulation.
(B) IFNβ1 regulated genes in treated A549 cells. IFNβ1 regulated genes are defined as any gene that show ≥2 fold difference (p < 0.001) between stimulated and unstimulated cells, regardless of siRNA treatment.
(C) Functional Classification of siPAF regulated genes. All siPAF dependent genes identified in (A) were used as the input for functional analyses in IPA software. The full defined list of higher order functions used for the analysis is shown.
Supplementary Table 8. Expression of housekeeping genes are not affected by siPAF mediated hPAF1 deficiency.
(A) Log2(normalized signal values) of probesets representing canonical housekeeping genes in cells that were not transfected (ut) or transfected with control (siCtrl) or hPAF1 targetting (siPAF) pools of siRNAs are shown
Acknowledgments
We thank P. deGross and A. Rudensky for the mass spectroscopy analysis of the NS1 binding proteins. A. Rojas Soto, D. Reinberg, M. Dobenecker and T. Zhanyun provided us with recombinant CHD1 (A.R.S., D.R.), recombinant Set7/9 (M.D.) and Set1C (T.Z.). F. Casadio, P. Lewis, O. Binda, O. Gozani, N. Levenkova, A. Mele, R. Darnell, L. Core, J. Lis and P. Palese gave us valuable technical advice and help with data analysis. We acknowledge the Rockefeller University Genomics Resource Center for technical support. We thank R. Cadagan, A. Santana, W. Huang, R. Chandramouli and H. Zebronsky for technical assistance, R. Rizzo for help with manuscript preparation and C. Nathan for discussion. L.M.K. for artwork. B.M is supported by NIH/ NIAID K99 Pathway to Independence award (1K99AI095320-01). A.G.-S. is partially supported by NIAID grants R01AI046954, U19AI083025 and by CRIP (Center for Research in Influenza Pathogenesis), an NIAID funded Center of Excellence for Influenza Research and Surveillance, HHSN266200700010C. R.G.R. is supported by NIH grant CA129325. J.K. is supported by Charles H. Revson Foundation. I.M. is supported by American Italian Cancer Foundation. J.H. is supported by the Agency for Science, Technology and Research (A*STAR), Singapore. A.T. is supported by the NIH grant R01AI068058 and by Starr Cancer Consortium.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions I.M. contributed to design, execution, analysis of the experiments and manuscript preparation. J.S.Y.H. studied the role of PAF1 in viral infection and assisted in manuscript preparation. J.K. and R.R. studied the impact of NS1 on hPAF1C and transcriptional elongation. B.M., R.A.A. engineered the recombinant influenza viruses and studied viral infectivity. U.S. was involved in gene expression studies. S.D. performed bioinformatic analysis. C.W.S. generated antibody against viral polymerase. K.L.J. gave technical assistance. R.K.P. and K.L. contributed to manuscript preparation and enabled ChIP-seq and RNA-seq. A.G.-S. supervised and discussed the work with infectious influenza viruses. A.T. conceived and supervised this study and wrote the final manuscript.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com/nature.
Readers are welcome to comment on the online version of this article at www.nature.com/nature.
References
- 1.Kornberg RD, Thomas JO. Chromatin structure—oligomers of histones. Science. 1974;184:865–868. doi: 10.1126/science.184.4139.865. [DOI] [PubMed] [Google Scholar]
- 2.Campos EI, Reinberg D. Histones: annotating chromatin. Ann Rev Genet. 2009;43:559–599. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 3.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nature Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Kelly AE, et al. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science. 2010;330:235–239. doi: 10.1126/science.1189505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandez-Capetillo O, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nature Cell Biol. 2002;4:993–997. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 7.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 8.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicodeme E, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishiyama A, et al. Intracellular delivery of acetyl-histone peptides inhibits native bromodomain-chromatin interactions and impairs mitotic progression. Febs Lett. 2008;582:1501–1507. doi: 10.1016/j.febslet.2008.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138:129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sampath SC, et al. Methylation of a histone mimic within the histone methyltransferase G9a regulates protein complex assembly. Mol Cell. 2007;27:596–608. doi: 10.1016/j.molcel.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 13.Elde NC, Malik HS. The evolutionary conundrum of pathogen mimicry. Nature Rev Microbiol. 2009;7:787–797. doi: 10.1038/nrmicro2222. [DOI] [PubMed] [Google Scholar]
- 14.Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008;89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Sastre A, et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- 16.Lu Y, Wambach M, Katze MG, Krug RM. Binding of the influenza-virus NS1 protein to double-stranded-RNA inhibits the activation of the protein-kinase that phosphorylates the Elf-2 translation initiation-factor. Virology. 1995;214:222–228. doi: 10.1006/viro.1995.9937. [DOI] [PubMed] [Google Scholar]
- 17.Gack MU, et al. Influenza A Virus NS1 targetsthe ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pichlmair A, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 19.Hale BG, Jackson D, Chen YH, Lamb RA, Randall RE. Influenza A virus NS1 protein binds p85b and activates phosphatidylinositol-3-kinase signaling. Proc Natl Acad Sci USA. 2006;103:14194–14199. doi: 10.1073/pnas.0606109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krug RM, Yuan WM, Noah DL, Latham AG. Intracellular warfare between human influenza viruses and human cells: the roles of the viral NS1 protein. Virology. 2003;309:181–189. doi: 10.1016/s0042-6822(03)00119-3. [DOI] [PubMed] [Google Scholar]
- 21.Nemeroff ME, Barabino SML, Li YZ, Keller W, Krug RM. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′ end formation of cellular pre-mRNAs. Mol Cell. 1998;1:991–1000. doi: 10.1016/s1097-2765(00)80099-4. [DOI] [PubMed] [Google Scholar]
- 22.Das K, et al. Structural basis for suppression of a host antiviral response by influenza A virus. Proc Natl Acad Sci USA. 2008;105:13093–13098. doi: 10.1073/pnas.0805213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satterly N, et al. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc Natl Acad Sci USA. 2007;104:1853–1858. doi: 10.1073/pnas.0610977104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 25.Hale BG, Barclay WS, Randall RE, Russell RJ. Structure of an avian influenza A virus NS1 protein effector domain. Virology. 2008;378:1–5. doi: 10.1016/j.virol.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 26.Xhemalce B, Kouzarides T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev. 2010;24:647–652. doi: 10.1101/gad.1881710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becker PB, et al. Site-specific acetylation of ISWI by GCN5. BMC Mol Biol. 2007;8 doi: 10.1186/1471-2199-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: Intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 29.Wang PF, et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol. 2009;29:6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guillemette B, et al. H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1001354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lachner M, O’Carroll N, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 32.Shi XB, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lan F, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wysocka J. Identifying novel proteins recognizing histone modifications using peptide pull-down assay. Methods. 2006;40:339–343. doi: 10.1016/j.ymeth.2006.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sims RJ, et al. Human but not yeast CHD1 binds directly and selectively to histone H3 methylated at lysine 4 via its tandem chromodomains. J Biol Chem. 2005;280:41789–41792. doi: 10.1074/jbc.C500395200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim J, Guermah M, Roeder RG. The human PAF1 complex acts in chromatin transcription elongation both independently and cooperatively with SII/TFIIS. Cell. 2010;140:491–503. doi: 10.1016/j.cell.2009.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramirez-Carrozzi VR, et al. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. 2009;138:114–128. doi: 10.1016/j.cell.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim KY, Levin DE. Mpk1 MAPK association with the Paf1 complex blocks Sen1-mediated premature transcription termination. Cell. 2011;144:745–756. doi: 10.1016/j.cell.2011.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen YX, et al. DSIF, the Paf1 complex, and Tat-SF1 have nonredundant, cooperative roles in RNA polymerase II elongation. Genes Dev. 2009;23:2765–2777. doi: 10.1101/gad.1834709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaehning JA. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Bioiphys Acta. 2010;1799:379–388. doi: 10.1016/j.bbagrm.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min IM, et al. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Gene Dev. 2011;25:742–754. doi: 10.1101/gad.2005511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mapendano CK, Lykke-Andersen S, Kjems J, Bertrand E, Jensen TH. Crosstalk between mRNA 39 end processing and transcription initiation. Mol Cell. 2010;40:410–422. doi: 10.1016/j.molcel.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 44.Loucaides EM, et al. Nuclear dynamics of influenza A virus ribonucleoproteins revealed by live-cell imaging studies. Virology. 2009;394:154–163. doi: 10.1016/j.virol.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Engelhardt OG, Smith M, Fodor E. Association of the influenza a virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J Virol. 2005;79:5812–5818. doi: 10.1128/JVI.79.9.5812-5818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jackson D, Hossain MJ, Hickman D, Perez DR, Lamb RA. A new influenza virus virulence determinant: The NS1 protein four C-terminal residues modulate pathogenicity. Proc Natl Acad Sci USA. 2008;105:4381–4386. doi: 10.1073/pnas.0800482105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 48.Turner BM. Histone acetylation and an epigenetic code. Bioessays. 2000;22:836–845. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 49.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 50.Yang Y, et al. The transmissibility and control of pandemic influenza A (H1N1) virus. Science. 2009;326:729–733. doi: 10.1126/science.1177373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Genes affected by Influenza Infection
FPKM values for genes that are upregulated (A) or downregulated (B) by greater than 2 fold (shown as natural log of fold change; ln(Fold Change)) between 0h and 12h post influenza infection are shown. RNA from virally infected cells was collected at 0h and 12h post infection and used to prepare libraries for RNA-sequencing.
Supplementary Table 2. List of genes used for the integrated profiles
(A) Virus-induced genes were identified by cross-referencing RNA expression and RNA Pol II and H3K4me3 ChIP-sequencing profiles. Expression of antiviral RNA levels were determined by RNA-sequencing (Suppl. Table 1)
(B) Number of aligned reads in ChIP-Seq and RNA-Seq experiments shown in Fig. 2c
(C) Number of aligned reads in ChIP-Seq experiments shown in Fig. 2d
(D) Number of aligned reads in GRO-Seq experiments shown in Fig. 3
Supplementary Table 3. siPAF dependent genes in PR8/ΔNS1 infected cells
siPAF regulated genes in PR8/ΔNS1 infected cells were identified by microarray analysis of infected A549 cells. The impact of siPAF on virally regulated genes was determined by identifying siPAF regulated genes (A) and cross referencing them with PR8/ΔNS1 regulated genes (B). siCtrl regulated genes are listed in (C). Functional analyses of siPAF dependent genes were also carried out with IPA software (D).
(A) siPAF regulated genes in PR8/ΔNS1 infected A549 cells. siPAF regulated genes are defined as any gene that show ≥2.0 fold difference (p <0.001) in response magnitude between siPAF treated cells and siCtrl upon stimulation.
(B) PR8/ΔNS1 regulated genes in infected A549 cells. PR8/ΔNS1 regulated genes are defined as any gene that show ≥2.0 fold difference (p < 0.001) between stimulated and unstimulated cells, regardless of siRNA treatment.
(C) siCtrl dependent genes in infected A549 cells. siCtrl dependent genes are defined as any gene that show ≥1.5 fold difference (p <0.001) in response magnitude between siCtrl treated cells and mock transfected cells (ut) upon stimulation.
(D) Functional Classification of siPAF regulated genes. All siPAF dependent genes identified in (A) were used as the input for functional analyses in IPA software. The full defined list of higher order functions used for the analysis is shown.
Supplementary Table 4. siPAF dependent genes in Influenza (H1N1) infected cells
siPAF regulated genes in Influenza (H1N1) infected cells were identified by microarray analysis of infected A549 cells. The impact of siPAF on virally regulated genes was determined by identifying siPAF regulated genes (A) and cross referencing them with Influenza (H1N1) regulated genes (B). siCtrl regulated genes are listed in (C). Functional analyses of siPAF dependent genes were also carried out with IPA software (D).
(A) siPAF regulated genes in Influenza (H1N1) infected A549 cells. siPAF regulated genes are defined as any gene that show ≥2.0 fold difference (p <0.001) in response magnitude between siPAF treated cells and siCtrl upon stimulation. Genes that show ≥2.5 fold difference, as displayed in Supplementary Fig. 12 due to space constraints, are highlighted.
(B) Influenza (H1N1) regulated genes in infected A549 cells. Influenza (H1N1) regulated genes are defined as any gene that show ≥2.0 fold difference (p < 0.001) between stimulated and unstimulated cells, regardless of siRNA treatment.
(C) siCtrl dependent genes in infected A549 cells. siCtrl dependent genes are defined as any gene that show ≥1.5 fold difference (p <0.001) in response magnitude between siCtrl treated cells and mock transfected cells (ut) upon stimulation.
(D) Functional Classification of siPAF regulated genes. All siPAF dependent genes identified in (A) were used as the input for functional analyses in IPA software. The full defined list of higher order functions used for the analysis is shown.
Supplementary Table 5. siPAF dependent genes in VSV infected cells
siPAF regulated genes in VSV infected cells were identified by microarray analysis of infected A549 cells. The impact of siPAF on virally regulated genes was determined by identifying siPAF regulated genes (A) and cross referencing them with VSV regulated genes (B). siCtrl regulated genes are listed in (C). Functional analyses of siPAF dependent genes were also carried out with IPA software (D).
(A) siPAF regulated genes in VSV infected A549 cells. siPAF regulated genes are defined as any gene that show ≥2.0 fold difference (p <0.001) in response magnitude between siPAF treated cells and siCtrl upon stimulation.
(B) VSV regulated genes in infected A549 cells. VSV regulated genes are defined as any gene that show ≥2.0 fold difference (p < 0.001) between stimulated and unstimulated cells, regardless of siRNA treatment.
(C) siCtrl dependent genes in infected A549 cells. siCtrl dependent genes are defined as any gene that show ≥1.5 fold difference (p <0.001) in response magnitude between siCtrl treated cells and mock transfected cells (ut) upon stimulation.
(D) Functional Classification of siPAF regulated genes. All siPAF dependent genes identified in (A) were used as the input for functional analyses in IPA software. The full defined list of higher order functions used for the analysis is shown.
Supplementary Table 6. siPAF dependent genes in Poly(I:C) transfected cells
siPAF regulated genes in Poly(I:C) transfected cells were identified by microarray analysis of transfected A549 cells. The impact of siPAF on virally regulated genes was determined by identifying siPAF regulated genes (A) and cross referencing them with Poly(I:C) regulated genes (B). siCtrl regulated genes are listed in (C). Functional analyses of siPAF dependent genes were also carried out with IPA software (D).
(A) siPAF regulated genes in Poly(I:C) transfected A549 cells. siPAF regulated genes are defined as any gene that show ≥2.0 fold difference (p <0.001) in response magnitude between siPAF treated cells and siCtrl upon stimulation.
(B) Poly(I:C) regulated genes in transfected A549 cells. Poly(I:C) regulated genes are defined as any gene that show ≥2.0 fold difference (p < 0.001) between stimulated and unstimulated cells, regardless of siRNA treatment.
(C) siCtrl dependent genes in transfected A549 cells. siCtrl dependent genes are defined as any gene that show ≥1.5 fold difference (p <0.001) in response magnitude between siCtrl treated cells and mock transfected cells (ut) upon stimulation.
(D) Functional Classification of siPAF regulated genes. All siPAF dependent genes identified in (A) were used as the input for functional analyses in IPA software. The full defined list of higher order functions used for the analysis is shown.
Supplementary Table 7. siPAF dependent genes in IFNβ1 treated cells
iPAF regulated genes in IFNβ1 treated cells were identified by microarray analysis of treated A549 cells. The impact of siPAF on IFNβ1 regulated genes was determined by identifying siPAF regulated genes (A) and cross referencing them with IFNβ1 regulated genes (B). No significantly (≥1.5 fold, p<0.001) siCtrl regulated genes were identified. Functional analyses of siPAF dependent genes were carried out with IPA software (D).
(A) siPAF regulated genes in IFNβ1 treated A549 cells. siPAF regulated genes are defined as any gene that show ≥1.5 fold difference (p <0.001) in response magnitude between siPAF treated cells and siCtrl upon stimulation.
(B) IFNβ1 regulated genes in treated A549 cells. IFNβ1 regulated genes are defined as any gene that show ≥2 fold difference (p < 0.001) between stimulated and unstimulated cells, regardless of siRNA treatment.
(C) Functional Classification of siPAF regulated genes. All siPAF dependent genes identified in (A) were used as the input for functional analyses in IPA software. The full defined list of higher order functions used for the analysis is shown.
Supplementary Table 8. Expression of housekeeping genes are not affected by siPAF mediated hPAF1 deficiency.
(A) Log2(normalized signal values) of probesets representing canonical housekeeping genes in cells that were not transfected (ut) or transfected with control (siCtrl) or hPAF1 targetting (siPAF) pools of siRNAs are shown