

Abstract
CdII has been used as a probe of zinc metalloenzymes and proteins because of the spectroscopic silence of ZnII. One of the most commonly used spectroscopic techniques is 113Cd NMR; however, in recent years 111mCd Perturbed Angular Correlation spectroscopy (111mCd PAC) has also been shown to provide useful structural, speciation and dynamics information on CdII complexes and biomolecules. In this article, we show how the joint use of 113Cd NMR and 111mCd PAC spectroscopies can provide detailed information about the CdII environment in thiolate-rich proteins. Specifically we show that the 113Cd NMR chemical shifts observed for CdII in the designed TRI series (TRI = Ac-G-(LKALEEK)4G-NH2) of peptides vary depending on the proportion of trigonal planar CdS3 and pseudotetrahedral CdS3O species present in the equilibrium mixture. PAC spectra are able to quantify these mixtures. When one compares the chemical shift range for these peptides (from δ = 570 to 700 ppm), it is observed that CdS3 species have δ 675–700 ppm, CdS3O complexes fall in the range δ 570–600 ppm and mixtures of these forms fall linearly between these extremes. If one then determines the pKa2 values for CdII complexation [pKa2 is for the reaction Cd[(peptide–H)2(peptide)]+→Cd-(peptide)3− + 2H+ and compares these to the observed chemical shift for the Cd(peptide)3− complexes, one finds that there is also a direct linear correlation. Thus, by determining the chemical shift value of these species, one can directly assess the metal-binding affinity of the construct. This illustrates how proteins may be able to fine tune metal-binding affinity by destabilizing one metallospecies with respect to another. More important, these studies demonstrate that one may have a broad 113Cd NMR chemical shift range for a chemical species (e.g., CdS3O) which is not necessarily a reflection of the structural diversity within such a four-coordinate species, but rather a consequence of a fast exchange equilibrium between two related species (e.g., CdS3O and CdS3). This could lead to reinterpretation of the assignments of cadmium–protein complexes and may impact the application of CdII as a probe of ZnII sites in biology.
Keywords: cadmium, metallopeptides, NMR spectroscopy, zinc proteins
Introduction
Zinc proteins are among the most abundant metalloproteins, with the metal center either having a catalytic role or a structural role. These ubiquitous metalloproteins are involved in extraordinarily diverse processes which extend from hydrolytic reactions in metabolism to gene regulation (transcription factors), DNA repair and post-transcriptional modification of proteins.[1–10] In addition, the advent of proteomics is increasing our recognition of the number of proteins that require ZnII either as a catalytic and/or structural element.[11] This utilization is not surprising since zinc is an abundant trace metal, found second only to iron in eukaryotic organisms. Therefore, to clarify fully metallobiochemistry, one needs a deep understanding of the structural aspects defining different zinc-binding sites and how the protein framework proficiently modulates the properties of this metal ion to perform, in each case, the required function.
Unlike other common metals in biology, zinc is promiscuous with respect to the types and number of ligands bound to it in proteins. One can observe structures that are low coordination number (e.g., 4), sulfur-rich centers to moderate coordination number (5 or 6) environments that are oxygen and nitrogen rich. Unfortunately, due to the poor spectroscopic properties of ZnII, a diamagnetic closed shell d10 ion, it is difficult to obtain information regarding the structure of these binding sites and, in the case of enzymes, of their mechanisms of action. To overcome this drawback, scientists have widely replaced ZnII with other metal ions that have useful spectroscopic properties.[12,13]
CdII has often been employed as a probe to understand ZnII-binding sites. This is because CdII-coordination preferences resemble the broad coordination number and ligand affinities of ZnII, but permits the use of 113Cd NMR spectroscopy—an extremely useful and accessible technique that is very sensitive to the type and number of ligands bound to CdII.[12,14,15] Less used, but gaining recognition, is 111mCd Perturbed Angular Correlation (PAC) spectroscopy.[16] Until recently, 113Cd NMR and 111mCd PAC spectroscopies were rarely used in combination to study natural metal-binding sites, and in fact, few examples exist in the literature.[17–19] Both methods are highly sensitive to the CdII first coordination sphere and provide complementary dynamics information because of the different timescale of the methods. Thus, the combined use of these two techniques could be a valuable strategy to probe CdII-substituted ZnII-binding sites in biomolecules; however, the expectation for establishing a proper structural interpretation can only be realized once a systematic correlation of the 113Cd NMR and 111mCd PAC signatures of CdII in well defined environments exists.
Our group has used a de novo design approach to define the chemistry of CdII in a thiolate-rich environment as might be found in CadC[20] or cadmium substituted ZnII proteins, such as aminolevulinic acid dehydratase (ALAD)[21] or the hepatitis C virus NS3 protease domain.[22] The TRI peptide family (TRI = Ac-G(LKALEEK)4G-NH2, Table 1)[23,24] utilizes a heptad repeat unit that contains hydrophobic residues, usually Leu, in the 1st (“a”) and 4th (“d”) positions, generating an amphiphatic a-helix that in aqueous solution associates above pH 6.0 to form three-stranded coiled coils. We have been studying the binding of CdII to different TRI peptides where a single Leu, either in position “a” or “d”, was replaced by a Cys to generate a thiol-rich binding site inside the hydrophobic core of these coiled coils.[25–29] These peptides are the perfect scaffolds as they are much simpler than the natural systems but retain enough structural complexity to study how the protein framework can modulate the geometries and physical properties of metal ions bound to thiol-rich centers. By combining 113Cd NMR and 111mCd PAC spectroscopies, we have shown how the peptide TRIL16C binds CdII as a mixture of pseudotetrahedral (CdS3O) and trigonal planar (CdS3) structures.[25] Subtle changes in the amino acid sequence of the parent peptide were sufficient to generate two peptides, TRIL12AL16C and TRIL16Pen, that bind CdII exclusively in the CdS3O and CdS3 geometries, respectively.[30] Furthermore, these techniques have driven our design strategy for the heterochromic peptide, GRANDL16PenL26AL30C, a single peptide containing two binding sites within the same three-stranded coiled coil which is capable of binding two CdII with different physical properties and showing site-selective CdII recognition.[31] The combination of the 113Cd NMR and 111mCd PAC spectroscopic data has been crucial to the characterization of these peptides to high chemical resolution.
Table 1.
Peptide sequence examples of the TRI peptide family and derivatives.
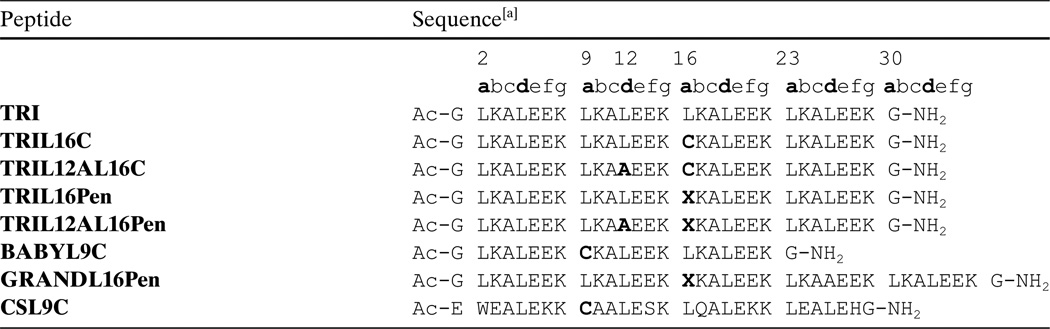 |
X = Penicillamine. Residues in bold indicate modifications.
The 113Cd NMR and 111mCd PAC spectroscopic data also provide useful information on the chemical exchange because of the different time scales probed by these techniques [NMR (0.01–10 ms); 111mCd PAC (0.1–100 ns).[25,28] For a peptide, such as TRIL16C, a single resonance (625 ppm) is observed suggesting that a single Cd structure is present; however, PAC studies revealed the existence of the CdS3O and CdS3 species. Based on these observations, we have been able to place a limitation on the exchange rate for the oxygen of the CdS3O species as being between 0.01–10 ms. More important, these data reveal an important caveat when using 113Cd NMR as the sole method of defining the metal-coordination sphere. In cases, where ligand exchange occurs, the chemical shift range for the 113Cd NMR may not alone be representative of the true speciation of a system.
Here we present a systematic study of CdS3O and CdS3 centers in selected peptides that are analyzed by 113Cd NMR, 111mCd PAC and UV/Vis spectroscopies. Important linear correlations are observed between 113Cd NMR and 111mCd PAC spectroscopies and the acid/base properties of the metal-binding site that allows one to identify the proportion of three- and four-coordinate sites in any peptide. These types of correlations are very hard to obtain in native systems since they require a system capable of accommodating different metal coordination geometries while keeping basically the same overall structure. Indeed, to the best of our knowledge, there are no reports in the literature where this type of data, 111mCd PAC–113Cd NMR–CdII binding acidity, has been correlated either for native systems or small molecules. Since numerous zinc proteins bind ZnII with thiolate ligands,[7,9,21,22,32–37] and CdII substitution is quite often used to study these binding sites, the results reported here can shed light into understanding better CdII-substituted thiolate-rich zinc proteins.
Results
111mCd Perturbed Angular Correlation (PAC) spectroscopy
111mCd PAC spectroscopy was used to determine the CdII coordination geometry of the different TRI peptides complexes. The 111mCd PAC spectra for different peptides of the TRI family are shown in Figure 1. The PAC data were analyzed as described in the Experimental Section. Each nuclear quadrupole interaction (NQI) was modelled using a separate set of parameters that includes ω0, η, Δω0/ω0, 1/τc and A. The parameters fitted to the PAC data for each peptide complex are reported in Table 2. All the NQIs have relatively low values of η, the asymmetry parameter, indicating that there is symmetry around the z axis which is the principal axis of the electric field gradient tensor with the largest eigenvalue. All the values of ω0, the parameter giving information regarding the first coordination sphere ligands, fall into two frequency regions, one around 0.450 rad ns−1 and the other around 0.340 rad ns−1. This indicates that two coordination geometries, trigonal planar (ω0 = 0.450 rad ns−1) and distorted tetrahedral (ω0 = 0.340 rad ns−1), may adequately describe the metal site structures in all these peptides. The PAC spectroscopic data for the peptides TRIL9AL16C and TRIL12AL16Pen give in both cases a well defined NQI with ω0 and η values of 0.3332 rad ns−1 and 0.19, and 0.339 rad ns−1 and 0.36, respectively. These values are similar to the value observed for the peptide TRIL12AL16C with a ω0 = 0.3405 rad ns−1 and η = 0.141,[30] and may correspond to a distorted tetrahedral CdS3O species. The NQI for the peptide containing the Pen substitution, TRIL12AL16Pen, has the highest value of η (0.36) in comparison with the other two peptides which is indicative of a larger perturbation of the geometry from axial symmetry. The peptides TRIL16-CL23A and TRIL12VL16Pen give PAC spectra showing two NQIs which are similar to those found for the peptide TRIL16C.[25] These results indicate that CdII binds to these peptides as a mixture of distorted tetrahedral CdS3O species (lower frequency species) and distorted trigonal planar CdS3 species (higher frequency species). For the peptide TRIL16-CL23A, there is a similar occupation of the two geometries since the amplitude of both frequencies is similar. However, for TRIL12VL16Pen, there is a dominating species and this belongs to the high-frequency group. The low-frequency component is difficult to fit due to the low amplitude of this signal. Thus, in order to limit the number of free parameters fitted to this NQI, the ω0, η, Δω0/ω0 and 1/τc values were fixed to the values of low frequency component found for the TRIL16C peptide. Different parameters fitted to the low frequency signal may give equally good χr2 values. A relative high 1/τc value is observed for the signals in all the peptides containing the Pen substitution species (Table 2, see last three rows). This is indicative of local dynamics at the metal ion binding site that occur on the nanosecond timescale. For TRIL16CL23A an experiment was carried out without TRIS buffer and no significant change of the spectrum was observed.
Figure 1.

111mCd PAC spectra of the different TRI peptides. All the samples contained 20 mm TRIS buffer, 300 µm peptide and a CdII/peptide ratio of 1:12. Left: Perturbation function (experimental data with error bars are shown in grey, fit shown as solid line); right: the Fourier transform (the thin line gives the Fourier transform of the experimental data, and the bold faced line gives the Fourier transform of the fit).
Table 2.
Parameters fitted to the PAC data.
| Peptide | pH | ω0 [rad ns−1] | η | Δω0/ω0 (× 100) |
1/τc [µs−1] | A (×100) | χr2 |
|---|---|---|---|---|---|---|---|
| TRIL16C[a] | 8.7 | 0.337 ± 0.002 | 0.23 ± 0.02 | 5.1 ± 0.7 | 8 ± 5 | 5.1 ± 0.6 | 1.10 |
| 0.438 ± 0.004 | 0.20 ± 0.03 | 5.4 ± 0.7 | 3 ± 3 | 3.6 ± 0.4 | |||
| TRIL9AL16C | 8.4 | 0.3332 ± 0.0005 | 0.19 ± 0.01 | 2.6 ± 0.2 | 10.6 ± 0.9 | 7.5 ± 0.2 | 1.26 |
| TRIL12AL16C[b] | 8.8 | 0.3405 ± 0.0003 | 0.141 ± 0.004 | 1.7 ± 0.1 | 5.4 ± 0.5 | 8.3 ± 0.2 | 1.06 |
| TRIL16CL23A | 8.7 | 0.337 ± 0.002 | 0.25 ± 0.01 | 7.6 ± 0.5 | 4.5 ± 0.5[c] | 4.3 ± 0.2 | 1.20 |
| 0.453 ± 0.002 | 0.14 ± 0.01 | 4.1 ± 0.3 | 4.5 ± 0.5[c] | 3.6 ± 0.2 | |||
| TRIL16Pen[b] | 8.7 | 0.454 ± 0.001 | 0.02 ± 0.10 | 1.6 ± 0.4 | 19 ± 2 | 6.9 ± 0.3 | 0.91 |
| TRIL12VL16Pen | 8.7 | 0.337[d] | 0.23[d] | 5.1[d] | 8[d] | 1.5 ± 0.2 | 0.99 |
| 0.459 ± 0.002 | 0.20 ± 0.02 | 2.2 ± 0.8 | 26 ± 5 | 5.4 ± 0.5 | |||
| TRIL12AL16Pen | 8.5 | 0.339 ± 0.001 | 0.36 ± 0.01 | 5.6 ± 0.7 | 16 ± 2 | 7.5 ± 0.3 | 1.11 |
UV/Vis spectroscopy
The pH dependence of CdII binding to the thiol groups of different peptides was followed by monitoring the formation of the characteristic ligand to metal charge transfer (LMCT) band at 235 nm.[27,29] For those experiments where CdII was binding to the peptides TRIL16Pen, GRANDL16Pen and TRIL12VL16Pen, longer equilibration times after addition of KOH were in general required. Figure 2 shows the pH profiles for several of the peptides of the TRI family. The experimental data was fit using the model published previously that corresponds to the simultaneous release of the two protons (see Experimental Section).[27] This model implies the formation of the species Cd[(peptide–H)2(peptide)]+ at low pH. The apparent acidity constant Ka2 has thus units of molar squared (M2) and the apparent pKa2 values determined for each peptide are reported in Table 3. The experimental data was also fit using an extended model that did not imply the full formation of the species Cd[(peptide–H)2(peptide)]+ and included different equilibria (see Supporting Information). Although the values obtained for pKa2 are slightly different, the information they provide is identical and the same trend is observed (see Discussion and Figure S1).
Figure 2.

pH dependence of CdII binding to TRI peptides that contain fully 4-coordinate and fully 3-coordinate binding sites: TRIL12AL16C (▲), TRIL12AL16Pen (○), TRIL16C (■) and TRIL16Pen (●). The solid lines represent the fit to the simultaneous deprotonation model.
Table 3.
Percentage of CdII species based on 111mCd PAC spectroscopic data, pKa2 values for CdII binding and 113Cd NMR chemical shifts for the different TRI peptides and derivates.
| Peptide | % CdII Species 4-coordinate S3(O/N) |
3-coordinate S3 | Apparent pKa2 | 113Cd NMR (ppm) |
|---|---|---|---|---|
| TRIL16C | 60 | 40 | 13.4 ± 0.2[a] | 625[b] |
| TRIL9AL16C | 100 | 0 | 13.3 ± 0.2 | 583[c] |
| TRIL12AL16C | 100 | 0 | 12.2 ± 0.2[d] | 574[c] |
| TRIL16CL23A | 55 | 45 | 13.5 ± 0.2 | 643 |
| TRIL16Pen | 0 | 100 | 15.8 ± 0.2[e] | 684[f] |
| TRIL12VL16Pen | 22 | 78 | 15.7 ± 0.2 | 687 |
| TRIL12AL16Pen | 100 | 0 | 12.7 ± 0.2 | 583 |
| TRIL9C | – | – | 13.4 ± 0.2[g] | 615[g] |
| TRIL2WL9C | – | – | 13.4 ± 0.2[g] | 618[g] |
| TRIL23C | – | – | 13.4 ± 0.2[g] | 612[g] |
| GRANDL9C | – | – | 13.0 ± 0.2 | 614 |
| GRANDL12AL16C | – | – | 11.3 ± 0.2 | 572 |
| GRANDL16Pen | – | – | 15.7 ± 0.2[d] | 685 |
| BABYL9C | – | – | 13.5 ± 0.2 | 618 |
| CSL9C | – | – | 13.2 ± 0.2[g] | 602[g] |
113Cd NMR spectroscopy
The 113Cd NMR spectra of the different CdII peptide complexes were recorded at pH values where, based on the pH titrations, the metal ion was fully bound (pH 8.5–9.0). In all cases, a single resonance is observed and the 113Cd chemical shifts obtained (Table 3) are consistent with thiol coordination to CdII.[14,38]
Discussion
Proteins harness the chemical properties of ZnII in order to perform a large variety of important cellular functions. The most common zinc binding sites are distorted tetrahedral ZnII centers where the metal ion is typically bound to nitrogen, oxygen and sulfur donor ligands. Usually the amino acids defining these binding pockets are His (N), Cys (S), Glu (O) and Asp (O), with the first two amino acids being the most prevalent donors.[1,39] Thiolate-rich binding centers are found in proteins implicated in very different biological activities, including gene regulation, methyl transfer reactions and protein modification.[7,9,10,32–34,40,41] Surprisingly, despite possessing a common tetrahedral geometry and similar binding pockets, these thiolaterich ZnII centers are accurately tuned to carry out their distinct biological function. Therefore, it has become clear that besides the chemical nature of the coordinating amino acids, there are additional features of the metal environment which control the properties of ZnII. Unfortunately, a clear knowledge of the structural, thermodynamic and kinetic factors that differentially modulate the chemical and physical properties of ZnII bound to these sites is sparse. This lack of information stems, to some extent, from the fact that ZnII is not amenable to most forms of spectroscopies, which complicates the direct study of the zinc-binding properties of these proteins. The use of CdII as a probe to study naturally containing ZnII proteins has been one of the most employed alternatives, since this metal replacement opens the door to the use of 113Cd NMR and 111mCd PAC spectroscopies.[12,14,16]
Our group is now in the position to start analyzing in great detail specific thiol-rich binding sites using de novo designed peptides. These peptides sequester CdII by enforcing a pseudotetrahedral S3O first coordination sphere. The fourth ligand, thought to be an exogenous water molecule, can be present or not, generating in the last case a trigonal planar S3 coordination site. The significance of studying these systems lays in the fact that in zinc enzymes, the ZnII center normally possesses three permanent coordinating positions occupied by amino acid ligands and the fourth site is filled either with an exogenous water/hydroxide molecule, which is very important for catalysis, a fourth amino acid or serves as the location of substrate binding.[1–3,22,34,35,42]
We have successfully designed two peptides, TRIL12AL16C and TRIL16Pen, capable of binding CdII exclusively in pseudotetrahedral (S3O) and trigonal planar (S3) geometries, respectively.[30] These CdII complexes, [Cd(TRIL12AL16C)3]− and [Cd(TRIL16Pen)3]−, possess very different 113Cd NMR and 111mCd PAC spectral signatures ([Cd(TRIL12AL16C)3]−: 574 ppm and ω0 = 0.3405 rad ns−1; [Cd(TRIL16Pen)3]−: 684 ppm and ω0 = 0.454 rad ns−1; Tables 2 and 3). We have proposed that these spectroscopic properties are the result of the different CdII-coordination numbers and geometries. As support for this model, the parent TRIL16C peptide, when interrogated with 111mCd PAC spectroscopy, appears to bind CdII as a mixture of both species (with two different signature signals (ω0 = 0.3405 rad ns−1 and ω0 = 0.454 rad ns−1), while a single coalesced 113Cd NMR resonance (625 ppm) is observed.[25]
Two important observations can be inferred from these data. First, that both species, a pseudotetrahedral CdS3O and trigonal planar CdS3, interconvert rapidly on the NMR timescale (0.01–10 ms) but slowly on the PAC timescale (0.1–100 ns).[25,28] The best model to describe this system is a fast water exchange that interconverts the three- and four-coordinate sites quickly. Considering the timescale of both techniques, this water exchange process occurs in the range of ms to ns, a value consistent with the water exchange rates reported for solvated CdII.[43] Second, this model predicts that there should be a linear correlation between the chemical shift in the 113Cd NMR spectrum and the ratio of CdS3O/CdS3 extracted from the 111mCd PAC spectrum. If the existence of this correlation is true, then the 113Cd NMR chemical shift (which experimentally is more generally available than PAC) could be used as an extremely sensitive indicator of the percentage of four-coordinate CdS3O and three-coordinate CdS3 species present in solution.
To assess this last point, we studied a wide range of peptides in the TRIL16C family (TRIL9AL16C, TRIL16CL23A and TRIL12VL16Pen) using 111mCd PAC spectroscopy. These peptides were chosen because their 113Cd NMR chemical shifts (Table 3) predicted either pure CdS3O or CdS3 species, or a mixture of both species co-existing. In all cases, the NQIs obtained (Table 2) have values of ω0 that fall into the two frequency regions obtained for the pure species [Cd(TRIL12AL16C)3]− (ω0 = 0.3405 rad ns−1) and Cd(TRIL16Pen)3]− (ω0 = 0.454 rad ns−1), indicating that these peptides can in fact be described using the same type of geometries. While the complex [Cd(TRIL9AL16C)3]− can be described as a 100% CdS3O species, the other two peptides, TRIL16CL23A and TRIL12VL16Pen, bind CdII as a mixture of CdS3O and CdS3. It is worthwhile to note here that the substitution of Leu to Ala above the plane of the Cys seems to selectively stabilize the CdS3O structure since in both cases, TRIL12AL16C and TRIL9AL16C, CdII binds exclusively as a four-coordinate CdS3O species. Figure 3 shows a plot of the percentage of the four-coordinate species CdS3O, calculated based on the 111mCd PAC spectroscopic data (Table 3), against the corresponding 113Cd NMR chemical shift for the different TRI peptides. The graphic obtained indicates that indeed there is a linear correlation between both spectroscopic signatures that follows the percentage speciation between the four- and three-coordinate species. This linear correlation predicts a theoretical 113Cd NMR chemical shift of 579 ppm for the four-coordinate CdS3O species and of 702 ppm for the three-coordinate CdS3 complex. This last value is very close to the 113Cd NMR chemical shift predicted previously (698 ppm).[30] Our more recent result supports this prediction since the peptide TRIL12LdL16C binds CdII with a 100% trigonal planar geometry, as determined by 111mCd PAC spectroscopy, and has a 113Cd NMR chemical shift of 697 ppm.[44] Using 113Cd NMR spectroscopy and this linear correlation one can calculate the CdII speciation present in solution for each of our peptidic systems with very high precision. If one considers that usually 111mCd PAC spectroscopy can determine the per cent of the different species in solution to within 10% accuracy, a 113Cd NMR chemical shift in the range of 570–600 ppm, and of 675–700 ppm, can be expected for CdII bound as a CdS3O and CdS3, respectively. This 113Cd NMR spectral window is consistent with that observed for similar thiol-rich environments.[14,15]
Figure 3.

Plot of the percentage of 4-coordinate CdII calculated based on the 111mCd PAC spectroscopic data vs the 113Cd chemical shift for different TRI peptides. The solid line represents the linear fit of the experimental data (r2 = 0.97414).
We know that the TRIL12AL16C and TRIL16Pen peptides possess a very different pH profile for CdII binding.[31] As Figure 2 shows, the complex [Cd(TRIL12AL16C)3]− is fully formed at pH 7.5 with a pKa2 value of 12.2, while a higher pH is required to obtain the complex [Cd(TRIL16Pen)3]−, which has a pKa2 value of 15.8. Therefore, CdII has a lower pKa2 of binding to the four-coordinate site (TRIL12AL16C) compared to the three-coordinate site (TRIL16Pen). This behavior was also observed in the heterochromic peptide GRANDL16PenL26AL30C.[31] Shown in the same Figure 2 there is the pH profile for CdII binding to the parent peptide TRIL16C that binds this metal ion as a mixture of four- and three-coordinate species. This pH profile is intermediate between those obtained for the formation of the complexes [Cd(TRIL12AL16C)3]− and [Cd(TRIL16Pen)3]−, suggesting that the pKa2 value for the formation of the different CdII complexes could also be reflecting the speciation present in solution. If this is the case, and one considers that the 113Cd NMR chemical shift is an indicator of the CdII speciation as explained above and shown in Figure 3, a linear trend can be expected if one plots the pKa2 values against the corresponding 113Cd NMR chemical shifts for the different CdII complexes of the TRIL16C peptide family. Figure 4 demonstrates that this expectation is satisfied. The pKa2 value is directly correlated with the 113Cd NMR chemical shifts and, thus, we have another indicator of the percentage of four- and three-coordinate CdII species, CdS3O and CdS3. More important, this correlation provides an independent confirmation of our proposed exchange mechanism.
Figure 4.

Plot of the pKa2 value for the formation of the complex Cd-(peptide)3− vs the 113Cd chemical shift for different peptides of the TRI family and derivatives: 1) TRIL16Pen; 2) TRIL12VL16Pen; 3) GRANDL16Pen; 4) TRIL16CL23A; 5) TRIL16C; 6) BABYL9C; 7) TRIL2WL9C; 8) TRIL9C; 9) TRIL23C; 10) GRANDL9C; 11) CSL9C; 12) TRIL9AL16C; 13) TRIL12AL16Pen; 14) TRIL12AL16C; 15) GRANDL12AL16C; 16) TRIL12LDL16C. The solid line represents the linear fit of the experimental data (r2 = 0.9468).
We next assessed the generality of this correlation by testing whether other designed peptides conformed to the same trends. Two groups of peptides were examined: 1) peptides of the TRI peptide family that contain the thiol-rich binding site in a different position from 16, and 2) GRAND and BABY peptides that are a heptad longer and shorter than TRI peptides, respectively. The pKa2 values and the corresponding 113Cd NMR chemical shifts of the different GRAND, BABY and TRI peptides containing the binding site at diverse positions were determined and included in the plot(Figure 4). The final graphic shows that these systems satisfy the same linear trend and, therefore, we conclude that the 113Cd NMR–pKa2 correlation seems to stand independently of the location of the Cys site or the length of the peptide. Furthermore, we have found that the CdII complex of the thiol derivative of the Coil–Ser peptide, [Cd-(CSL9C)3]−, which has a significantly different sequence also follows this linear dependence.[29]
The strategy used in our design to obtain a fully three-coordinate CdS3 species was the replacement of Leu by the non-natural amino acid penicillamine (Pen), which increases the local bulk directly around the CdII center. This strategy was successful and leads to peptides with large 113Cd NMR chemical shifts and high pKa2 values. While the chemical shift values are a direct representation of the coordination number of the metal, one might expect that the pKa2 values could reflect both the coordination number of the metal as well as the chemical properties of the coordinated ligands. Both Cys and Pen have thiol ligands; however, Pen has two methyl groups at the Cβ in comparison to two protons for Cys. This substitution is predicted to change the acidity of the thiol group and, thus, potentially the pH of CdII sequestration. The basicities of both amino acids are similar, pKa of 8.0 for Pen vs 8.2 for Cys.[45,46] Therefore, the higher pH necessary to bind CdII to the Pen site would appear to be the result of the intrinsic coordination geometry of the binding site. To verify that this is the case, it was necessary to obtain a fully four-coordinate CdII binding site using Pen. By analogy with TRIL12AL16C, we thought that opening a hole above the Pen layer might generate the desired four-coordinate CdII and answer this question. A 113Cd NMR chemical shift of 583 ppm was observed at pH 8.5 for the complex [Cd(TRIL12AL16Pen)3]−. Based on the 113Cd NMR—111mCd PAC correlation (Figure 3), this result indicates that we have obtained a four-coordinate CdS3O species using Pen as a ligand. Conclusive proof for this assignment comes from the 111mCd PAC data since a single NQI with a ω0 value of 0.339 rad ns−1 was obtained. The results obtained for [Cd(TRIL12AL16Pen)3]− fit nicely in the 113Cd NMR—111mCd PAC correlation (see Figure 3). This CdII complex has a higher value of η (0.36) in comparison with the other two complexes containing a 100% CdS3O, [Cd(TRIL12AL16C)3]− (η = 0.141) and [Cd(TRIL9AL16C)3]− (η = 0.19). The parameter η is the asymmetry parameter and gives information regarding the symmetry of the CdII center. Its value ranges from 0 to 1, where 0 represents pure axial symmetry. A higher value of η is indicative of a higher degree of distortion in the binding site and, thus, a larger deviation from a tetrahedral geometry. This higher distortion of the four-coordinate Pen site is most likely due to the presence of the methyl groups at the Cβ that introduces local hindrance around the thiol donors.
We next determined the pH dependence of CdII binding to TRIL12AL16Pen. As shown in Figure 2 (open circles) the pH profile obtained reveals that the coordination geometry, and not the ligand basicity, is the factor controlling the site acidity. The pKa2 value drops from 15.8 to 12.7 in moving from a three-coordinate to a four-coordinate Pen metal-binding site. Consistent with these observations, a pKa2 of 15.1 is obtained for CdII binding to TRIL12LDL16C, a peptide that binds CdII as a fully 100% CdS3 and contains Cys as a coordinating ligand.[44]
Figure 4 demonstrates that all four-coordinate sites, independently of the nature of the thiol group (Cys or Pen), bind CdII fully under lower pH conditions than do the three-coordinate sites. Another feature is apparent upon closer examination of this correlation. The pKa2 values obtained for peptides that bind CdII exclusively as three-coordinate CdS3 species have little variation, whereas those peptides sequestering CdII as a four-coordinate CdS3O species exhibit a wide range for the pKa2 values (11.3 to 13.3). These data indicate that moving from TRI to GRAND peptides or by introducing double substitutions, the pH dependence of CdII binding to the three-coordinate site is essentially unaffected, while these modifications significantly perturb the four-coordinate site. This variation most likely is a reflection of different self-association affinities and pH-dependent aggregation state preferences of the peptides themselves. For example, the GRAND peptides show a higher self-association affinity than TRI peptides, ≈5 kcal mol−1 per coiled coil aggregate. On the other hand, the disruption of a Leu layer by smaller nonpolar amino acids, such as Val and Ala, can destabilize the final coiled coil up to 6–7 kcal mol−1, reflecting poorer hydrophobic packing.[26] The greater stability of the GRAND peptides result in the formation of three-stranded coiled coils at pH conditions lower than 6.0. This enhanced aggregate stability provides a scaffold which can complex CdII at lower pH values than the corresponding TRI peptides. At the other extreme, coiled coils that have been destabilized by disrupting the core packing require higher pH to form the assembly which is necessary to sequester the metal properly. In support of this model, we observe that an increase in the concentration of the TRI peptide with respect to the amount of CdII shifts the pH profile for formation of the complex to lower pH values. This suggests that the formation/stabilization of the four-coordinate CdII may be achieved at more acidic pH values than those observed here by increasing the self-association energy of the three-stranded coiled coil. This explanation also rationalizes the small variance in pKa2 values for three-coordinate CdS3 peptides containing penicillamine. The Pen substitution stabilizes the three-stranded coiled coil significantly and, since high pH is required to form the trigonal structure, these peptides all exhibit high self-affinity under these conditions. The data indicate that the lower limit for the pKa2 value of this type of four-coordinate site is determined in our systems by the actual formation of the binding site. The data included in the supporting information corroborates all these observations.
This behavior is very different from that observed for the HgII complexes of the TRI peptide family where the formation of a three-coordinate thiolate HgII center does not depend on the stability of the coiled coil.[47–49] In this case, there are two relevant equilibria. The first is the conversion of a two-stranded coiled coil to a three-stranded coiled coil aggregate. This equilibrium appears to be metal independent having a pKa for TRI, with and without HgII, of approximately 5.5. The second equilibrium is between a three-stranded coiled coil containing linear HgII as HgS2 and the trigonal planar HgII species HgS3. As long as one examines peptides that have Cys substitution in the “a” heptad position, one observes the conversion of Hg-(peptide)2(peptide−H) to Hg(peptide)3− plus loss of a proton with a pKa = 7.7. This is true regardless of whether the peptide used is TRIL16C, TRIL9C, BABYL9C or GRANDL9C.[47, 48]
At this point, we have obtained two important correlations between CdS3O centers (ω0 ≈ 0.34 rad ns−1) and CdS3 centers (ω0 ≈ 0.45 rad ns−1) that allow the determination, based only in the 113Cd NMR chemical shift, of the percentage of four-coordinate CdS3O and three-coordinate CdS3 species (Figure 3) and the pH required for full CdII binding (Figure 4). Both correlations track the ratio of both species in solution. Often one sees quoted in references that a specific coordination geometry (e.g., CdS3O) has a broad 113Cd NMR chemical shift range without an explanation for the factors that cause the large variation. To our knowledge, this study provides the first systematic explanation for why the 113Cd NMR chemical shift range can vary over what might be perceived to be 125 ppm. Furthermore, we have demonstrated for the first time that 113Cd NMR can be used as a highly sensitive probe of the equilibrium contributions of two rapidly interchanging protein bound species when the two limiting species have been clearly identified. The next question that we must ask is how general this correlation is.
To address this point we must recognize that a four-coordinate CdII with three endogenous sulfur ligands can vary its structure in several ways within our designed peptides. First, is the issue of metal binding to “a” versus “d” type sites. Both CdII and HgII show, for example, different pKa values for formation of MS3 species when the site is in an “a” versus “d” position. Modelling studies suggest that the orientation of Cys in these two sites is markedly different with Cys sulfurs in an “a” site positioned towards the central axis of the helix whereas “d” sites orient the Cys towards the helical interfaces. Second, the position of the metal binding site may be displaced either towards the N- or C-terminal ends of the peptides. As an example of this discrepancy we can consider our recent crystallographic study of As-(CSL9C)3, an analogous peptidic system to TRI, that has provided the first X-ray structure of AsIII coordinated to three Cys residues in a protein environment (Figure 5A).[50] The AsIII was found to be in a trigonal pyramidal geometry with As–S bond lengths (2.28 ± 0.05 Å) and S-As-S angles (92°) in good agreement with small molecule complexes and the EXAFS distances previously determined for AsIII coordination to TRIL12C, TRIL16C, ArsR and ArsD.[51–54] Most important, the AsIII ion was found to be coordinated below the plane of the β-methylene protons of Cys in an endo conformation and not in an exo conformation capping the Cys residues as was previously assumed by most workers in this field.[55] On the other hand, PbII, which also prefers a trigonal pyramidal thiol-rich binding site, shows an exo configuration being coordinated above the thiolate plane when bound to the protein aminolevulinic acid dehydratase (Figure 5B).[56] This binding mode is consistent with the larger size of PbII in comparison to AsIII, with typical PbII–S distances of 2.62–2.67 Å for three-coordinate PbS3 structures.[33, 57–60] Consistent with these data, our EXAFS spectroscopic studies demonstrate that PbII binds to TRIL16C and TRIL12C peptides as a PbS3 structure with PbII–S distances of 2.63 Å.[27] CdII is intermediate in size between AsIII and PbII and has the potential to bind to peptides in either the endo or exo conformations as is demonstrated in Figure 5C and D. However, unlike both AsIII and PbII which contain stereochemically active lone pairs filling the fourth coordination sites, CdII can bind a water molecule in this fourth position.
Figure 5.

a) Side-view of AsIII coordinated to the Cys residues in CSL9C.[50] b) Drawing generated using the X-ray crystal structure of PbII bound to ALAD (pdb: 1QNV).[56] Models, based on the X-ray crystal structure of As(CSL9C)3,[50] of CdS3O bound c) to TRIL12AL16C in the exo conformation and d) to TRIL16CL19A in the endo conformation. All figures were generated using PyMOL.
From the 113Cd NMR and 111mCd PAC spectroscopic data we know that the TRIL16C peptide binds CdII as a mixture of four- and three-coordinate CdII species (CdS3O and CdS3).[25] Certainly, the CdS3 polyhedron is trigonal planar; however, one must consider whether the CdII in the CdS3O structure is in an exo or endo conformation. The replacement of the Leu layer above the metal thiol-rich binding site by Ala has proven sufficient to generate two peptides, TRIL12AL16C[30] and TRIL12AL16Pen, capable of binding CdII solely with a four-coordinate CdS3O distorted tetrahedral geometry. This mutation should generate sufficient space above the thiol ligands to allow water access to the hydrophobic interior. This observation would suggest that CdII should bind to these sites in a way that resembles most closely the PbII exo conformation rather than the AsIII endo. Such an assignment is consistent with an NMR structure of CdII bound to the HIV-I integrase H12C mutant.[61] The observation that when the second Leu layer above the Cys is replaced with Ala (TRIL9AL16C) the peptide binds CdII as solely a CdS3O species, whereas the equivalent mutation below the Cys layer (TRIL16CL23A) has essentially no effect on the ratio of CdS3O and CdS3 as compared to TRIL16C, further supports an exo conformation. Our best interpretation of these results is that opening a hole at position 9 could affect the hydrophobic packing of the Leu at position 12 allowing the entrance of water to the hydrophobic interior and thus its coordination to CdII. It should be noted that these three peptides (TRIL12AL16C, TRIL12AL16Pen and TRIL9AL16C) all exhibit PAC spectra with ω0 ≈ 0.34 rad ns−1. In fact, every peptide complex reported in the correlations of Figure 3 and 4 which contain a CdS3O species also shows ω0 ~0.34 rad ns−1. Thus, we conclude that the correlations that we have discovered are directly applicable to understanding equilibria between CdS3 and CdS3O species bound in “a” site peptides with an exo metal conformation.
One might then ask what are the properties of CdS3O sites oriented in an endo conformation or bound within a “d” site environment. Unfortunately, sufficient data do not yet exist for such constructs to evaluate these possibilities fully; however, we have been able to isolate one system that may begin to address one of these questions. The peptide TRIL16CL19A engineers a water-binding pocket below the metal binding site which might induce a shift in metal environment to the endo conformation. The 113Cd NMR chemical shift for [Cd(TRIL16CL19A)3]− is 605 ppm and the PAC spectrum indicates that this peptide contains 100% CdS3O but now with an ω0 = 0.274 rad ns−1. This chemical shift value and composition is inconsistent with the trend found in Figure 3. Furthermore, the pKa2 for this peptide is 11.2 a value which again is inconsistent with the 16 points reported in Figure 4. The tempting conclusion is that CdS3O sites with ω0 > 0.33 rad ns−1 correspond to exo conformations of the CdII whereas ω0 < 0.28 rad ns−1 may indicate the endo conformers. Furthermore, a change in the exo versus endo conformation may lead to different trend lines for the scaling of the CdS3O/CdS3 ratio with NMR and the acidity of the corresponding metal binding site. These intriguing possibilities must await further study to be confirmed; however, the concepts presented by these data raise interesting general questions for CdII binding to proteins, the use of CdII as a probe for ZnII and the interpretation of 113Cd NMR for CdII-substituted ZnII sites in biology.
The pioneers investigating CdII binding to proteins, especially as a spectroscopically amenable substitute for ZnII, were confined to those systems that became available and which may or may not have been structurally characterized. This meant that broad chemical shift ranges could be mapped; however, detailed questions relating to specific geometries or the impact of protein dynamics were difficult to explore. In this article, we have demonstrated how the conjunction of protein design and 111mCd PAC spectroscopy allows for a more refined level of understanding for biological 113Cd NMR. The systematic variation of protein constructs using de novo designed peptides allows the control of most structural variables for a metalloprotein. Thus, one may make a subtle sequence change that perturbs a highly defined metal chromophore in an interpretable manner. In this way it becomes straightforward to prepare a suite of proteins that can be used to interrogate defined physical properties of interest (e.g., 113Cd NMR shift or pKa). PAC spectroscopy provides a faster timescale to interrogate the metal center. In this case, it revealed that what may have been interpreted as a single species based on 113Cd NMR was in fact an equilibrium between two fast exchanging species. Furthermore, by extracting the NQI parameters, one may obtain useful information about the metal site such as whether a pseudotetrahedral metal binds in an endo or an exo conformation. Therefore, one is now able to systematically vary the properties of well defined binding sites so that correlations as presented here may be deduced.
The first of these correlations provides decisive proof for the CdS3 to CdS3O equilibrium model and demonstrates that one may use 113Cd NMR as an accurate and convenient method to assess the ratios of these two species. Moreover, it illustrates that CdII bound to a protein with fast ligand exchange can exhibit intermediary physical properties between the two exchanging species. Certainly this is true for the coalesced 113Cd NMR chemical shift, but more importantly, the pH at which the complex converts to having all sulfurs bound to the metal (either as CdS3O or CdS3) is dependent upon the ratio of these two components. For this system, if one knows the 113Cd chemical shift, then one can predict with a high degree of accuracy the pKa2 for complex formation. Since the binding affinity of the metal to the protein is enhanced when all three sulfurs bind, this suggests that the thermodynamically more stable constructs will be enriched in the CdS3O component. CdII binding to heterochromic polypeptides exhibits just this specificity and selectivity.[31]
To date, there has been the tendency to assign a 113Cd chemical shift to a single, specific coordination structure and to support this assignment by indicating that the chemical shift value falls within a broad range of shifts consistent with the assigned structure. Our studies indicate that one should consider a dynamic model for metal binding which may include more than one species. Furthermore, these observations suggest that a protein may be able to fine tune the affinity of CdII binding by developing a metal site that controls the ratio of CdS3O to CdS3 (or in principle two other species of different affinity which are in fast exchange).
One example where such a process may be operable is the SmtB type metalloregulatory protein CadC.[20] Site-directed mutagenesis and Cd EXAFS spectroscopy support the assignment of the CdII binding site as CdS4. The 113Cd chemical shift is 625 ppm, a value that is comfortably within literature values for CdS4 (typically reported as 590 to 750 ppm).[12, 15] However, it is surprising that the CdS4 range is so large and overlaps so significantly with CdS3-type complexes. Typically, the addition of each sulphur to the CdII-coordination sphere causes a significant downfield shift, yet CdS4 overlaps CdS3 shifts for over half their range (from 590 to 700 ppm). We have noticed that the pure species CdS3 or CdS3O have relatively narrow chemical shift ranges. With pure CdS3 species, we observe a range of chemical shifts of ≈25 ppm (from 675 to 700 ppm). The same can be said for CdS3O which appears to span 570–600 ppm. The 125 ppm breadth that occurs is a result of the equilibrium between these two species. It is tempting to suggest that the even broader range reported for CdS4 (160 ppm) could also be a consequence of mixed sites. If this reasoning is correct, then a more appropriate value for a pure CdS4 species would likely be closer to 750 ppm, with a lower limit of ≈720 ppm. If a pure CdS4 in a protein environment truly has this chemical shift range, how then could one explain the 625 ppm resonance of CadC? The simplest explanation would be that CdII might bind as a mixture of CdS4 and CdS3O, with water as the fourth ligand. This makes the CadC system an ideal target for PAC studies to test this hypothesis.
The situation becomes even more interesting when considering proteins designed to bind ZnII. Important examples for our studies are the ZnS3O site of hepatitis C virus NS3 proteinase and an HIV-1 integrase mutant which also forms a ZnS3O structure.[22, 61] While it has become commonplace to substitute CdII for ZnII in proteins like these, the replacement is not completely benign. CdII is larger and has longer bond lengths than ZnII (e.g., for CdS3O the Cd–S bond is approximately 2.54 Å whereas the corresponding ZnII distance is 2.32 Å).[42, 62] Furthermore, CdII is somewhat softer than ZnII and prone to form higher coordination number complexes. In the case of substitution into an identical protein environment the most likely concern is size. Size can shift the preference of a metal from an exo to an endo conformation as has been reported for As(CSL9C)3.[50] The differential size may also restrict the ability for an exogenous ligand such as water to bind to the metal. Thus, in metal-binding sites such as those found in the TRI peptides, steric constraints appear to favour the formation of a significant percentage of CdS3 centers (e.g., TRIL16C). The 0.2 Å shorter bond lengths of ZnS3O should not be as influenced by this steric constraint, suggesting that ZnII might bind to all, or most, of these designed peptides as a four coordinate complex with bound water. What these data would suggest is that in cases where an equilibrium process is in effect for CdII in a protein, cadmium substitution may not provide fully appropriate information about the zinc environment.
We have proposed a number of tantalizing questions which may have profound impact for understanding biological cadmium coordination chemistry, the interpretation of 113Cd NMR spectra and the application of CdII substitution as a reporter for the highly prevalent ZnII class of metalloproteins. However, there are some caveats that must be considered at this stage. While we have a reasonable explanation for the large chemical shift range for at least one coordination environment (ostensibly CdS3O) and have enumerated several factors that contribute to the chemical shift variation, our correlation is, in fact, limited to CdS3 trigonal planar to CdS3O pseudotetrahedral structures found in “a” type sites, presumably with the metal in an exo configuration. One might expect some variation to our reported trends if the metal is located in “d” sites or in an endo conformation. Furthermore, other factors such as the change in orientation of thiolate donors on converting from parallel to antiparallel helices, and hydrogen bonding could further broaden the chemical shift ranges discussed here. Because of the importance of zinc metalloproteins and the applications of CdII for understanding the metal center structure and function, it is clear that additional studies addressing each of these points is essential. We have shown how de novo designed peptides can be powerful vehicles to examine these important issues. We expect that future studies with variants of these peptides will further elucidate this fascinating and important problem.
Experimental Section
Peptide synthesis and purification
The TRI peptides and derivatives (see Table 1 for sequence nomenclature) were synthesized on an Applied Biosystems 433 A peptide synthesizer using standard F-moc protocols,[63] and purified and characterized as described previously.[47]
PAC spectroscopy
All perturbed angular correlation (PAC) experiments were performed with a setup using six detectors and a temperature of 1 ± 2 °C, which was controlled by a Peltier element. The radioactive cadmium was produced on the day of the experiment at the University Hospital cyclotron in Copenhagen and extracted as described previously,[64] except for the HPLC separation of zinc and cadmium which was omitted. This procedure may lead to zinc contamination of the sample, but the level of contamination should not interfere with the experiment. The 111mCd solution (10–40 µL) was mixed with nonradioactive cadmium acetate and TRIS buffer. The peptide, dissolved in ion-exchanged water, was then added and the sample was left to equilibrate for 10 min to allow for metal binding. Finally, sucrose was added to produce a 55% w/w solution. The pH of the solution was adjusted with H2SO4 or KOH. In order to avoid chloride contamination, a small volume of sample was removed from the solution and the pH measured. The pH reported in Table 2 was measured at room temperature the following day and corrected to the pH at 1 °C as follows. The pH of solutions buffered by TRIS is temperature dependent; therefore the pH of the solutions at 1 °C was calculated using the following equation pH(1 °C) 0.964[pH(25 °C)] + 0.86.[65] The samples were either used immediately after preparation or left on ice for up to 2 h until the measurement was started. All buffers were purged with Ar and treated so as to lower metal contamination. The final volume of the samples ranged between 0.05 and 0.5 mL with concentrations of 300 µm peptide, 20 mm TRIS buffer and a CdII/peptide ratio of 1:12. All fits were carried out with 400 data points, disregarding the 3–5 first points due to systematic errors in these. For the TRIL12AL16Pen peptide, 300 points were used in the fit.
Each nuclear quadrupole interaction (NQI) was modelled using a separate set of parameters that includes ω0, η, Δω0/ω0, 1/τc and A. The parameter ω0 [ω0 = 12π | eQVzz | /(40 h), where Q is the nuclear electric quadrupole moment and Vzz is the numerically largest eigenvalue of the electric field gradient tensor] is associated with the strength of the interaction between the surrounding electronic environment and the Cd nucleus, η is the so-called asymmetry parameter which is 0 in an axially symmetric complex and has a maximal value of 1; Δω0/ω0 describes static structural variations from one CdII site to the next, and is as such a measure of the structural variability; τc is the rotational correlation time; and A is the amplitude of the signal (see ref. [25] for a more detailed description). The parameters fitted to the PAC data are presented in Table 2. The percentage of each species reported in Table 3 were calculated based on the A value.
113Cd NMR spectroscopy
All the spectra were collected at room temperature on a Varian Inova 500 spectrometer (110.92 MHz for 113Cd) equipped with a 5 mm broadband probe. 113Cd NMR spectra were externally referenced to a 0.1 m Cd(ClO4)2 solution in D2O. A spectral width of 847 ppm (93 897 Hz) was sampled using a 5.0 µs 90° pulse and 0.05 s acquisition time with no delay between scans. Samples were prepared under a flow of argon or nitrogen by dissolving 30–35 mg of the lyophilized and degassed peptides in 450–500 µL 15% D2O solution. The peptide concentrations were determined by using either the Ellman’s[66] or 4,4′-dipyridyl disulphide tests,[67] and the concentrations range from 12 to 18 mm peptide, which corresponds to 4–6 mm three-stranded coiled coil. The final samples were prepared by the addition of the appropriate amount of 250 mm 113Cd(NO3)2 solution (prepared from 95% isotopically enriched 113CdO obtained from Oak Ridge National Laboratory) and the adjustment of the pH with KOH or HCl solutions. The final pH value for each peptide was chosen based on its pH titration curves. This pH value correspond to the fully formation of the CdII complex. An Argon or Nitrogen atmosphere was maintained when possible but the samples came in contact with O2 while the pH was adjusted. The actual final concentrations for each experiment are indicated in the text and the Figure captions. The data were analyzed using the software MestRe-C.[68] All free induction decays (FID’s) were zero filled to double the original points and were processed by application of 50 Hz line broadening prior to Fourier transformation.
UV/Vis spectroscopy
All the UV/Vis pH titration experiments were carried out at room temperature on an Ocean Optics SD 2000 fiber optic spectrometer. Fresh stock solutions of the purified peptides were prepared for each experiment using doubly distilled water and were purged with argon to minimize the chances of oxidation. The peptide concentration was determined as described in the 113Cd NMR section.[66, 67] pH Titrations were performed by adding small aliquots of KOH (1 mm to 1 m stock solutions) to unbuffered solutions containing CdCl2 salt (20 µm) and peptide (60–120 µm). All the doubly distilled water used in these experiments was bubbled with Ar before addition of CdCl2 and peptide. The change in pH was monitored using an Accumet gel-filled pencil-thin Ag/AgCl single-junction electrode with an Orion Research digital pH millivolt meter 611. In all cases, equilibration time was allowed before reading the final pH and it was found that binding of CdII to TRIL16Pen, TRIL12VL16Pen and GrandL16Pen was slower than binding the other peptides. To verify the reversibility of the process, reverse titrations were carried out subsequently in all the experiments by adding small aliquots of 1 mm to 1 m solutions of HCl. The pH-dependent absorption spectra were fit using the same models and procedures as those used in our previous studies.[27, 29] Specifically, the experimental data were analyzed by non-linear least square fitting to the following equation:
Cd[(peptide−H)2(peptide)]+ = Cd(peptide)3− + 2H+ Abs = (c[M]t*Δε)/(10(pKa2−2pH)+1)
where Abs is the observed absorbance after addition of KOH, c[M]t is the total metal concentration, Δε is the extinction coefficient of the metal complex, and pKa2 the acidity constant for the simultaneous release of two protons. The data were also fit using an extended model including additional species in solution (see Supporting Information for a full description).
Acknowledgements
V.L.P. thanks the National Institute of Health for support of this research (R01 ES0 12236), O.I. thanks the Margaret and Herman Sokol Foundation for a Postdoctoral Award and L.H. thanks The Danish Natural Science Research Council for support.
Footnotes
PAC Spectroscopy = perturbed angular correlation spectroscopy Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.200802105: Models and equations for extended model calculations in order to fit the UV/Vis pH titration curves; pK for several equilibria determined for several peptides: 1) TRIL16Pen, 3) GRANDL16Pen, 4) TRIL16CL23A, 6) BABYL9C, 10) GRANDL9C, 12) TRIL9AL16C, 13) TRIL12AL16Pen, 14) TRIL12AL16C.
References
- 1.Vallee BL, Auld DS. Biochemistry. 1990;29:5647–5659. doi: 10.1021/bi00476a001. [DOI] [PubMed] [Google Scholar]
- 2.Lipscomb WN, Sträter N. Chem. Rev. 1996;96:2375–2434. doi: 10.1021/cr950042j. [DOI] [PubMed] [Google Scholar]
- 3.Coleman JE. Curr. Opin. Chem. Biol. 1998;2:222–234. doi: 10.1016/s1367-5931(98)80064-1. [DOI] [PubMed] [Google Scholar]
- 4.Berg JM, Shi Y. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 5.Klug A. J. Mol. Biol. 1999;293:215–218. doi: 10.1006/jmbi.1999.3007. [DOI] [PubMed] [Google Scholar]
- 6.McCall KA, Huang CC, Fierke CA. J. Nutr. 2000;130:1437S–1446S. doi: 10.1093/jn/130.5.1437S. [DOI] [PubMed] [Google Scholar]
- 7.Tobin DA, Pickett JS, Hartman HL, Fierke CA, Penner-Hahn JE. J. Am. Chem. Soc. 2003;125:9962–9969. doi: 10.1021/ja035927o. [DOI] [PubMed] [Google Scholar]
- 8.Sousa SF, Fernandes PA, Ramos MJ. J. Biol. Inorg. Chem. 2005;10:3–10. doi: 10.1007/s00775-004-0612-6. [DOI] [PubMed] [Google Scholar]
- 9.Myers LC, Terranova MP, Nash HM, Markus MA, Verdine GL. Biochemistry. 1992;31:4541–4547. doi: 10.1021/bi00134a002. [DOI] [PubMed] [Google Scholar]
- 10.Penner-Hahn J. Curr. Opin. Chem. Biol. 2007;11:166–171. doi: 10.1016/j.cbpa.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 11.Andreini C, Banchi L, Bertini I, Rosato A. J. Proteome Res. 2006;5:196–201. doi: 10.1021/pr050361j. [DOI] [PubMed] [Google Scholar]
- 12.Ecoleman J. Methods Enzymol. 1993;227:16–43. doi: 10.1016/0076-6879(93)27004-z. [DOI] [PubMed] [Google Scholar]
- 13.Maret W, Vallee BL. Methods Enzymol. 1993;226:52–71. doi: 10.1016/0076-6879(93)26005-t. [DOI] [PubMed] [Google Scholar]
- 14.Summers MF. Coord. Chem. Rev. 1988;86:43–134. [Google Scholar]
- 15.Öz G, Pountney DL, Armitage IM. Biochem. Cell Biol. 1998;76:223–243. doi: 10.1139/bcb-76-2-3-223. [DOI] [PubMed] [Google Scholar]
- 16.Hemmingsen L, Nárcisz K, Danielsen E. Chem. Rev. 2004;104:4027–4061. doi: 10.1021/cr030030v. [DOI] [PubMed] [Google Scholar]
- 17.Hemmingsen L, Damblon C, Antony J, Jensen M, Adolph HW, Wommer S, Roberts GCK, Bauer R. J. Am. Chem. Soc. 2001;123:10329–10335. doi: 10.1021/ja0112240. [DOI] [PubMed] [Google Scholar]
- 18.Kofod P, Bauer R, Danielsen E, Larsen E, Bjerrum MJ. Eur. J. Biochem. 1991;198:607–611. doi: 10.1111/j.1432-1033.1991.tb16057.x. [DOI] [PubMed] [Google Scholar]
- 19.Bauer R, Bjerrum MJ, Danielsen E, Kofod P. Acta Chem. Scand. 1991;45:593–603. doi: 10.3891/acta.chem.scand.45-0593. [DOI] [PubMed] [Google Scholar]
- 20.Busenlehner LS, Cosper NJ, Scott RA, Rosen BP, Wong MD, Giedroc DP. Biochemistry. 2001;40:4426–4436. doi: 10.1021/bi010006g. [DOI] [PubMed] [Google Scholar]
- 21.Erskine PT, Norton E, Cooper JB, Lambert R, Coker A, Lewis G, Spencer P, Sarwar M, Wood SP, Warren MJ, Shoolingin-Jordan PM. Biochemistry. 1999;38:4266–4276. doi: 10.1021/bi982137w. [DOI] [PubMed] [Google Scholar]
- 22.Stempniak M, Hostomska Z, Nodes BR, Hostomsky Z. J. Virol. 1997;71:2881–2886. doi: 10.1128/jvi.71.4.2881-2886.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, O’Halloran TV, Penner-Hahn JE, DeGrado WF, Pecoraro VL. J. Am. Chem. Soc. 1997;119:6195–6196. [Google Scholar]
- 24.a) Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL. J. Mol. Biol. 1998;280:897–912. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]; b) Peacock AFA, Iranzo O, Pecoraro VL. Dalton Trans. 2009 doi: 10.1039/b818306f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matzapetakis M, Farrer BT, Weng T-C, Hemmingsen L, Penner-Hahn JE, Pecoraro VL. J. Am. Chem. Soc. 2002;124:8042–8054. doi: 10.1021/ja017520u. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh D, Lee K-H, Demeler B, Pecoraro VL. Biochemistry. 2005;44:10732–10740. doi: 10.1021/bi0506674. [DOI] [PubMed] [Google Scholar]
- 27.Matzapetakis M, Ghosh D, Weng K-H, Penner-Hahn JE, Pecoraro VL. J. Biol. Inorg. Chem. 2006;11:876–890. doi: 10.1007/s00775-006-0140-7. [DOI] [PubMed] [Google Scholar]
- 28.Lee K-H, Matzapetakis M, Mitra S, Marsh ENG, Pecoraro VL. J. Am. Chem. Soc. 2004;126:9178–9179. doi: 10.1021/ja048839s. [DOI] [PubMed] [Google Scholar]
- 29.Iranzo O, Ghosh D, Pecoraro VL. Inorg. Chem. 2006;45:9959–9973. doi: 10.1021/ic061183e. [DOI] [PubMed] [Google Scholar]
- 30.Lee K-H, Cabello C, Hemmingsen L, Marsh ENG, Pecoraro VL. Angew. Chem. 2006;118:2930–2934. doi: 10.1002/anie.200504548. Angew. Chem. Int. Ed.2006, 45, 2864 –2868. [DOI] [PubMed] [Google Scholar]
- 31.Iranzo O, Cabello C, Pecoraro VL. Angew. Chem. 2007;119:6808–6811. doi: 10.1002/anie.200701729. Angew. Chem. Int. Ed.2007, 46, 6688 –6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matthews RG, Goulding CW. Curr. Opin. Chem. Biol. 1997;1:332–339. doi: 10.1016/s1367-5931(97)80070-1. [DOI] [PubMed] [Google Scholar]
- 33.Erskine PT, Senior N, Awan S, Lambert R, Lewis G, Tickle IJ, Sarwar M, Spencer P, Thomas P, Warren MJ, Shoolingin-Jordan PM, Wood SP, Cooper JB. Nat. Struct. Biol. 1997;4:1025–1031. doi: 10.1038/nsb1297-1025. [DOI] [PubMed] [Google Scholar]
- 34.González B, Pajares MA, Martínez-Ripoll M, Blundell TL, Sanz-Aparicio J. J. Mol. Biol. 2004;338:771–782. doi: 10.1016/j.jmb.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 35.Kim JL, Morgenstern KA, Lin1 TFC, Dwyer MD, Landro JA, Chambers SP, Markland W, Lepre CA, O’Malley ET, Harbeson SL, Rice CM, Murcko MA, Caron PR, Thomson JA. Cell. 1996;87:343–355. doi: 10.1016/s0092-8674(00)81351-3. [DOI] [PubMed] [Google Scholar]
- 36.Rice WG, Supko JG, Malspeis L, Clanton RWBD, Jr, Bu M, Graham L, Schaeffer CA, Turpin JA, Domagala J, Gogliotti R, Bader JP, Halliday SM, Coren L, S RC, II, Arthur LO, Henderson LE. Science. 1995;270:1194–1197. doi: 10.1126/science.270.5239.1194. [DOI] [PubMed] [Google Scholar]
- 37.Cai M, Zheng R, Caffrey M, Craigie R, Clore GM, Gronenborn AM. Nat. Struct. Biol. 1997;4:567–577. doi: 10.1038/nsb0797-567. [DOI] [PubMed] [Google Scholar]
- 38.Santos RA, Gruff ES, Koch SA, Harbison GS. J. Am. Chem. Soc. 1991;113:469–475. [Google Scholar]
- 39.Alberts IL, Nadassy K, Wodak SJ. Protein Sci. 1998;7:1700–1716. doi: 10.1002/pro.5560070805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coyne HJ, III, Ciofi-Baffoni S, Banci L, Bertini I, Zhang L, George GM, Winge DR. J. Biol. Chem. 2007;282:8926–8934. doi: 10.1074/jbc.M610303200. [DOI] [PubMed] [Google Scholar]
- 41.Castro C, Millian NS, Garrow TA. Arch. Biochem. Biophys. 2008;472:26–33. doi: 10.1016/j.abb.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parkin G. Chem. Rev. 2004;104:699–767. doi: 10.1021/cr0206263. [DOI] [PubMed] [Google Scholar]
- 43.Docommun Y, Merbach AE, et al. In: Inorganic High Pressure Chemistry: Kinetics and Mechanisms. Eldik Rv., editor. Amsterdam: Elsevier; 1986. p. 70. [Google Scholar]
- 44.Peacock AFA, Hemmingsen L, Pecoraro VL. Proc. Natl. Acad. Sci. USA. 2008;105:16566–16571. doi: 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boggess RK, Absher JR, Morelen S, Taylor LT, Hughes JW. Inorg. Chem. 1983;22:1273–1279. [Google Scholar]
- 46.Cheesman BV, Arnold AP, Rabenstein DL. J. Am. Chem. Soc. 1988;110:6359–6364. [Google Scholar]
- 47.Farrer BT, Harris NP, Balchus KE, Pecoraro VL. Biochemistry. 2001;40:14696–14705. doi: 10.1021/bi015649a. [DOI] [PubMed] [Google Scholar]
- 48.Ghosh D, Pecoraro VL. Inorg. Chem. 2004;43:7902–7915. doi: 10.1021/ic048939z. [DOI] [PubMed] [Google Scholar]
- 49.Iranzo O, Thulstrup PV, Ryu S-B, Hemmingsen L, Pecoraro VL. Chem. Eur. J. 2007;13:9178–9190. doi: 10.1002/chem.200701208. [DOI] [PubMed] [Google Scholar]
- 50.Touw DS, Nordman CE, Stuckey JA, Pecoraro VL. Proc. Natl. Acad. Sci. USA. 2007;104:11969–11974. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farrer BT, McClure CP, Penner-Hahn JE, Pecoraro VL. Inorg. Chem. 2000;39:5422–5423. doi: 10.1021/ic0010149. [DOI] [PubMed] [Google Scholar]
- 52.Shaikh TA, Bakus RC, II, Parkin S, Atwood DA. J. Organomet. Chem. 2006;691:1825–1833. [Google Scholar]
- 53.Shaikh TA, Parkin S, Atwood DA. J. Organomet. Chem. 2006;691:4167. [Google Scholar]
- 54.Shi W, Dong J, Scott RA, Ksenzenko MY, Rosen BP. J. Biol. Chem. 1996;271:9291–9297. doi: 10.1074/jbc.271.16.9291. [DOI] [PubMed] [Google Scholar]
- 55.Carter TG, Healey ER, Pitt MA, Johnson DW. Inorg. Chem. 2005;44:9634–9636. doi: 10.1021/ic051522o. [DOI] [PubMed] [Google Scholar]
- 56.Erskine PT, Duke EMH, Tickle IJ, Senior NM, Warren MJ, Cooper JB. Acta Crystallogr. Sect. D. 2000;56:421–430. doi: 10.1107/s0907444900000597. [DOI] [PubMed] [Google Scholar]
- 57.Dean PAW, Vittal JJ, Payne NC. Inorg. Chem. 1984;23:4232–4236. [Google Scholar]
- 58.Christou G, Folting K, Huffman JC. Polyhedron. 1984;3:1247–1253. [Google Scholar]
- 59.Magyar JS, Weng T-C, Stern CM, Dye DF, Rous BW, Payne JC, Bridgewater BM, Mijovilovich A, Parkin G, Zaleski JM, Penner-Hahn JE, Godwin HA. J. Am. Chem. Soc. 2005;127:9495–9505. doi: 10.1021/ja0424530. [DOI] [PubMed] [Google Scholar]
- 60.Busenlehner LS, Weng T-C, Penner-Hahn JE, Giedroc DP. J. Mol. Biol. 2002;319:685–701. doi: 10.1016/S0022-2836(02)00299-1. [DOI] [PubMed] [Google Scholar]
- 61.Cai M, Huang Y, Caffrey M, Zheng R, Craigie R, Clore GM, Gronenborn AM. Protein Sci. 1998;7:2669–2674. doi: 10.1002/pro.5560071221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Govindaswamy N, Moy J, Millar M, Koch SA. Inorg. Chem. 1992;31:5343–5344. [Google Scholar]
- 63.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. New York: Oxford University Press; 2000. [Google Scholar]
- 64.Hemmingsen L, Bauer R, Bjerrum MJ, Zeppezauer M, Adolph HW, Formicka G, Cedergren-Zeppezauer E. Biochemistry. 1995;34:7145–7153. doi: 10.1021/bi00021a028. [DOI] [PubMed] [Google Scholar]
- 65.Hemmingsen LB, Bjerrum R, Adolph M, Zeppezauer H, Cedergren-Zeppezauer ME. Eur. J. Biochem. 1996;241:546–551. doi: 10.1111/j.1432-1033.1996.00546.x. [DOI] [PubMed] [Google Scholar]
- 66.Ellman GL. Arch. Biochem. Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 67.Mantle M, Stewart G, Zayas G, King M. Biochem. J. 1990;266:597–604. [PMC free article] [PubMed] [Google Scholar]
- 68.Cobas C, Cruces J, Sardina FJ. MestRe-C Version 2.3. Spain: Universidad de Santiago de Compostela; 2000. [Google Scholar]