

Abstract
Polymer nanoparticles (PNPs) possessing a high density of drug payload have been successfully stabilized against aggregation in biological buffers after amine modification, which renders these PNPs positively charged. The resulting charge-stabilized PNPs retain their original narrow particle size distributions and well-defined spherical morphologies. This stabilization allows these PNPs to have an improved anti-proliferative effect on MDA-MB-231-Br human breast cancer cells compared to non-functionalized PNPs. As a non-cytotoxic control, similar surface-modified PNPs containing cholesterol in place of doxorubicin did not inhibit cell proliferation, indicating that the induced cytotoxic response was solely due to the doxorubicin release from the PNPs.
Introduction
In recent years, drug-loaded nanoparticles have been investigated extensively for anticancer therapy due to their promise as carriers for chemotherapeutics,1 proteins,2 and peptides.3 However, the lack of stability in ionic aqueous media has proven to be a major challenge for several of these delivery systems, including polycaprolactone nanoparticles,4 iron-oxide nanoparticles,5 carbon nanotubes,6 and graphene oxide,7 rendering them impractical for in vivo8 and clinical9 studies. In our own work, the aggregation of doxorubicin-containing polymer nanoparticles (PNPs) under physiologically relevant conditions (i.e., in PBS buffer where [NaCl] ~150 mM) has greatly limited their application as an in vivo therapeutic delivery platform,10 even though they have been shown to have good in vitro activity against cancer cells.11 Thus, we set out to explore modification strategies (Scheme 1) that can improve the dispersion stability of our PNPs in ionic, biological media.
Scheme 1.

Schematic illustration of possible steric and electrostatic strategies for stabilizing doxorubicin-containing PNPs in biological buffers: Steric stabilization through: (a, top) increasing the length of the hydrophilic block and (a, bottom) using polymer stabilizer Pluronic F68. Electrostatic stabilization through: (b, top) introducing charged groups in the hydrophilic block prior to PNP formation and (b, bottom) using amino surface modifiers.
The dispersibility of nanoparticles in solution is strongly dependent on the ability of the media to stabilize them against attractive interparticle interactions.12 Specifically, the Derjaguin, Landau, Verwey, and Overbeek (DLVO) theory suggests that nanoparticles will stay dispersed in media if repulsive interactions between the nanoparticles dominate over attractive van der Waals forces.13 To this end, both steric7, 14, 15 and electrostatic7, 16, 17 repulsion have been used to stabilize several types of nanoparticles in buffer solutions. As shown herein, the flexibility of our PNP fabrication method18 allows us to easily implement both of these strategies in our quest to prevent PNP aggregation. Given that positively charged nanoparticles are known to exhibit enhanced uptake in cell studies,19 we were particularly interested in the possibility to additionally improve the cellular uptake, and hence cytotoxicity, of our doxorubicin-containing PNPs upon engendering them with cationic groups. In this paper, we report the stable dispersion of positively charged doxorubicin-containing PNPs, synthesized via the modification of tosyl-functionalized PNPs with primary amines. Notably, these modified PNPs exhibited a better anti-proliferative effect in comparison to the parent PNPs.
Results and Discussion
As illustrated in Scheme 1, our doxorubicin-containing PNPs can be modified either before or after nanoparticle formation. In the pre-nanoparticle-formation protocol, the length of the poly(ethylene glycol) (PEG) side chain in the hydrophilic component of the amphiphilic doxorubicin-containing block copolymer can be increased to improve the dispersibility of the resulting PNPs in buffer (Scheme 1a, top). Similarly, a charged block copolymer can be synthesized by modifying the ends of the PEG side chains (Scheme 1b, top).
In the post-nanoparticle-formation case (Scheme 1a, bottom), an amphiphilic block copolymer such as Poloxamer 188 can be physically adsorbed onto the surface of the PNPs after their formation. The resulting adsorbed polymer layer has been known to impart a steric barrier and prevent nanoparticle destabilization.9 More attractive, however, is the introduction of tunable cationic groups to the surface of the PNPs via the displacement of a surface leaving group with amines: a wide range of available amine nucleophiles, with different substituents and degrees of substitution, can be used to modify a single PNP material (Scheme 1b, bottom). Such tunability would allow us to quickly determine the cationic groups (CGs) that provide the best stability to our PNPs in ionic media.
As additional design criteria, it is essential that the modification process maintains the high drug-to-mass ratio (~50 wt%)18 of the doxorubicin-containing PNPs without increasing their sizes and densities. High drug loadings are deemed desirable in nanoscale delivery platforms, especially in extended-release applications.20 Not increasing the size or density of the PNPs is also important because larger21–23 and heavier24–27 particles have been known to adversely affect cellular uptake profiles.
Previously, we demonstrated that monodisperse drug-containing amphiphilic block copolymers such as methoxy-terminated 215-b-315 (Scheme 2a) can undergo directed assembly into therapeutically active core-shell PNPs with uniform, tunable diameters.18, 28 Because 215-b-315 could not be modified further, it cannot be used as a single starting material for evaluating the effectiveness of the aforementioned modification methods in maintaining the dispersibility of our doxorubicin-containing PNPs. For such a purpose, we need to employ a single copolymer material that can be used to create several PNP formulations. Hence, we incorporated a third tosyl-functionalized monomer 4 into our amphiphilic copolymer, wherein the tosylate groups can be easily displaced by nucleophilic reagents to create surface cationic groups.
Scheme 2.
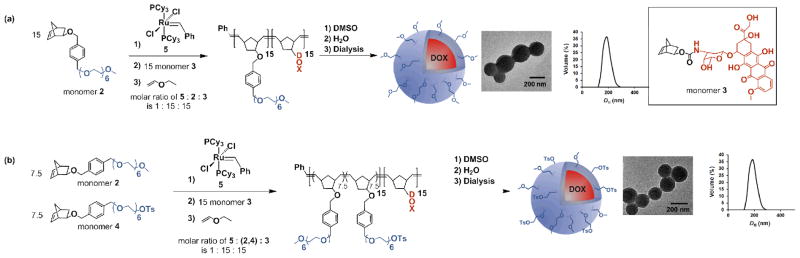
(a) The preparation of PNPs from methoxy-terminated 215-b-315 block copolymer. (b) The preparation of PNPs from tosyl-functionalized (2-co-4)15-b-315 block copolymer. At the end of each sequence, the first image is a representative TEM image of the PNP (see Fig. S4 in the ESI† for an expanded TEM image of the PNPs prepared from (2-co-4)15-b-315) and the second image is a plot of its DH distribution, as measured by DLS.
Preparation of tosyl-functionalized doxorubicin-containing PNPs
Monomers 2 and 4 (for characterization data, see Fig. S1 in the ESI† and the associated discussion) were first copolymerized using the 1st-generation Grubbs’ catalyst 5, followed by the sequential addition and polymerization of monomer 3 (Scheme 2b).10 The resulting block copolymer, (2-co-4)15-b-315 (for characterization data, see Fig. S2 in the ESI† and the associated discussion), contains a block of the hydrophilic comonomers 2 and 4 and a second block of the hydrophobic monomer 3 (first block theoretical Mn = 9000, observed Mn = 10000, polydispersity index (PDI) = 1.11; final polymer theoretical Mn = 19000, observed Mn = 19000, PDI = 1.13). As previously observed, 18 copolymerization reactions initiated with the hydrophobic monomer 3 did not proceed to completion (see Fig. S3 in the ESI† and the associated discussion), necessitating the need for the hydrophilic monomers to be copolymerized first. To maintain an optimal balance between the ability to synthesize a monodisperse, well-defined copolymer and the charge-stabilization of the final PNP (Section III in the ESI†), we employed a 1:1 stoichiometric ratio of monomers 2 and 4.
Following our previously established strategy,28 an aqueous suspension of tosyl-functionalized, doxorubicin-containing PNPs was obtained by dropwise addition of water to a DMSO solution of copolymer (2-co-4)15-b-315, followed by exhaustive dialysis against ultrapure deionized water (Scheme 2b). Dynamic light scattering (DLS) measurements indicated a narrow size distribution (PDI = 0.036) of particles centered at DH = 211 nm (Scheme 2b, right inset). Transmission electron microscopy (TEM, Scheme 2b, left inset) also confirmed the homogeneous spherical morphology of these PNPs with diameters of ~200 nm.
PNP aggregation and attempted steric stabilization route
As mentioned in the introduction, we previously observed the aggregation of methoxy-terminated, doxorubicin-containing PNPs derived from copolymer 215-b-315 in cell-culture media, which resulted in a non-uniform cellular uptake.11 Attempts to disperse these PNPs, or the tosyl-functionalized, doxorubicin-containing PNPs derived from copolymer (2-co-4)15-b-315, in PBS buffer (10 mM, pH 7.4, 150 mM [NaCl]) also led to PNP aggregation (Table 1, cf entries 1–2, Fig. S4 in the ESI†).
Table 1.
The size distributions, PDI, and surface zeta potentials of the various surface-modified PNPs suspended in PBS buffer.
| Entry | Modified PNP | Size in PBS buffer (nm) | PDI | Zeta potential, ζ (mv) | |
|---|---|---|---|---|---|

|
1 | PEO | 534 ±27 | 0.430 | −17±1.8 |
| 2a | Unmodified | 895 ± 98 | 0.904 | −16±1.4 | |
| 2b | Unmodified | (211 ± 13)* | (0.036) | - | |
| 3 | NEt3 | 191 ±15 | 0.004 | +32 ± 0.8 | |
| 4 | HNEt2 | 203 ± 21 | 0.040 | +43±1.1 | |
| 5 | H2NEt | 210±22 | 0.034 | +55 ±1.6 | |
| 6 |
|
219±27 | 0.080 | +52 ±1.7 | |
| 7 |
|
209 ±15 | 0.024 | +54±1.3 | |
|
| |||||

|
8a | Unmodified | 118±9 | 0.024 | +58 ±3.2 |
| 8b | Unmodified | (113 ±13)* | (0.017) | - | |
| 9 |
|
1310 ±130 | 0.277 | +41 ±1.5 | |
| 10 |
|
2356± 115 | 0.356 | −17± 1.3 | |
in ultrapure deionized water
We first investigated the use of a surface stabilizer to reduce the aggregation of doxorubicin-containing PNPs and maintain their dispersion stability in PBS buffer.29–31 Poloxamer 188 (Pluronic F68),31 a well-known biocompatible triblock copolymer, was successfully incorporated into both methoxy- and tosyl-functionalized PNP matrices (215-b-315 and (2-co-4)15-b-315, respectively), either during the PNP fabrication or post-particle formation (Fig. S5 in the ESI†). While the resulting Poloxamer-incorporated PNPs were stable in water, re-suspending them in PBS buffer afforded only aggregated structures, as observed by both DLS and TEM (Figs. S6–S8 in the ESI†). Even at 100 wt% Poloxamer incorporation, which drastically decreases the drug loading capacity per particle (50 wt% to 25 wt%), the steric protection was inadequate to prevent aggregation.
We also attempted to achieve steric stabilization of PNPs by replacing the PEG group of hydrophilic monomer 2 with a much longer side chain (PEG 1000 instead of PEG 282;32 for characterization data, see Figs. S9–S11 in the ESI† and the associated discussion). Unfortunately, doxorubicin-containing PNPs prepared with this new monomer 7 are also unstable in PBS buffer. TEM analysis suggests that these PNPs have highly irregular surface features (Fig. S11 in the ESI†), which may contribute to their inability to resist aggregation in buffer.
Electrostatic stabilization of PNPs
We then turned to electrostatic stabilization as a means to stabilize PNP dispersion. Nanoparticles with |zeta potential| >30 mV have been found to maintain good colloidal stability in buffers and can be used for in vitro studies.33 While the surface of our PNPs can be modified with either cationic or anionic charged groups, negatively charged nanoparticles are known to suffer from poor cellular uptake due to their low affinity towards the negatively charged cell membrane.34 In contrast, cationic nanoparticles would have stronger electrostatic interactions with the cell membrane and a better chance to translocate.35 Indeed, positively charged nanoparticles have been shown to have an enhanced ability to penetrate the cell membrane and undergo cellular internalization.36–39
To introduce positive charges on the PNP surface, we initially synthesized a cationic polymer from the parent tosyl-bearing copolymer (2-co-4)15-b-315 by converting its tosylate groups to cationic ammonium moieties (for characterization data, see Fig. S12 in the ESI† and the associated discussion). The reaction of excess diethylamine with (2-co-4)15-b-315 afforded ~80% ammonium groups. While this ionic copolymer can be processed into PNPs, the polydispersity is high (0.22, see Fig. S13 in the ESI†), prompting us to pursue an alternate path that would engender the PNP surface with cationic moieties.
Since doxorubicin-containing PNPs with exposed tosyl groups on the surface can be made easily with low dispersity,10 we reasoned that exposing these to amines could result in new PNPs with cationic surface ammonium groups that are buffer-stable. Indeed, treating the aforementioned tosyl-bearing PNPs, derived from block copolymer (2-co-4)15-b-315, with aqueous solutions of triethylamine, diethylamine, ethylamine, 2-methoxyethylamine, or 2-methylthioethylamine over a period of 72 h (Scheme 3) resulted in materials that do not visibly aggregate in PBS buffer (Figs. S14–S18 in the ESI†), in contrast to that observed for the parent PNPs. TEM image (Scheme 3a; see also Figs. S14a-S18a in the ESI†) and size distribution plot as obtained by DLS (Scheme 3b; see also Figs. S14b-S18b in the ESI†) demonstrates the retention of the discrete and spherical morphology of these PNPs with diameters of ~200 nm and low PDI (0.004–0.080). The zeta potentials of these PBS-resuspended PNPs are positive (Table 1, cf entries 3–7), suggesting that the nucleophilic amines were covalently linked to the PNP surface by displacement of the tosyl leaving groups, resulting in PNPs functionalized with cationic ammonium moieties.
Scheme 3.

The modification of tosyl-functionalized, doxorubicin-containing PNPs with amines. (a) a representative TEM image and (b) the DH distribution measured by DLS upon re-suspending the ethylamine-modified PNPs in PBS buffer (10 mM, pH 7.4, 150 mM [NaCl]). See Figs. S14–S18 in the ESI† for the data for other amine-modified materials, including expanded TEM images.
As shown in Table 1, the presence of charged groups on the ammonium-modified PNPs induces interparticle repulsion and increases their dispersibility in buffer solutions. The observed decrease in the surface zeta potential as the steric environment at the amine nitrogen becomes more hindered (Table 1, cf entries 3–5) is suggestive of a smaller number of charged groups on the PNP surface. That the three most stable doxorubicin-containing PNPs (see below) possess surface zeta potentials that are close to that of indomethacin-containing PNPs (Table 1, entry 8a), known to be dispersible in PBS buffer,40 is perhaps not coincidental. When compared to the surface zeta potential of the parent tosyl-functionalized PNPs (−16 mV),41 these values (52 to 55 mV) mirror the stark contrast in dispersibilities between the ammonium-modified materials and their parent PNPs: the low surface charge associated with the latter is perhaps insufficient to ensure its dispersibility in PBS buffers. Supporting this hypothesis is the observation that when the indomethacin-containing PNPs were modified with negative surface groups to reduce their intrinsically positive surface zeta potential, their dispersibilities are dramatically reduced (Table 1, cf entries 9–10).42
To estimate the number of surface cationic charges on the PNP surface, the parent tosyl-functionalized doxorubicin-containing PNPs were modified with three different propargylamines in increasing degree of substitutions (1°, 2°, 3°). The resultant “clickable” PNPs were then treated with folate-PEG-azide43 following a previously published protocol44 (Fig. 1, for characterization data, see Fig. S20 in the ESI† and the associated discussion). As expected, the propargylamine-modified PNPs were the most readily modified, with approximately 1200 coupled folate groups. As the steric environment around the amine nucleophile becomes more crowded, the number of coupled folate groups steadily decreased (Fig. 1), with ~650 for the methylpropargylamine-modified PNP and ~320 for the dimethylpropargylamine-modified PNP. Since the propargylamine-modified PNPs have similar zeta potentials (Fig. S20 in the ESI†) and are sterically and electronically analogous to those that have been modified with ethylamines, we assumed that the degree of surface amine modification is similar in both series of PNPs.
Fig. 1.

Top: A schematic illustration of the modification of alkyne-functionalized PNPs with folate-PEG-azide in the presence of a Cu/NaAsc catalyst system. Bottom: A comparison of the degree of modification of the different alkyne-functionalized PNPs with folate-PEG-azide, showing that maximum modification is achieved with propargylamine-modified PNPs. This is indicative of propargylamine being the most efficient in tosylate displacement on the PNP surface.
The aforementioned difference in conjugation efficiency for the propargyl amines can be attributed to the steric environment at the amine nitrogen: a hydrophobic, bulky trialkylamine is less likely to displace tosylate groups on the PNP surface under aqueous conditions compared to a more hydrophilic, smaller primary amine. As such, the use of trialkylamine modifiers may lead to insufficient coverage of charged groups on the PNP surface and cause premature aggregation. Additionally, the secondary and tertiary amine-modified PNPs would also be more hydrophobic overall in comparison to the primary amine-modified PNPs (there are more alkyl groups per charge), making them less stable in buffer.
To determine whether the number of surface ammonium groups affects PNP dispersibility, the size distributions of these modified PNPs in buffers were analyzed by DLS over a period of time. Indeed, it was found that the primary amine-modified formulations, with 1200 charged ammonium groups/nanoparticle, can maintain PNP stability in biological buffers for at least 96 h (Figs. S22c-S22e in the ESI†) while the other lower-charge formulations were not as stable (Figs. S22a and S22b in the ESI†).
Importantly, the amine modification does not significantly change the size and mass of our parent NPs, or their drug loading capacities. Although the primary amine modification afforded the highest number of ammonium groups on the PNP surface, it is only 1% of the total number of tosyl groups and added merely 0.02 wt% of mass to the PNP. Such an inconsequential increase in the overall PNP mass did not affect the original per-particle drug loading and is expected to result in only small changes in the total number of surface charged groups, thus minimizing charge-induced toxicities (see below). Together, these features make amine modification the most attractive stabilization strategy that we have explored in this study.
Determination of effect on cell viability and cellular localization of surface-modified doxorubicin-containing PNPs
The cytotoxicities of the parent tosyl-functionalized doxorubicin-containing PNPs and their ammonium derivatives were evaluated against MDA-MB-231-Br, a highly metastatic variant of MDA-MB-231 breast cancer cells.45 The relative cell viabilities were measured using MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay over a period of 72 h. The half-maximum inhibitory concentrations (IC50) of the PNPs were determined by plotting cell viability as a function of equivalent doxorubicin concentration and fitting the data to a model of dose-dependent inhibition of growth. The ethylamine-, 2-methoxyethylamine-and 2-methylthioethylamine-modified PNPs were effective in inhibiting cell growth with IC50 values of 19.1 ± 3.0 μM, 13.1 ± 3.3 μM, 12.9 ± 3.5 μM, respectively (Fig. 2). Under the same conditions, the cell viability plots for diethylamine- and triethylamine-modified PNPs (Fig. S26 in the ESI†) suggest that these formulations have no significant cytotoxicity, presumably due to their lower stabilities in the cell culture media (see above). We note that IC50 values could not even be obtained for the parent tosyl-modified PNPs or the methoxy-terminated PNPs due to their fast aggregation in cell culture media.
Fig. 2.
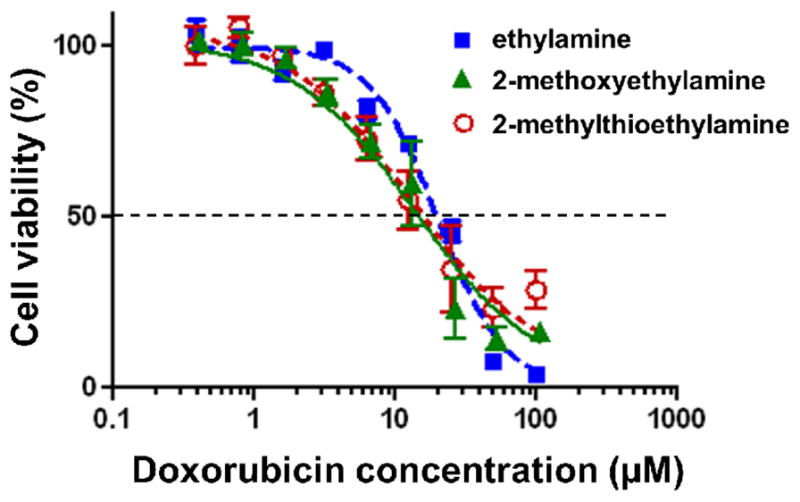
In vitro cyctotoxicity profiles of: (
 ) ethylamine- (IC50 = 19.1 ± 3.0 μM), (
) ethylamine- (IC50 = 19.1 ± 3.0 μM), (
 ) 2-methoxyethylamine- (IC50 = 13.1 ± 3.3 μM), and (
) 2-methoxyethylamine- (IC50 = 13.1 ± 3.3 μM), and (
 ) 2-methylthioethylamine-modified PNPs (IC50 = 12.9 ± 3.5 μM) against MDA-MB-231-Br cells. Cells were exposed to the PNPs for 72 h at 37 °C.
) 2-methylthioethylamine-modified PNPs (IC50 = 12.9 ± 3.5 μM) against MDA-MB-231-Br cells. Cells were exposed to the PNPs for 72 h at 37 °C.
Positively charged nanoparticles have been known to have some in vitro cytotoxicity due to their surface charges.46 In addition, the size and the type of nanoparticle may also induce any observed toxicity of the material.47 To determine whether the cationic nature of the charge-stabilized, doxorubicin-containing PNPs was causing the inhibitory effect in cell growth, a series of analogous ammonium-modified, cholesterol-containing PNPs were synthesized (for characterization data, see Figs. S24 and S25 and Table S1 in the ESI† and the associated discussion). Because we have previously employed cholesterol as an inert drug payload in the PNP platform,18 the use of these materials as a control should reveal whether a moderately charged PNP such as ours would have any cytotoxicity. As shown in the ESI† (Fig. S26), none of the cholesterol-containing PNP formulations exhibit cytotoxic effects over the tested concentration range (0.39–100 μM), indicating that doxorubicin was the active agent responsible for the observed cytotoxic response of ethylamine-, 2-methoxyethylamine-, a n d 2-methylthioethylamine-modified PNPs. This low toxicity is similar to those observed for other cationic nanoparticle systems with comparable surface zeta potentials.48, 49
That the anti-proliferative effect of the primary amine-modified PNPs may be attributed to their better dispersibilities in cell culture media, and presumably better cellular localization, is supported by confocal laser-scanning fluorescent microscopy (CLSM) experiments. As shown in Fig. 3 (see also Fig. S27 in the ESI†), CLSM images of MDA-MB-231-Br cells exposed to these formulations reveal that the surface-stabilized PNPs appeared to not only localize at the cancer cells, but also had uniform distribution throughout the cellular population (Fig. 3b), in clear contrast to the parent tosyl-functionalized PNPs (Fig. 3a).
Fig. 3.

Representative differential interference contrast and fluorescence microscopy images, based on the fluorescence of doxorubicin, of MDA-MB-231-Br cells incubated with (a) tosyl-functionalized PNPs and (b) ethylamine-modified PNPs showing the ability of the latter to localize at cancer cells more effectively than the former.
Conclusions
In summary, we have demonstrated that the dispersibility of doxorubicin-containing PNPs in biologically relevant media can be greatly increased through an electrostatic stabilization route that is easily implemented after nanoparticle formation. While the flexibility of our PNPs fabrication allows us to explore both electrostatic and steric stabilization through many different modification protocols, post-nanoparticle-formation modification is the most convenient protocol because it eliminates the additional synthesis of new monomers/polymers and potential challenges encountered in the formation of PNP from new polymer compositions.
Our data clearly indicates that per an added-mass basis the levels of stabilization afforded by charge repulsion were significantly better than those provided by steric hindrance. While significant PNP aggregation in ionic media still occurs in the presence of large amounts of Poloxamer (100 wt% of the drug-containing copolymer), good stabilization can be achieved with minimal amine modification of tosyl-functionalized PNP (only 0.02 wt% of mass was added in the case of ethylamine). Most importantly, the small number of charged groups introduced on the PNP surface via this method did not trigger charge- induced toxicity.
Among the amines explored for surface modification in this study, primary amines were the most effective in stabilizing PNPs. Presumably, the smaller steric demand of primary amines results in their higher coverage on the PNP surface and better PNP stabilization. These primary-amine-modified PNPs exhibited improved and uniform cellular localizations in cancer cells, leading to an enhanced inhibition of cell proliferation. Together with the aforementioned low charge-induced toxicity, this data suggests that post-synthesis modification with charged groups can be a highly useful strategy for increasing the biocompatibility and therapeutic potential of nanoparticle delivery systems.
Materials and methods
1-[4-({Bicyclo[2.2.1]hept-5-en-2-yloxy}methyl)phenyl]-2,5,8,11,14,17-hexaoxanonadecan-19-ol (1),40 1-[4-({bicyclo[2.2.1]hept-5-en-2-yloxy}methyl)phenyl]-2,5,8,11,14,17,20-heptaoxahenicosane (2),18 and bicyclo[2.2.1]hept-5-en-2-yl-N-(3-hydroxy-2-methyl-6-{[3,5,12-trihydroxy-3-(2-hydroxyacetyl)-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl]oxy}oxan-4-yl)carbamate (3)18 were prepared according to literature procedures.
Synthesis of 1-[4-({bicyclo[2.2.1]hept-5-en-2-yloxy}methyl)phenyl]-2,5,8,11,14,17-hexaoxanonadecan-19-yl 4 methylbenzene-1-sulfonate (4)
4-Toluenesulfonyl chloride (1.17 gm, 15.2 mmol) was added as a solid to a solution of 1-[4-({bicyclo[2.2.1]hept-5-en-2-yloxy}methyl)phenyl]-2,5,8,11,14,17-hexaoxanonadecan-19-ol (1, 150 mg, 0.303 mmol) in dry DCM (25 mL) in a 50-mL Schlenk flask. After the addition of triethylamine (0.93 mL, 6.67 mmol) using a gas-tight syringe, the reaction mixture was allowed to stir for 15 min at 0 °C. The resulting solution was allowed to stir at room temperature overnight before being concentrated and purified by flash chromatography (95:5 v/v CH2Cl2:MeOH) to afford monomer 4 as a dark yellow oil (178 mg, 90% yield). See the ESI† for 1H NMR, 13C NMR, and ESIMS data.
Synthesis of block copolymer (2-co-4)15-b-315
In an inert-atmosphere glovebox, monomers 2 (11.8 mg, 0.0231 mmol) and 4 (15 mg, 0.0231 mmol) were dissolved in an anhydrous mixture of CHCl3:MeOH (9:1 v/v, 2 mL) in a 20-mL scintillation vial equipped with a magnetic stirring bar. A stock solution of catalyst 5 (5 mg) in CH2Cl2 (5 mL) was prepared separately and a portion of which (2.54 mL, 3.08 μmol) was added to the vial containing the mixture of monomers 2 and 4 under vigorous stirring. The resulting reaction mixture was stirred at room temperature for 30 min, at which time an aliquot (100 μL) was removed and quenched with excess ethyl vinyl ether. A portion of this quenched aliquot was evaporated to dryness, redissolved in CDCl3, and analyzed by 1H NMR spectroscopy, which indicated complete consumption of the monomer. The remaining portion was evaporated to dryness, dissolved in HPLC-grade THF, and subjected to GPC analysis (Mn = 10000 (theoretical Mn = 9000), PDI = 1.11).
Immediately after aliquot removal, a solution of monomer 3 (30.4 mg, 0.0447 mmol) in a mixture of CHCl3:MeOH (9:1 v/v, 1.5 mL) was added to the reaction vial and the resulting polymerization mixture was stirred for an additional 45 min before being terminated with the addition of ethyl vinyl ether (1 mL). The reaction mixture was added quickly into vigorously stirred cold (−10 °C) pentanes (200 mL), and the resulting precipitate was isolated via vacuum-filtration and washed thoroughly with fresh pentanes to afford the product copolymer quantitatively as a dark red solid (GPC: Mn = 19000 (theoretical Mn = 19000), PDI = 1.13). See the ESI† for 1H NMR data..
General procedure for the preparation of nanoparticle dispersions
Aqueous suspensions of the polymer nanoparticles were prepared by dialysis following a modification of the published procedure.28 An aliquot (2.5 mL) of a stock solution of the block copolymer (2-co-4)15-b-315 (0.01 wt%) in DMSO was transferred to a 4-mL scintillation vial and set to stir vigorously. Ultrapure deionized water was added to this stirring copolymer solution at a rate of 1 drop (10 μL, 0.35 wt%) every 10 s using a 2–20 μL micropipette until the mixture contained 18 wt% water. The resulting cloudy mixture was placed in a 3-mL dialysis cassette and dialyzed against ultrapure deionized water (500 mL), with the dialate changed every 2 h. Complete absence of DMSO in the dialate after 48 h was verified by UV-vis spectroscopy as indicated by the disappearance of the UV cut-off for DMSO at 268 nm. DLS analysis of the final PNP aqueous suspension revealed narrowly dispersed PNPs with an average diameter (DH) of 211 ± 18 nm and a corresponding PDI of 0.036 (Scheme 2b right inset). TEM analysis indicated a uniform size distribution for the PNPs in the solid-state, with an average diameter of ~200 nm that is consistent with the DLS data (Scheme 2b left inset). See Fig. S23 in the ESI† for doxorubicin release profile.
General procedure for tosylate displacement on PNPs
In a 1.5-mL safe-lock Eppendorf tube, an aliquot (10 μL ) of the appropriate amine (NEt3, HNEt2, H2NEt, 2-methoxyethylamine, or 2-methylthioethylamine) was added to an aliquot (500 μL) of the PNPs derived from block copolymer (2-co-4)15-b-315. The resulting mixture was incubated on a platform shaker (Thermomixer R, Eppendorf, Hauppauge, NY, USA)) and allowed to agitate (1150 rpm) at room temperature for 72 h. The reaction mixture was then centrifuged for 30 min (Eppendorf centrifuge 5804R set at 10K rpm) to a solid pellet. The supernatant, containing majority of the excess amine, was removed and the PNPs were resuspended in PBS buffer (500 μL, 10 mM, pH = 7.4, [NaCl] = 150 mM). See the ESI† for DLS, TEM, and stabilization investigation studies.
Cell Culture
MDA-MB-231-Br cells (both GFP-producing and nonproducing), highly metastatic variants of MDA-MB-231 cells, were provided as a generous gift to Prof. Vincent L. Cryns (Dept. of Medicine, University of Wisconsin at Madison) by Dr. Patricia S. Steeg (National Cancer Institute, National Institutes of Health). Cells were maintained in Dulbecco’s Modified Eagle’s Medium, supplemented with Fetal Bovine Serum (10 vol%), L-glutamine (4 mM), and penicillin/streptomycin (100 units/mL). All media, sera, and supplements were purchased from Invitrogen (a division of Life Technologies, Grand Island, NY, USA). Cells were maintained at 37 °C in a humidified atmosphere with 5% (v/v) CO2 during these experiments.
Cellular toxicity investigation
To evaluate the inhibitory effects of the charge-stabilized PNPs, MDA-MB-231-Br cells (1.5 × 104 cells/mL) were seeded in 96-well tissue culture plates (Grenier Bio One North America, Monroe, NC, USA) for 24 h prior to drug treatment. The adhered cells were then incubated with serial dilutions of the doxorubicin-containing PNPs for 72 h, starting at 100 μM doxorubicin. After treatment, relative cell viability was determined using CellTiter 96 AQueous Non-Radioactive Cell Proliferation (MTS) assay (Promega Corporation, Madison, WI, USA). The data was fitted to a model of dose-dependent inhibition of growth (GraphPad Prism, GraphPad Software, Inc., La Jolla, CA).
Supplementary Material
Acknowledgments
This work is financially supported by the NIH (NCI Center of Cancer Nanotechnology Excellence Grant C54CA151880, Cancer Nanotechnology Platform Partnerships Grant U01CA151461, and Core Grant P30CA060553 to the Robert H. Lurie Comprehensive Cancer Center of Northwestern University). Instruments in the Northwestern University Integrated Molecular Structure Education and Research Center (IMSERC), Keck, and Northwestern University Atomic and Nanoscale Characterization Experimental (NUANCE) facilities were purchased with grants from NSF-NSEC, NSF-MRSEC, the Keck Foundation, the State of Illinois, and Northwestern University. We thank Dr. Abhijit A. Date for helpful discussions and Prof. Vincent L. Cryns and Dr. Patricia S. Steeg for the MDA MB-231-Br cell line used in this work.
Footnotes
Electronic Supplementary Information (ESI) available: General descriptions of materials and instrumentation, characterization data for the monomers and polymers, data from control experiments, and additional data as referenced in the text. See DOI: 10.1039/b000000x/
Notes and references
- 1.Aryal S, Hu CMJ, Zhang L. ACS Nano. 2010;4:251–258. doi: 10.1021/nn9014032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bale SS, Kwon SJ, Shah DA, Banerjee A, Dordick JS, Kane RS. ACS Nano. 2010;4:1493–1500. doi: 10.1021/nn901586e. [DOI] [PubMed] [Google Scholar]
- 3.Crombez L, Morris MC, Deshayes S, Heitz F, Divita G. Curr Pharm Des. 2008;14:3656–3665. doi: 10.2174/138161208786898842. [DOI] [PubMed] [Google Scholar]
- 4.Otman O, Boullanger P, Drockenmuller E, Hamaide T. Beilstein J Org Chem. 2010;6:58/1–7. doi: 10.3762/bjoc.3766.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amstad E, Textor M, Reimhult E. Nanoscale. 2011;3:2819–2843. doi: 10.1039/c1nr10173k. [DOI] [PubMed] [Google Scholar]
- 6.Raja PMV, Connolley J, Ganesan GP, Ci L, Ajayan PM, Nalamasu O, Thompson DM. Toxicol Lett. 2007;169:51–63. doi: 10.1016/j.toxlet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Hong BJ, Compton OC, An Z, Eryazici I, Nguyen ST. ACS Nano. 2012;6:63–73. doi: 10.1021/nn202355p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taratula O, Garbuzenko OB, Kirkpatrick P, Pandya I, Savla R, Pozharov VP, He H, Minko T. J Controlled Release. 2009;140:284–293. doi: 10.1016/j.jconrel.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lourenco C, Teixeira M, Simões S, Gaspar R. Int J Pharm. 1996;138:1–12. [Google Scholar]
- 10.Smith D. PhD Thesis. Northwestern University; Evanston, IL, USA: 2010. [Google Scholar]
- 11.Smith D, Clark SH, Bertin PA, Mirkin BL, Nguyen ST. J Mater Chem. 2009;19:2159–2165. doi: 10.1039/B817511J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sperling RA, Parak WJ. Philos Trans R Soc, A. 2010;368:1333–1383. doi: 10.1098/rsta.2009.0273. [DOI] [PubMed] [Google Scholar]
- 13.Laaksonen T, Ahonen P, Johans C, Kontturi K. ChemPhysChem. 2006;7:2143–2149. doi: 10.1002/cphc.200600307. [DOI] [PubMed] [Google Scholar]
- 14.Dickson JL, Shah PS, Binks BP, Johnston KP. Langmuir. 2004;20:9380–9387. doi: 10.1021/la048564u. [DOI] [PubMed] [Google Scholar]
- 15.Schiffelers RM, Ansari A, Xu J, Zhou Q, Tang Q, Storm G, Molema G, Lu PY, Scaria PV, Woodle MC. Nucleic Acids Res. 2004;32:e149/1–10. doi: 10.1093/nar/gnh140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hang J, Shi L, Feng X, Xiao L. Powder Technol. 2009;192:166–170. [Google Scholar]
- 17.Hirsjaervi S, Peltonen L, Hirvonen J. Int J Pharm. 2008;348:153–160. doi: 10.1016/j.ijpharm.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 18.Bertin PA, Smith D, Nguyen ST. Chem Commun. 2005:3793–3795. doi: 10.1039/b504643b. [DOI] [PubMed] [Google Scholar]
- 19.He C, Hu Y, Yin L, Tang C, Yin C. Biomaterials. 2010;31:3657–3666. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 20.Smith D, Pentzer EB, Nguyen ST. Polym Rev. 2007;47:419–459. [Google Scholar]
- 21.Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S. Adv Mater. 2009;21:419–424. doi: 10.1002/adma.200801393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu F, Wu S-H, Hung Y, Mou C-Y. Small. 2009;5:1408–1413. doi: 10.1002/smll.200900005. [DOI] [PubMed] [Google Scholar]
- 23.Oh E, Delehanty James B, Sapsford Kim E, Susumu K, Goswami R, Blanco-Canosa Juan B, Dawson Philip E, Granek J, Shoff M, Zhang Q, Goering Peter L, Huston A, Medintz Igor L. ACS Nano. 2011;5:6434–6448. doi: 10.1021/nn201624c. [DOI] [PubMed] [Google Scholar]
- 24.Das RK, Kasoju N, Bora U. Nanomedicine. 2010;6:153–160. doi: 10.1016/j.nano.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh D, Pramanik A, Sikdar N, Pramanik P. Biotechnol Bioprocess Eng. 2011;16:383–392. [Google Scholar]
- 26.Ghosh D, Pramanik P. Int J Pharm Sci Drug Res. 2010;2:31–34. [Google Scholar]
- 27.Kumari A, Yadav SK, Yadav SC. Colloids Surf, B. 2010;75:1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 28.Bertin PA, Watson KJ, Nguyen ST. Macromolecules. 2004;37:8364–8372. [Google Scholar]
- 29.Kumar V, Wang L, Riebe M, Tung H-H, Prud’homme RK. Mol Pharm. 2009;6:1118–1124. doi: 10.1021/mp900002t. [DOI] [PubMed] [Google Scholar]
- 30.Xing J, Deng L, Dong A. J Appl Polym Sci. 2010;117:2354–2359. [Google Scholar]
- 31.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. J Controlled Release. 2001;70:1–20. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 32.The number following the PEG notation indicates the molecular weight.
- 33.Zhang L, Zhang L. Nano LIFE. 2010;1:163–173. [Google Scholar]
- 34.Villanueva A, Cañete M, Roca AG, Calero M, Veintemillas-Verdaguer S, Serna CJ, Morales MdP, Miranda R. Nanotechnology. 2009;20:115103–115111. doi: 10.1088/0957-4484/20/11/115103. [DOI] [PubMed] [Google Scholar]
- 35.Verma A, Stellacci F. Small. 2010;6:12–21. doi: 10.1002/smll.200901158. [DOI] [PubMed] [Google Scholar]
- 36.Xia T, Kovochich M, Liong M, Meng H, Kabehie S, George S, Zink JI, Nel AE. ACS Nano. 2009;3:3273–3286. doi: 10.1021/nn900918w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin AL, Bernas LM, Rutt BK, Foster PJ, Gillies ER. Bioconjugate Chem. 2008;19:2375–2384. doi: 10.1021/bc800209u. [DOI] [PubMed] [Google Scholar]
- 38.Abbasi S, Paul A, Shao W, Prakash S. J Drug Delivery. 2012 doi: 10.1155/2012/686108. article ID 686108/1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmed M, Deng Z, Liu S, Lafrenie R, Kumar A, Narain R. Bioconjugate Chem. 2009;20:2169–2176. doi: 10.1021/bc900350c. [DOI] [PubMed] [Google Scholar]
- 40.Bertin PA, Gibbs JM, Shen CK-F, Thaxton CS, Russin WA, Mirkin CA, Nguyen ST. J Am Chem Soc. 2006;128:4168–4169. doi: 10.1021/ja056378k. [DOI] [PubMed] [Google Scholar]
- 41.This value is obtained immediately (within 5 minutes) upon adding an aqueous PNP solution into PBS buffer (DH = 198 ± 19 nm, PDI = 0.07). The PNPs aggregate significantly after 15 minutes (DH = 837 ± 482 nm, PDI = 0.69).
- 42.We note that while zeta potential is a major parameter that determines particle aggregation, the nature of the drug also plays an important role: doxorubicin-containing PNPs are much more likely to aggregate in PBS buffer while indomethacin-containing PNPs remain dispersed (Table 1, cf entries 2a and 8a) and can be readily uptaken by SKBR3 cells, as described in reference 40.
- 43.Lee S-M, Chen H, O’Halloran TV, Nguyen ST. J Am Chem Soc. 2009;131:9311–9320. doi: 10.1021/ja9017336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krovi SA, Smith D, Nguyen ST. Chem Commun. 2010;46:5277–5279. doi: 10.1039/c0cc00232a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palmieri D, Bronder JL, Herring JM, Yoneda T, Weil RJ, Stark AM, Kurek R, Vega-Valle E, Feigenbaum L, Halverson D, Vortmeyer AO, Steinberg SM, Aldape K, Steeg PS. Cancer Res. 2007;67:4190–4198. doi: 10.1158/0008-5472.CAN-06-3316. [DOI] [PubMed] [Google Scholar]
- 46.De Jong WH, Borm PJA. Int J Nanomed. 2008;3:133–149. doi: 10.2147/ijn.s596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewinski N, Colvin V, Drezek R. Small. 2008;4:26–49. doi: 10.1002/smll.200700595. [DOI] [PubMed] [Google Scholar]
- 48.Basarkar A, Devineni D, Palaniappan R, Singh J. Int J Pharm. 2007;343:247–254. doi: 10.1016/j.ijpharm.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldstein D, Gofrit O, Nyska A, Benita S. Cancer Res. 2007;67:269–275. doi: 10.1158/0008-5472.CAN-06-2731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.