

Abstract
Objective
Septal defects and coronary vessel anomalies are common congenital heart defects, yet their ontogeny and the underlying genetic mechanisms are not well understood. Here, we investigated the role of chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII, NR2F2) in cardiac organogenesis.
Methods and Results
We analyzed embryos deficient in COUP-TFII and observed a spectrum of cardiac defects, including atrioventricular septal defect, thin-walled myocardium, and abnormal coronary morphogenesis. We show by expression analysis that COUP-TFII is expressed in the endocardium and the epicardium but not in the myocardium of the ventricle. Using endothelial-specific COUP-TFII mutants and molecular approaches, we show that COUP-TFII deficiency resulted in endocardial cushion hypoplasia. This was attributed to the reduced growth and survival of atrioventricular cushion mesenchymal cells and defective epithelial-mesenchymal transformation (EMT) in the underlying endocardium. In addition, the endocardial EMT defect was accompanied by downregulation of Snai1, one of the master regulators of EMT, and upregulation of vascular endothelial-cadherin. Furthermore, we show that although COUP-TFII does not play a major role in the formation of epicardial cell cysts, it is critically important for the formation of epicardium. Ablation of COUP-TFII impairs epicardial EMT and coronary plexus formation.
Conclusion
Our results reveal that COUP-TFII plays cell-autonomous roles in the endocardium and the epicardium for endocardial and epicardial EMT, which are required for proper valve and coronary vessel formation during heart development.
Keywords: atrioventricular septal defect, cardiac morphogenesis, chicken ovalbumin upstream promoter-transcription factor II, epicardium, epithelial-mesenchymal transformation
Congenital heart defects are the most common type of birth defects, occurring in 8 of 1000 newborns. It often results from abnormal development of the inner curvatures of the looping heart, including the inflow tract, atrioventricular (AV) cushion (AVC), and the outflow tract (OFT), as well as the malformation of coronary vessels.1
During cardiac morphogenesis, the endothelial cells lining the endocardial cushions respond to the signals emitted by the myocardium within AVC and OFT and undergo an endothelial-mesenchymal transformation (EMT). The transformed cells then migrate into the underlying extracellular matrix, eventually contributing to the leaflets of the mitral and tricuspid valves, as well as to the interatrial and interventricular septa.2 The reciprocal signals between the endocardium and the myocardium were shown to induce EMT. Many genetic pathways/signals, transcription and growth factors, adhesion molecules, and proteases have been implicated in the formation of valves and septum.2
The epicardium is the primordial cell sheet in which coronary vessels derive from. It originates from the proepicardium (PE), which is a protrusion of the septum transversum near the venous pole of the developing heart.3 At around mouse embryonic day (E) 9.5, proepicardial cells either migrate directly from the PE to the developing atrium or detach as cell cysts and float freely into the pericardial cavity to reach the myocardium. Soon after the attachment, the proepicardial cells flatten, spread, and eventually form a continuous epithelial sheet, the epicardium. In mice, the heart is entirely covered by the epicardium by E11. Similar to the process that occurs in the endocardium, between E11.5 and E12.5, a subpopulation of epicardial cells undergoes EMT soon after contacting the underlying myocardium. These epicardium-derived mesenchymal cells invade into the subepicardial space, which is rich in extracellular matrix proteins. A subset of these cells further migrates into the compact zone of the myocardium. The epicardium-derived mesenchymal cells are thought to be the sources of cardiac fibroblasts and coronary vascular smooth muscle cells.4 Coronary vessels begin to form when PE-derived angioblasts or hemangioblasts differentiate into primitive vascular plexus in the subepicardial space and the myocardium. This process is called vasculogenesis. The vascular network then undergoes remodeling, an angiogenic process, to give rise to the mature coronary vascular tree. Several transcription factors regulating coronary vessel development have been characterized, including Wilms tumor 1 (Wt1), GATA4, and friend of GATA (FOG2).5–7
Chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) is an orphan member of the nuclear receptor superfamily. Genetic analysis in the mouse uncovered essential roles of the transcription factor COUP-TFII in the development of numerous organ systems.8 Previously, we have demonstrated that COUP-TFII plays an important role in angiogenesis and heart development.9 Targeted disruption of COUP-TFII leads to embryonic lethality at midgestation, with severe hemorrhage and edema.9 However, an intrinsic function of COUP-TFII during cardiac development is yet to be defined.
Here, we present data demonstrating novel roles of COUP-TFII in the formation of AVC and coronary vessels. Inactivation of COUP-TFII results in a variety spectrum of cardiac defects, including cardiac valve malformation, compact zone hypoplasia, and defective coronary vasculature. We discovered that COUP-TFII in the endocardium is important for appropriate EMT and is involved in Snai1–vascular endothelial (VE)-cadherin signaling to regulate endocardial cushion development. Furthermore, COUP-TFII in the epicardium is required for normal epicardial formation, epicardial EMT, and epicardium-derived mesenchymal cell migration. Collectively, our results indicate that endocardial COUP-TFII is important for AVC formation and that epicardial COUP-TFII is required for coronary vascular development.
Methods
Animal Experiments
COUP-TFIIF/F mice, COUP-TFII+/− mice, COUP-TFII−/− embryos, and Tie2Cre; COUP-TFIIF/F embryos were generated as previously reported.10–12 The Gata5CreTg mouse strain was provided by Dr Pilar Ruiz-Lozano,13 and the ROSA26CRE-ERT2 knock-in mouse strain was provided by Dr Thomas Ludwig.14 All mouse strains were maintained in a mixed genetic background (129/Sv×C57BL/6) and received standard rodent chow. A vaginal plug was set as E0.5. For inducible deletions during embryonic stages, tamoxifen (TAM) was dissolved in corn oil (Sigma; 10 mg/mL), and 3 mg of TAM was injected intraperitoneally into pregnant females starting at E9.5 to E15.5. For all studies, littermate controls were used. Experimental animals and studies were approved by the Institutional Animal Care and User Committee of Baylor College of Medicine.
Histological Analysis
Embryos were dissected and fixed in 4% paraformaldehyde in PBS overnight, dehydrated, embedded in paraffin, and then sectioned and stained with hematoxylin and eosin. For the frozen section, the dissected embryos were slightly fixed in 2% paraformaldehyde/PBS for 30 minutes at 4°C. After PBS wash, the embryos were cryoprotected in 30% sucrose/PBS solution overnight and embedded in optimal cutting temperature compound (Tissue Tek). Serial sections at 7 to 10 μm were made for X-gal staining, performed as described below, and counterstained with Nuclear Fast Red (Vector).
Immunohistochemistry
Immunohistochemistry analysis was performed, as described previously.15 Antibodies to smooth muscle α-actin (Sigma, clone 1A4, 1: 3000), endoglin (R&D systems, 1:200), pan-cytokeratins (Sigma, 1:2000), α-sarcomeric actin (Sigma, 1: 3000), phospho-Histone H3 (Upstate, 1:500), Wt1 (Santa Cruz, 1:500), COUP-TFII (Perseus Proteomics, 1:1000), GATA4 (Santa Cruz, 1:200), cleaved Notch1 (Val1744; Notch1 intracellular domain [NICD], Cell Signaling, 1:20), platelet-endothelial cell adhesion molecule (PECAM; Abcam, 1:200), and platelet-derived growth factor receptor β (PDGFRβ; Abcam, 1:1000) were used.
Whole-Mount PECAM Immunohistochemistry
Hearts were isolated and fixed in 4:1 methanol:dimethyl sulfoxide overnight and incubated in 4:1:1 methanol:dimethyl sulfoxide:hydrogen peroxide for 5 hours, rehydrated, and blocked in 2% milk/PBS/0.5% Triton X-100 (PBSMT). Tissues were stained with rat anti-mouse PECAM antibody (BD Biosciences, 1:200), washed 5× with PBSMT, followed by biotinylated donkey anti-rat IgG secondary antibody (Jackson Immmunoresearch). Signals were amplified by Vectastain ABC-peroxidase reagent (Vector) and visualized by 3,3′-diaminobenzidine staining (Vector). After PECAM staining, hearts were photographed and analyzed using Image J software (http://rsbweb.nih.gov/ij/). Quantification of the percentage of the ventricle covered by blood vessels was examined by dividing the area covered by blood vessels by the total ventricular area. At least 3 hearts were analyzed. After photography, PECAM-stained hearts were paraffin embedded and sectioned. Paraffin sections (7 μm) were then dewaxed, rehydrated, counterstained with hematoxylin (Vector), and mounted.
Whole-Mount X-gal Staining
Embryos or tissues were fixed in fresh 2% paraformaldehyde/PBS for 1 hour at 4°C, rinsed 3× with buffer A (100 mmol/L sodium phosphate at pH 8, 2 mmol/L MgCl2, 0.01% deoxycholate, and 0.02% NP-40) at room temperature, and stained with solution B (5 mmol/L potassium ferricyanide, 5 mmol/L potassium ferrocyanide, 1 mg/mL X-gal in rinse buffer) at 37°C. Postfixation was performed in fresh 4% paraformaldehyde/PBS at 4°C overnight.
Whole-Mount In Situ Hybridization
Whole-mount in situ hybridization was performed, as previously described.9
AVC Canal and Epicardial Explant Cultures
AVC explants and epicardial explants cultures were established as described.13,16 Briefly, endocardial cushions in the AV canal of E9.5 embryos were explanted and placed on 1 mg/mL rat tail collagen surface (BD Biosciences). The AVC was then sliced open with tweezers along the junction line of the 2 adjacent cushions in the canal. The endocardial layer was placed in direct contact with the surface of the collagen gel. Opti-MEM medium with 1% FCS and antibiotics was added to the wells 1 hour after explantation. The explants were then incubated in 5% CO2 at 37°C. Upon 24 to 48 hours of incubation, mesenchymal cells migrating from the endocardial cushions were counted using an inverted microscope that allows focusing on the mesenchymal cells present on different planes of the collagen gel.
Quantitative Reverse Transcription Polymerase Chain Reaction Assay
Total RNAs were extracted from ventricles and cultured cells using the RNeasy Mini Kit (QIAGEN) and TRIzol (Invitrogen), respectively. First-strand cDNA was synthesized by SuperScriptIII reverse transcriptase using random hexanucleotide primers according to the manufacturer’s instructions (Invitrogen). Quantitative reverse transcription polymerase chain reaction (PCR) analyses were carried out with an ABI PRISM 7500 Fast Real-Time PCR System (Applied Biosystems) using the SYBR Green PCR master mix (Applied Biosystems). The expression data from heart tissues and cultured cells were normalized to β-actin and 18S rRNA, respectively. The primer sequences are listed in Table I in the online-only Data Supplement.
Chromatin Immunoprecipitation and PCR
Chromatin immunoprecipitation (ChIP) assays were carried out with an EZ ChIP Kit (Millipore) following the manufacturer’s protocol. The primer sequences are listed in Table I in the online-only Data Supplement.
Results
Expression Pattern of COUP-TFII in the Embryonic Mouse Heart
To dissect the role of COUP-TFII during heart development, COUP-TFII/LacZ knock-in mouse17 and anti–COUP-TFII antibody were used to analyze the expression patterns of COUP-TFII in the developing hearts. X-gal staining of COUP-TFIILacZ/+ mouse showed that COUP-TFII is expressed at the posterior cardiac crescent and the lateral plate mesoderm at E8 (Figure 1A and 1B). As development proceeds, high expression of COUP-TFII was detected in the head folds, the somites, the sinus horn region, and the lateral plate mesoderm at the 6-somite stage (Figure 1C). By E9.5, the expression domains of COUP-TFII were greatly expanded throughout the embryos (Figure 1D). High levels of COUP-TFII expression were detected in the brain vesicle and sinus venosus, and medium levels of expression were observed in the common atrium (Figure 1D). Transverse sections of embryonic heart at E9.5 showed that high levels of COUP-TFII were observed in the endocardium and the myocardium of the atrium (Figure 1E). COUP-TFII was also detected in the endocardium of the AVC next to the start of the atrium and in the endocardium of the ventricle at this age (Figure 1F). The expression level of COUP-TFII was lower in the cushion mesenchymes compared with the endocardium at E10.5 (Figure 1G; Figure IA in the online-only Data Supplement). In addition, COUP-TFII was not detected in the endocardium of the OFT (Figure 1G; Figure IB in the online-only Data Supplement). Unlike the expression pattern in the atrium, COUP-TFII was only expressed in the endocardium but not detected in the myocardium of the ventricle (Figure 1H–1J). The colocalization of COUP-TFII with Tie2 in the endocardium (Figure 1H, arrow), but not with smooth muscle α-actin in the myocardium (Figure 1I), further demonstrated that COUP-TFII is specifically expressed in the endocardium but not in the myocardium of the ventricle. The expression of COUP-TFII was also detected in the epicardium (Figure 1H–1J, arrow in J), where COUP-TFII–positive cells were colocalized with the epicardium marker anti–pan-Cytokeratins (data not shown). Finally, X-gal staining of the embryonic heart at E15.5 further indicated that COUP-TFII is expressed in the endocardium and the epicardium of the ventricle (Figure 1J). The expression in both atrium and ventricles was maintained throughout development (Figure 1K). The expression patterns of COUP-TFII in the endocardium and the epicardium suggest that COUP-TFII may play important roles in the formation of the cardiac valves and coronary vasculature.
Figure 1.
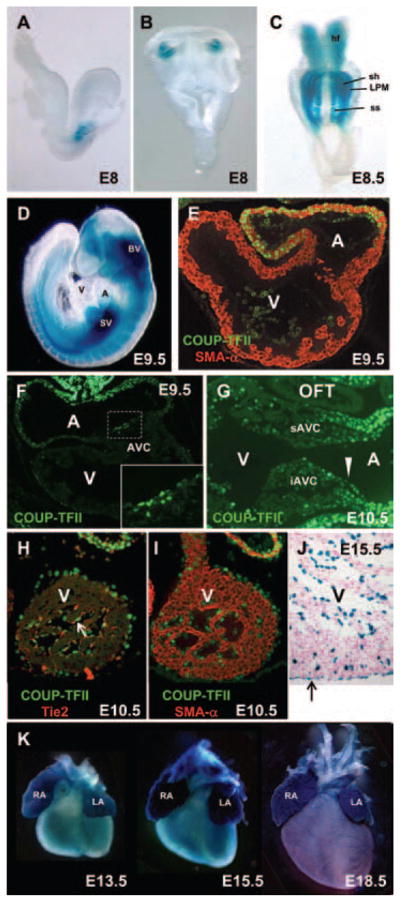
The expression domains of chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) during cardiac development. A and B, Using COUP-TFII/lacZ knock-in mice, COUP-TFII (blue) is first noted at the posterior cardiac crescent of E8 embryo shown in lateral (A) and ventral (B) views. C, The expression of COUP-TFII is largely localized in the sinus horn region at the 6-somite stage. Its expression is also shown in lateral plate mesoderm and somites. D, At E9.5, COUP-TFII is highly expressed in the sinus venosus and the atrium. E, High expression level of COUP-TFII (green) is detected in the atrium and is colocalized with the myocardium marker smooth muscle actin-α (SMA-α; red) at E9.5 (21 somites). F, COUP-TFII (green) begins to express in the endocardial atrioventricular cushion (AVC) toward the atrium later at E9.5. Area surrounding AVC (dotted rectangle) is magnified in the bottom panel. G, Sagittal section from E10.5 embryo shows high expression level of COUP-TFII (green) in the endocardium (arrowhead), atrial myocardium, and epicardium, whereas weak signal is detected in the myocardium underlying AVC. H, COUP-TFII (green) is detectable in the endocardium of the ventricle (arrow) as it is colocalized with the endothelial marker Tie2 (red). I, COUP-TFII (green) is not colocalized with the myocardial marker SMA-α in the ventricle (red). J, COUP-TFII is expressed in the endocardium and epicardium (arrow) of the ventricle at E15.5. K, From E13.5 to E18.5, high levels of COUP-TFII expression are observed in the atrium, whereas weak COUP-TFII expression is detected in the ventricle. A indicates atrium; Hf, head fold; iAVC, inferior atrioventricular cushion; LA, left atrium; LPM, lateral plate mesoderm; OFT, out-flow tract; RA, right atrium; sAVC, superior atrioventricular cushion; sh, sinus horn; ss, somites; SV, sinus venosus; V, ventricle.
Cardiac Abnormalities of COUP-TFIIF/− Embryos
To circumvent early embryonic lethality of COUP-TFII null mutants and to investigate the roles of COUP-TFII in the heart, we generated compound heterozygous mutants COUP-TFIIF/− by crossing COUP-TFIIF/F mice with COUP-TFII+/− mice. The resulting COUP-TFIIF/− mutants died by E16 and exhibited severe peripheral hemorrhage and subcutaneous edema at E14.5, suggesting the impending heart failure (Figure 2A). The mutant hearts at E14.5 showed a small left atrium and round apexes (Figure 2A). The facts that COUP-TFII+/− mice can survive postnatally and that COUP-TFIIF/− mutants die by E16 suggest that floxed COUP-TFII is a hypomorphic allele. Analysis of COUP-TFII transcripts in E12.5 ventricles revealed that COUP-TFII mRNA levels were reduced in the COUP-TFII hypomorphic mutant ventricles in comparison with the control ventricles (data not shown). Histological analysis revealed that COUP-TFIIF/− embryos at E15.5 exhibited AV septal defect (Figure 2B). Furthermore, in the control embryos, the cardiomyocytes of the developing myocardium undergo extensive growth, differentiation, and maturation to form the muscularized compact zone, whereas the COUP-TFIF/− mutant hearts remain extensively trabeculated with a thin compact zone (Figure 2C). These results suggest that a decrease in COUP-TFII dosage in the heart contributes to congenital heart abnormalities.
Figure 2.
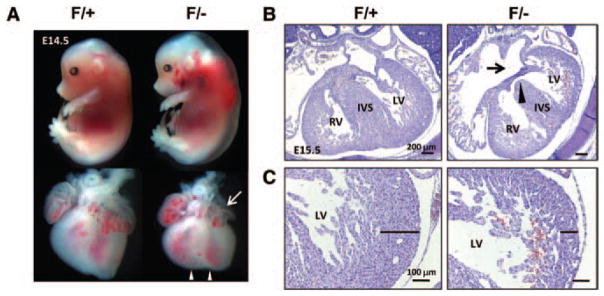
Cardiac abnormalities in chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) hypomorphic mutant embryos. A, Gross appearance and whole hearts of COUP-TFIIF/+ control and COUP-TFIIF/− mutants at E14.5. COUP-TFIIF/− mutants exhibit extensive peripheral hemorrhage and edema. Mutant heart has round apexes of the ventricle (arrowheads) and reduced left atrium (arrow). B, Hematoxylin and eosin (H&E) stained transverse sections from control and COUP-TFIIF/− mutant hearts at E14.5. Fusion of atrial septum and atrioventricular cushion is completed in COUP-TFIIF/+ littermate control. COUP-TFIIF/− mutants display an atrioventricular septal defect with openings between left and right atria (arrow) and left and right ventricles (arrowhead). The atrial septum is absent, and the central mesenchymal mass is suspended in the mutant embryo. C, H&E-stained transverse sections of control and COUP-TFIIF/− mutant hearts at E15.5. Black line indicates the thickness of the compact zone. Control embryos show normal compact zone of myocardium, whereas compact zone hypoplasia is observed in COUP-TFIIF/− mutants. IVS indicates interventricular septum; LV, left ventricle, RV, right ventricle.
Endocardial Cushion Hypoplasia in Endothelial-Specific COUP-TFII Mutants
During the formation of endocardial cushion, a subset of endocardial cells in the region of AV canal delaminates and transforms into mesenchymal cells.2 Subsequently, the mesenchymal cells undergo extensive proliferation to generate the AVC. Through remodeling and maturation processes, the primitive cushion then develops into septum and valves. Thus, EMT is a critical step for valve leaflet formation during cardiac development. To study the roles of COUP-TFII on AVC morphogenesis, we inactivated COUP-TFII in the endocardium by crossing the endothelial-specific Tie2Cre transgenic mouse line18 with the floxed COUP-TFII mice.17 Tie2Cre efficiently inactivates target genes in the endothelium and its derivatives, and its activity is evident in endothelial cells by E8.0 (data not shown). The endothelial-specific COUP-TFII null mutants (Tie2Cre; COUP-TFIIF/F) die by E12 with a variety of cardiac defects, including pericardial effusion and widespread hemorrhage.11 Histological analysis of E9.5 and E10.5 embryos showed that the number of mesenchymal cells in AVC was reduced by 50% in the endothelial-specific COUP-TFII null mutants (Figure 3A, arrows and data not shown). The AVC mesenchymal cells are identified based on the cell shape and location. In contrast, endocardial cushions in the OFT were not affected because of lack of COUP-TFII expression in the OFT (data not shown). The hypoplastic endocardial cushions present in the COUP-TFII mutants could be attributable to a failure in EMT, a decrease in cell proliferation, and an increase in apoptosis within the mesenchymal cells of AVC. To directly address whether COUP-TFII is required for normal EMT, we conducted in vitro collagen gel analysis. Normally, the endocardial cells undergo EMT and invade into and migrate through the collagen gel. As shown in Figure 3B, AVC explants from E9.5 control embryos had well-formed mesenchymal cells. In contrast, fewer mesenchymal cells were evident in the mutant explants (Figure 3B, arrows), suggesting that endocardial COUP-TFII is necessary for normal EMT.
Figure 3.

Endothelial-specific chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) null mutants exhibited endocardial cushion hypoplasia. A, Hematoxylin and eosin–stained sagittal sections of control and Tie2Cre; COUP-TFIIF/F embryos at E10.5 and the quantification of mesenchymal cells in atrioventricular cushions (AVCs). n=3 for control and n=4 for mutants. The data indicated that the mesenchymal cells in the AVCs are hypoplastic (arrows). B, AVC explants were harvested at E9.5 and cultured on 3-dimensional collagen gels for 24 hours. The number of transformed cells invading the collagen gel (arrows) is significantly reduced in Tie2Cre; COUP-TFIIF/F mutants compared with littermate controls. C, Increased expression of Notch1 intracellular domain (NICD) in AVCs of Tie2Cre; COUP-TFIIF/F mutant hearts at E10.5 compared with littermate control COUP-TFIIF/F hearts. D and E, Whole-mount in situ hybridization analysis with Snai1 (D) and vascular endothelial (VE)-cadherin (E) riboprobes at E9.5 and E9.75, respectively. Snai1 expression is down-regulated, whereas VE-cadherin expression persists in AVC endocardium. Arrows donate hybridization signal in AVC. F, Quantitative reverse transcription polymerase chain reaction analysis of COUP-TFII and Snai1 transcripts in RNA isolated from COUP-TFII or scrambled small interfering RNA–transfected human umbilical venous endothelial cells (HUVECs; normalization to 18S rRNA). G, Chromatin immunoprecipitation analysis of COUP-TFII binding to the putative Sp1-binding sites of human Snai1 promoter in HUVECs. A indicates atrium; Ex, exon; V, ventricle. Error bars indicate SD; *P<0.05; **P<0.01; ***P<0.001.
To ask whether decreased cell proliferation and increased cell apoptosis also contribute to the hypocellular endocardium cushions present in the COUP-TFII mutants, we measured proliferation and apoptotic rates by phosphorylated histone H3 immunostaining and terminal deoxynucleotidyl transferase dUTP nick end labeling assay, respectively. Our results showed that the proliferation of endocardial and cushion mesenchymal cells of the AVC region was significantly decreased in the mutants compared with the control embryos at E9.5 (Figure IIA in the online-only Data Supplement). Similarly, an enhanced apoptosis was observed in the mutant hearts at E10.5 (Figure IIB in the online-only Data Supplement). Therefore, loss of COUP-TFII in the endocardium affects not only EMT but also cell proliferation and cell apoptosis in the endocardium as well as in the underlying mesenchyme of AVC.
Notch signaling is involved in cell proliferation, survival, apoptosis, and differentiation, which affects the development of many organs.19 It was also shown to be involved in cardiac EMT.20,21 Our previous study demonstrated that COUP-TFII suppressed Notch signaling to specify venous identity during vascular development11,22; we then asked whether COUP-TFII also regulates Notch signaling during cardiac EMT. We used an antibody against NICD, the activated form of the Notch1 receptor. As shown in Figure 3C, NICD expression was increased in the AVC of Tie2Cre; COUP-TFIIF/F embryos, supporting our previous finding that COUP-TFII negatively regulates Notch1 expression.
Snai1, a zinc-finger transcription factor, is expressed in the AV endocardium and has been implicated in the endocardial EMT and EMT in cancer metastasis.21,23 We examined the expression level of Snai1. In contrast to the robust expression of Snai1 in the endocardium of control embryos, Snai1 expression was reduced in the Tie2Cre; COUP-TFIIF/F mutant AV canal at E9.5 (23-somite stage; Figure 3D). Snai1 is known as a negative regulator of epithelial-cadherin in epithelial cancer cells,24,25 prompting us to examine the expression of VE-cadherin in the AV endocardium. As EMT proceeds, Snai1 is normally upregulated and VE-cadherin is downregulated. As expected, the VE-cadherin expression is downregulated in controls by E9.75. However, in endothelial-specific COUP-TFII mutants, the expression of endocardial VE-cadherin persisted in AVC at 26-somite stage (Figure 3E), suggesting that COUP-TFII might be involved in Snai1–VE-cadherin pathway to regulate endocardial cushion formation.
To further test the above hypothesis, we examined the expression of Snai1 and VE-cadherin in COUP-TFII–depleted endothelial cells by quantitative reverse transcription PCR. Because COUP-TFII is expressed in the venous but not arterial endothelium,11 we used human umbilical venous endothelial cell as a cell model. Human umbilical venous endothelial cells were treated with specific small interfering RNA against COUP-TFII for 48 hours. Consistent with the in vivo result, when COUP-TFII was downregulated, the expression of Snai1 was reduced (Figure 3F). To determine whether Snai1 is a direct downstream target of COUP-TFII, we performed ChIP assays. We and others have previously demonstrated that when COUP-TFs act as a positive regulator to enhance its target gene expression, COUP-TFs interact with Sp1 on Sp1-binding sites to positively regulate their target genes.8 To assess whether COUP-TFII regulates Snai1 expression, we first searched for potentially conserved Sp1-binding sites in the human, mouse, and rat sequences and found at least 3 conserved sites in the promoter and intron 2 of the Snai1 gene locus (Figure 3G, red boxes). ChIP analysis showed that COUP-TFII was preferentially recruited to 3 regions containing Sp1-binding sites, site 1 in the promoter and sites 2 and 3 in the second intron, whereas COUP-TFII is not recruited to the region lacking Sp1-binding sites (Figure 3G). To further test whether COUP-TFII binding to the Snai1 promoter leads to activation of transcription, we performed luciferase reporter assays using a 0.9-kb human Snai1 promoter fragment that included the first conserved Sp1-binding sites (site 1). We transiently transfected a luciferase reporter containing the Snai1 promoter (pGL3-hSnai1) in COUP-TFI–overexpressing and control human embryonic kidney 293 cells. Relative luciferase reporter activity driven by the Snai1 promoter reporter was significantly increased in COUP-TFII–transduced cells compared with control cells (Figure III in the online-only Data Supplement). Collectively, these results strengthened our model that endogenous COUP-TFII is recruited to the Snai1 promoter to modulate Snai1 expression, implicating that COUP-TFII in the endocardium of the AVC region regulates proper cushion formation via Snai1–VE-cadherin pathway.
Myocardial Hypoplasia in COUP-TFII Mutant Embryos
In addition to AV septal defect phenotype, COUP-TFII hypomorphic mutants also display a thin compact zone (Figure 2D). It is well established that cardiac growth, contractile performance, and rhythmicity are determined by the cross talks between cardiac endothelial cells (endocardium) and cardiomyocytes and between epicardial cells and cardiomyocytes. Because COUP-TFII expression is not detected in the ventricular myocardium, the compact zone hypoplasia phenotype of COUP-TFII hypomorphic mutants is likely caused by indirect mechanisms. The epicardium is known to potentially direct the ventricular myocardium development and morphogenesis. Therefore, we deleted COUP-TFII in the epicardium using Gata5Cre mouse line. Gata5Cre activity is evident at E9 in PE and epicardium.13 Although Gata5Cre fails to completely ablate COUP-TFII in the epicardium (Figure IV in the online-only Data Supplement), nevertheless, the Gata5Cre; COUP-TFIIF/F mutants still developed a compact zone hypoplasia at E13.5 (Figure 4A), suggesting that COUP-TFII expression in the epicardium and its derivatives is required for normal development of the compact myocardium.
Figure 4.
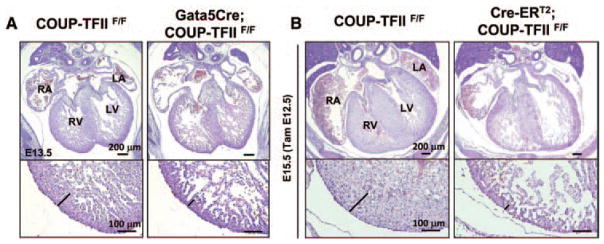
A myocardial hypoplasia in epicardial-specific chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) mutants and inducible COUP-TFII mutant embryos. A, Hematoxylin and eosin (H&E) stained transverse sections from control and Gata-5Cre; COUP-TFIIF/F mutant hearts at E13.5. Mutant hearts exhibit a thinned compact zone of myocardium. B, H&E staining of transverse sections of control COUP-TFIIF/F and littermate CRE-ERT2; COUP-TFIIF/F mutant embryos at E15.5 (with tamoxifen administration at E12.5). Black line indicates the thickness of the compact zone. Induced COUP-TFII deletion at E12.5 causes a compromised compact zone phenotype. LA indicates left atrium; LV, left ventricle; RA, right atrium, RV, right ventricle.
To investigate the time window in which inactivation of COUP-TFII caused a thin myocardium, TAM-inducible mouse model was used. As previously reported, a single dosage of 3 mg of TAM efficiently deletes COUP-TFII in embryos 2 days after administration of TAM to the pregnant female.15 The untreated ROSA26CRE-ERT2; COUP-TFIIF/F mutant embryos were indistinguishable from COUP-TFIIF/F control embryos, regardless of the developmental stage.15 Induced COUP-TFII deletion at E12.5 resulted in a pronounced hypoplastic compact zone at E15.5 (Figure 4B), suggesting, during a time window between E12.5 and E15.5, that COUP-TFII is necessary for the normal development of compact myocardium. Compact zone hypoplasia and defective coronary development usually occur together in many experimental mouse models. Our results showed that COUP-TFII is highly expressed in the epicardium (Figure 1H–1J), which is necessary for the formation of coronary vessel. As will be described later, COUP-TFII mutants indeed have defective coronary vessel formation. It has been speculated that functional coronary vasculature may in fact be required for the myocardium growth and morphogenesis via the delivery of oxygen and nutrients. Therefore, the thin myocardium phenotype observed in the Gata5Cre; COUP-TFIIF/F mutants might reflect a primary failure in coronary vascular development.
Loss of COUP-TFII Does Not Affect the Formation of PE
Coronary vascular system is derived from the epicardium, and the epicardium is derived from the PE. Migration of proepicardial cells to the naked heart tube begins with the formation of cell cysts with a central cavity. The cysts bud out from the pericardial surface of the septum transversum and either migrate directly or float freely to reach the myocardial surface of the hearts.26 After attachment to the myocardial surface, the cells migrate laterally to form an epicardial sheet to envelope the entire heart. The high levels of COUP-TFII in the PE (Figure 5A) and epicardium (Figure 1H–1J) suggest that COUP-TFII may be involved in the normal function of epicardial cells. To test the above hypothesis, we performed the histological analysis on COUP-TFII null embryos at E9.5. COUP-TFII null embryos showed no obvious defects in the formation of heart chambers and OFT.9 PE cell cysts were formed near the PE in E9.5 control and mutant embryos (Figure 5B and 5C, arrows). To investigate the epicardial development at the molecular level, we analyzed the expression of marker genes at E9.5. GATA4 is an essential factor for the formation of PE, and it is also expressed in the myocardium and the endocardium.27 Wt1 is a marker of PE and epicardial cells. In both control and mutant embryos, the PE and PE-derived cells were positive for GATA4, suggesting that the epicardial cell fate is determined (Figure 5D). Wt1-positive epicardial cell derivatives budded out from the PE and attached to the myocardial surface in both controls and mutants, indicating cell cyst formation is unaffected (Figure 5E, arrows). Taken together, the initiation of epicardial development is likely unaffected in the absence of COUP-TFII.
Figure 5.

Loss of chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) does not affect the formation of proepicardium. A, Immunofluorescent staining for COUP-TFII (green) in wild-type and COUP-TFII−/− littermates at E9.5. Nuclei were counterstained with 4′,6-diamidine-2′-phenylindole dihydrochloride. B and C, Hematoxylin and eosin staining of sagittal sections obtained from wild-type and COUP-TFII−/− mutants at E9.5. C, Higher magnification of the regions shown in B. In both wild-type and COUP-TFII−/− mutants, a substantial number of epicardial progenitors cell cysts are formed and bud out from the proepicardium (PE). D, Immunohistochemistry for GATA4 in sagittal sections of E9.5 wild-type and COUP-TFII−/− embryos. All tissues are labeled with GATA4. E, Immunohistochemistry reveals Wilms tumor 1 expression in the PE and epicardial cells in wild-type and COUP-TFII−/− littermates at E9.5. A indicates atrium; OFT, outflow tract; V, ventricle.
COUP-TFII Deficiency Causes Abnormal Epicardial Morphogenesis
To ask whether inactivation of COUP-TFII affects epicardial development, the morphology of epicardium was compared between control and COUP-TFII hypomorphic mutants. By E12.5, when the epicardium completely formed in control embryos and as the heart increases in size, the subepicardial spaces become less apparent and the epicardium is more uniform over the myocardium surface, as evidenced by hematoxylin and eosin staining (Figure VA in the online-only Data Supplement). Although the epicardium largely covered the entire heart in E12.5 mutant embryos, there is an increase of subepicardial spaces in the mutant embryos, suggesting that the epicardium is malformed (Figure VA in the online-only Data Supplement, arrows). Similar results were observed in E11.5-inducible COUP-TFII knockout mutants when TAM was administrated to the pregnant dams at E9.5 and in Gata5Cre; COUP-TFIIF/F mutants at E11.5 (Figure VB and VC in the online-only Data Supplement, arrows). Quantification of the number of Wt1-positive cells within a certain distance of epicardium was significantly reduced in E11.5-inducible COUP-TFII knockout mutants (Figure 5D in the online-only Data Supplement). In addition, scanning electron microscopy also showed that a bubble-like distension of the epicardium was evident in the COUP-TFII hypomorphic mutants at E12.5 (Figure VIA and VIB in the online-only Data Supplement). Histological analysis showed that large numbers of proepicardial cells were retained in the sinoatrial region of the hypomorphic mutant hearts at E15.5, implying that lack of COUP-TFII affects the proper migration of proepicardial cells from septum transversum mesenchyme to ensheathe the entire heart (Figure VIC in the online-only Data Supplement) Together, these results indicated the aberrant development of the epicardium in the mice lacking COUP-TFII.
Inactivation of COUP-TFII Leads to Abnormal Coronary Angiogenesis
To directly address whether COUP-TFII is required for coronary development, we performed whole-mount PECAM staining of E13.5 to E14.5 controls and the COUP-TFII hypomorphic mutant hearts to visualize the coronary vasculature. Analysis of E13.5 hearts demonstrated that although control hearts developed a vascular plexus that ensheathed the ventricle and was further remodeled to form a coronary vessel, the vascular plexus of COUP-TFIIF/− mutants failed to fully extend to the apices of the hearts (Figure 6A). Quantification of the percentage of coronary vessel coverage at E13.5 revealed that COUP-TFII mutants were ≈30% less in comparison with control littermates (Figure 6B). Analysis of coronary morphogenesis at E14.5 control hearts showed normal vascular plexus network in the ventricles (Figure 6C). A vessel with 4 major branches formed on the dorsal side of the control ventricles (Figure 6C, arrows). In contrast, COUP-TFII mutant ventricles exhibited abnormal coronary morphogenesis, with defective remodeling and maturation processes where the majority of vessels remain primitive plexus, suggesting an impairment in angiogenesis (Figure 6C). To further corroborate the above findings, E13.5 whole-mount stained hearts were sectioned, and multiple subepicardial (Figure 6D, asterisk) and intramyocardial coronary vessels (Figure 6D, arrows) were observed in the ventricles of the control embryos. Also, weak PECAM signals were detected in the endocardium (Figure 6D, arrowhead). In contrast, COUP-TFIIF/− mutant ventricles contained little or no subepicardial coronary vessels, but with normal endocardium (Figure 6D, arrowhead), suggesting COUP-TFII is necessary for coronary angiogenesis. To study whether COUP-TFII is expressed in PECAM-positive vessels, COUP-TFII and PECAM antibodies were used. Immunohistochemistry showed that COUP-TFII is observed in PECAM-positive subepicardial vessels at E13.5 hearts (Figure 6E, asterisks). COUP-TFII is also expressed in the smooth muscle cells surrounding the intramyocardial vessel,8 suggesting that COUP-TFII plays cell-autonomous functions on coronary angiogenesis. Collectively, these results indicate that loss of COUP-TFII results in defective coronary vasculature.
Figure 6.
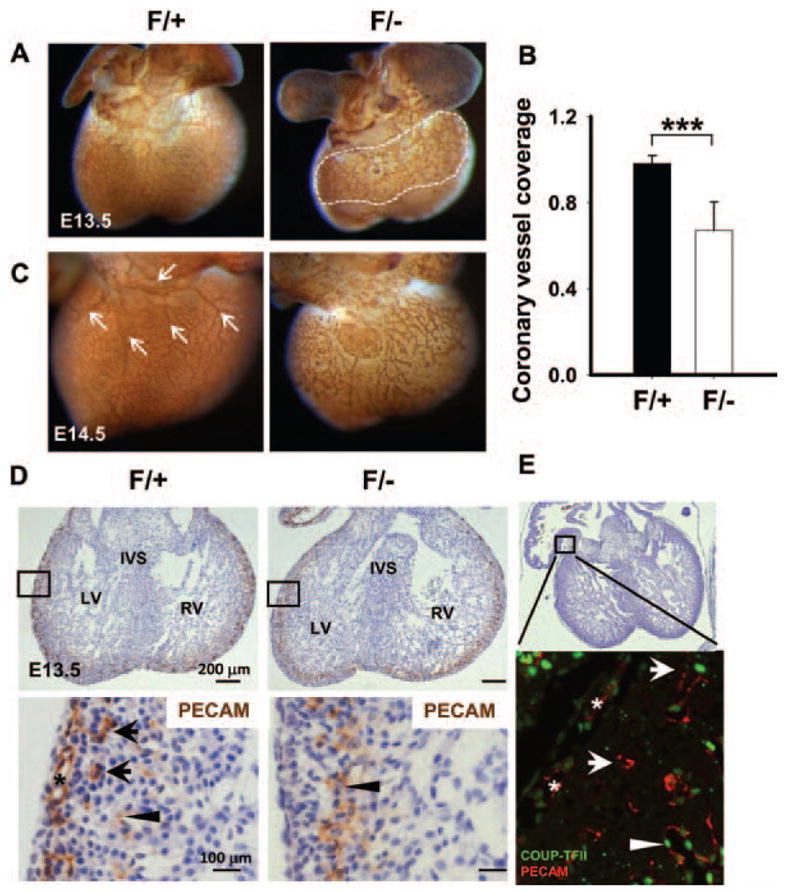
Inactivation of chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) leads to abnormal coronary angiogenesis. A and C, Whole-mount immunostaining of E13.5 and E14.5 mouse hearts with platelet-endothelial cell adhesion molecule (PECAM) illustrates vascular endothelium. A, A representative littermate control heart shows the normal vascular plexus in the ventricles. The vascular plexus of COUP-TFII mutant ventricle fails to extend to the apex of the heart. B, The coronary plexus coverage (ratio of region within white dotted line to total projected ventricular area) at E13.5 was significantly decreased in mutants. Error bars indicate SD; **P<0.01. C, A vessel with 4 major branches (arrows) forms on the dorsal side of the control ventricle, whereas COUP-TFIIF/− mutant ventricles at E14.5 present abnormal coronary morphogenesis, with less extensive vascular network and defective branching. D, Histological sections of PECAM-stained control and mutant hearts at E13.5 indicating that COUP-TFIIF/− mutants lack subepicardial vessels (asterisk) and intramyocardial vessels (arrows). Arrowheads denote endocardial PECAM staining. E, Hematoxylin and eosin and immunofluorescence stainings of wild-type mouse hearts at E13.5 reveal that COUP-TFII is expressed in subepicardial vessels (asterisks), intramyocardial vessels (arrows), and endocardium (arrowhead). LV indicates left ventricle; RV, right ventricle; IVS, interventricular septum.
Epicardial EMT and Epicardial Migration Are Perturbed in Conditional COUP-TFII Mutant Hearts
Epicardial EMT has an important role in the formation of coronary vascular precursor cells. To test whether the coronary vascular defect of COUP-TFII hypomorphic mutants was a result of defective epicardial EMT, an ex vivo epicardial explant assay was performed. When E12.5 control epicardial explants were cultured on a collagen gel, a subset of cells delaminated, formed spindle-like cells that are 4′,6-diamidine-2′-phenylindole dihydrochloride positive, and migrated into the collagen gel (Figure 7A; Figure VII in the online-only Data Supplement). In contrast, epicardial explants from COUP-TFII mutant embryos formed less spindle-like mesenchymal cells, indicating that epicardial EMT was severely compromised in COUP-TFII mutant embryos. Next, we asked whether the migration of epicardial cell derivatives into the myocardium was also perturbed; Wt1 was used as a marker for epicardial cell derivatives. We observed a drastic reduction in the number of Wt1-positive cells inside the myocardium of E14.5-inducible COUP-TFII knockout mutant hearts and COUP-TFII hypomorphic mutant hearts (Figure 7B, arrows, and data not shown) compared with controls. Intriguingly, the expression of Wt1 was noticeably decreased in the epicardium of the mutant hearts compared with controls (Figure 7B, arrowheads). In addition to Wt1 signaling, PDGFRβ signaling has also been implicated in epicardial migration and coronary vessel maturation.28 As expected, we observed a decrease in PDGFRβ-expressing cells in the myocardium of COUP-TFII hypomorphic mutant hearts compared with the control hearts at E13.5 (Figure 7C). To exclude systemic and other contribution, we examined the expression of Wt1 and PDGFRβ in E14.5 Gata5Cre; COUP-TFIIF/F mutant hearts. Consistent with the above findings, there was a reduction of Wt1-expressing or PDGFRβ-expressing cells in the myocardium of Gata5Cre; COUP-TFIIF/F mutant hearts. It is known that Notch signaling in the epicardium is important not only for coronary vascular morphogenesis but also for normal compact myocardium growth.29 Interestingly, we also observed the ectopic expression of NICD in the myocardium of Tie2Cre; COUP-TFIIF/F heart (Figure VIII in the online-only Data Supplement), suggesting that alteration of Notch signaling may contribute to the thin myocardium in COUP-TFII mutants. These data also imply that the reduction of epicardial cell derivatives within the myocardium was attributable to the impaired epicardial EMT and epicardial migration.
Figure 7.
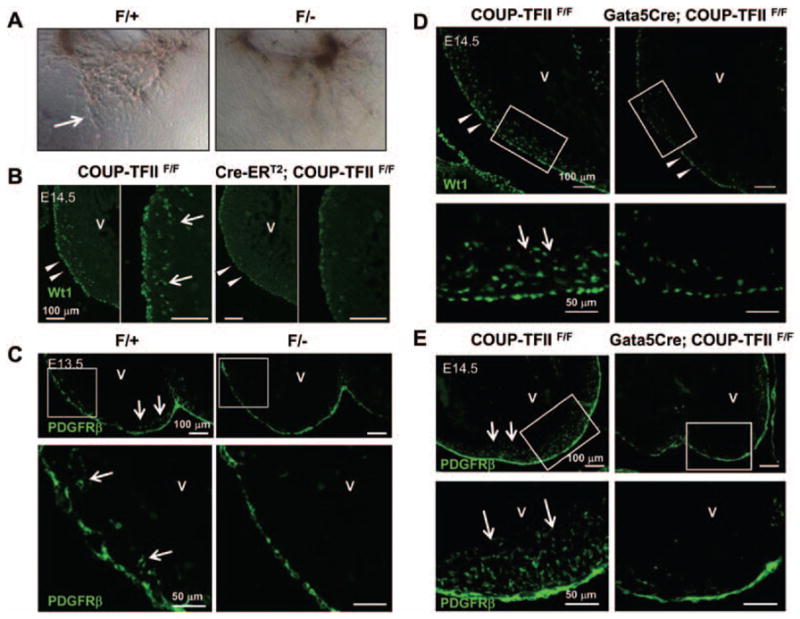
Epicardial-mesenchymal transformation (EMT) and epicardial migration are perturbed in chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) mutant hearts. A, Impaired EMT in COUP-TFIIF/− mutant epicardium. Representative figures of ex vivo heart explants on collagen gel. Cells migrate out of the control heart explants and transform to spindle-shaped mesenchymal cells (arrow), whereas little or no mesenchymal cells are observed in mutant heart explants. B and D, Decreased expression of Wilms tumor 1 (Wt1) in the epicardium of Cre-ERT2; COUP-TFIIF/F mutant hearts at E14.5 (with tamoxifen administration at E11.5) (B) or Gata5Cre; COUP-TFIIF/F mutant hearts at E14.5 (D) compared with littermate control COUP-TFIIF/F hearts. Numerous Wt1-positive epicardial derivatives were detected in the myocardium of controls (arrows), whereas decreased number of Wt1-positive epicardial derivatives was found in mutant hearts (white arrows). C and E, A decreased number of platelet-derived growth factor receptor β (PDGFRβ)–expressing epicardial-derived cells in the myocardium of COUP-TFIIF/− mutant hearts at E13.5 (C) or Gata5Cre; COUP-TFIIF/F mutant hearts at E14.5 (E) compared with littermate control hearts. Numerous PDGFRβ-positive epicardial derivatives were detected in the myocardium of controls (arrows), whereas a decreased number of PDGFRβ-positive epicardial derivatives were found in mutant hearts. V indicates ventricle.
Discussion
Cardiac development is regulated by an evolutionarily conserved transcriptional network throughout development. Aberrant transcriptional regulation contributes to the pathogenesis of congenital and acquired forms of heart diseases. Therefore, a better understanding of the function of members of this transcriptional network may provide insights into molecular pathways underlying cardiac diseases. Here, we report crucial roles for COUP-TFII in the endocardial cushion EMT and the subsequent AVC formation. COUP-TFII promotes EMT at least, in part, by directly regulating the expression of Snai1, an important EMT factor. In addition, we demonstrate that COUP-TFII in the epicardium is required for ventricular morphogenesis and development of coronary vessels. Exploring underlying mechanisms, we show that epicardial COUP-TFII promotes coronary angiogenesis by regulating epicardial EMT and epicardial cell migration.
In this study, we found that endothelial-specific disruption of COUP-TFII showed markedly reduced cellularization from E9.5 in the AVC, a decrease in cell proliferation, and an increase in apoptosis in the endocardium as well as in the underlying mesenchyme of AVC. Concordant with the previous finding, we demonstrated that Notch1, an important regulator of heart development, is also a target of COUP-TFII during cardiac morphogenesis. In addition to the increased expression of NICD in the AVC of Tie2Cre; COUP-TFIIF/F heart (Figure 3C), we also observed the ectopic expression of NICD in the myocardium of Tie2Cre; COUP-TFIIF/F ventricles (Figure VIII in the online-only Data Supplement). Evidences from gene targeting studies in the mice highlighted the importance of the Notch pathway in the cardiovascular development and disease. Either disruption or constitutive activation of Notch signaling disrupted cardiac development, indicating that the level of Notch signaling influences different microenvironmental factors that may affect cell differentiation, induce cell cycle arrest, and inhibit cell proliferation, and result in the loss of survival signals and an increase in cell apoptosis, and eventually elicit abnormal cardiac phenotypes.21,30–32 Interestingly, the AVC phenotypes of COUP-TFII mutants are similar to Notch inactivation.21 Notch-promoting EMT is attributable to the fact that Notch is shown to enhance Snai1 expression.21 However, here, we showed that COUP-TFII directly regulated Snai1, and the expression of Snai1 was downregulated in the absence of COUP-TFII. Therefore, we proposed that loss of COUP-TFII overrides the gain of Notch, resulting in reduced EMT, even when Notch is enhanced. Our present study suggests the possibility that activated Notch signaling contributes to cardiac abnormalities of Tie2Cre; COUP-TFIIF/F mutants. Clearly, the COUP-TFII–Notch1 transcription hierarchy in regulating cell differentiation, proliferation, and apoptosis during cardiac development will require additional investigation.
How COUP-TFII regulates epicardial EMT and coronary angiogenesis needs further investigation. Recent studies have established novel roles of Notch signaling in epicardial development and coronary vessel morphogenesis.29,33 Ectopic expression of Notch1 in epicardium caused defective epicardial integrity, a thinning of ventricular myocardium, and premature coronary smooth muscle cell differentiation of epicardial cells.29,33 Given that either epicardial-specific COUP-TFII mutants or ectopic Notch1 activation mutants exhibited similar coronary phenotypes and that COUP-TFII suppresses Notch signaling to govern venous identity and to control progenitor Leydig cell differentiation,11,34 it is reasonable to speculate that COUP-TFII may function upstream of the Notch signaling to regulate coronary angiogenesis. Indeed, we found the ectopic expression of NICD in endothelial-specific COUP-TFII–deleted ventricles. Intriguingly, knocking down COUP-TFII in endothelial cells resulted in the increased expression of not only genes involved in Notch signaling but also smooth muscle lineage markers,22 which correlated with the published results that Notch signaling promotes smooth muscle differentiation of epicardial cells. Additional experiments will be necessary to fully elucidate the regulation of Notch signaling by COUP-TFII.
The epicardium is a critical tissue that directs several aspects of heart development, including formation of coronary vasculature, as well as ventricular morphogenesis and maturation.35 In this study, we identified not only the origin (the epicardium) but also the time window (E12.5–E15.5) of COUP-TFII, which is critical for ventricular myocardial morphogenesis. It is well established that the epicardium secretes mitogenic factors to promote compact zone cardiomyocyte proliferation. This raises an intriguing question on whether epicardial COUP-TFII regulates secreted mitogen(s) to induce myocardial growth. Members of fibroblast growth factor (FGF) family, 9, 16 and 20, have been implicated in promoting ventricular cardiomyocyte proliferation in vivo.36 Although COUP-TFII was colocalized with FGF9, FGF16, and FGF20 in both endothelium and epicardium, we found no differences in the expression of FGF9, FGF16, and FGF20 between control and COUP-TFII hypomorphic ventricles at E12.5 as determined by quantitative reverse transcription PCR (data not shown). Similarly, the expression of erythropoietin was unchanged in COUP-TFII mutant ventricles (data not shown). These results suggest that COUP-TFII is unlikely a major regulator of the subgroup of FGFs and erythropoietin in the ventricle. Other secreted factors, for example, retinoic acid, Wnts, and PDGFs, remain to be further investigated. It has also been shown that valve defects or vascular deficiency could cause hemodynamic changes that result in the appearance of a thin-walled ventricular chamber. However, epicardial-specific knockout of COUP-TFII by Gata5Cre exhibits similar thin compact zone without apparent valve defects (Figure 4A), indicating that hemodynamic changes are unlikely to play significant role in this defect.
Despite the importance of epicardium itself in ventricular morphogenesis, the cross talks between both coronary endothelial cells and epicardium with the myocardium also affect the growth of cardiomyocytes. Functional coronary vasculature is required for the myocardium growth and morphogenesis via the delivery of oxygen and nutrients. Thus, failure in myocardial compaction in the COUP-TFII mutants is likely a manifestation of the disruption of these cross talks.
As we have shown, the decreased numbers of Wt1- and PDGFβ-positive cells within the compact myocardium in the COUP-TFII mutant hearts indicated an impairment of epicardial EMT and migration and, consequently, affected the maturation of intramyocardial vessels of COUP-TFII hypomorphic mutants and of epicardial deletion of COUP-TFII mutants. These results strongly suggest that COUP-TFII contributes to coronary angiogenesis. Like the expression domain in other vascular systems, COUP-TFII is expressed in the endothelial cells of the subepicardial vessels (cardiac veins) and the smooth muscle cells of the intramyocardial vessels (cardiac arteries).8 Although the origin of coronary endothelial cells is controversial, recent lineage tracing experiments in avian and mouse models have demonstrated that the majority of coronary vascular smooth muscle cells arise from the epicardium.37–40 Thus, based on the expression of COUP-TFII in specific cell types, we speculate that COUP-TFII affects coronary morphogenesis by at least 3 mechanisms: first, by recruiting COUP-TFII–positive vascular endothelial cells to promote the assembly of newly formed blood vessels; second, by promoting epithelial-mesenchymal transition to generate vascular smooth muscle cells and fibroblasts for the formation of vessel; third, by affecting the mobilization of the epicardial-derived cells from the epicardium into the myocardium. However, we could not distinguish the second and third views because the reduction of Wt1- and PDGFRβ-positive epicardial cell derivatives in the myocardium of COUP-TFII mutant hearts could be simply a result of the reduction of Wt1-positive subepicardial cells rather than reflecting their migration defects.
A study has identified that a male patient with a 6.5-Mb deletion covering chromosome 15q26.2 displayed several malformations, including congenital heart defects (left atrial hypoplasia, ventral septal defect, mitral valve atresia, aorta hypoplasia) and congenital diaphragmatic hernia.41 Intriguingly, COUP-TFII is one of the known genes residing within this critical region, and genetic studies showed that mouse lacking COUP-TFII in foregut mesenchyme exhibits Bochdalek-type congenital diaphragmatic hernia.10 Strikingly, the clinical manifestations of the male patient are consistent with the phenotypes displayed by the COUP-TFII hypomorphic mouse mutants, such as a small left atria, AV septal defect, and valve defects. Supporting the above finding, the patients with deletion at a similar region but with intact COUP-TFII gene did not have cardiac abnormalities.42 Therefore, it is likely that the malformation of heart and diaphragm in the human patients is attributable to hemizygosity/haploinsufficiency of the COUP-TFII gene.
Studies of transcription factors and signaling molecules in heart development will enable us to understand the complexity and underlying mechanisms of the pathogenesis of congenital heart defect in humans. Further information regarding COUP-TFII mutations in humans will also facilitate the genetic counseling on affected individuals. The goal of future study will be to integrate COUP-TFII, together with preexisting molecules, into a comprehensive model of the regulatory hierarchy of cardiac development.
Supplementary Material
Acknowledgments
We thank Dr Henry M. Sucov for the constructive comments on the manuscript, W. Chen, W. Qian, X.-F. Tong for their technical help, J. Hebert for proof reading of the article, and Dr Ching-Pin Chang for technical advice on the EMT assay. We also thank Dr Pilar Ruiz-Lozano for the Gata5CreTg mouse strain, Dr Thomas Ludwig for the ROSA26CRE-ERT2 knock-in mouse strain, and Dr Antonio Garcia de Herreros for the Snai1 promoter–driven reporter construct.
Sources of Funding
This work was supported by National Science Council of Taiwan grant NSC98-2320-B-010-010-MY3 and a grant from Ministry of Education, Aim for the Top University Plan (to L.-R. You), National Heart, Lung, and Blood Institute grant R01 HL076448 (to S.Y. Tsai), National Institute of Child Health and Human Development grant R01 HD17379, and National Institute of Diabetes and Digestive and Kidney Disease grant R01 DK45641 (to M.-J. Tsai), P01 DK059820 and U19 DK062434 (to S.Y. Tsai and M-J. Tsai), and core facilities of Diabetes and Endocrinology Research Center (DK079638).
Footnotes
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.112.300255/-/DC1.
Disclosures
None.
References
- 1.Srivastava D, Olson EN. A genetic blueprint for cardiac development. Nature. 2000;407:221–226. doi: 10.1038/35025190. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res. 2004;95:459–470. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ratajska A, Czarnowska E, Ciszek B. Embryonic development of the proepicardium and coronary vessels. Int J Dev Biol. 2008;52:229–236. doi: 10.1387/ijdb.072340ar. [DOI] [PubMed] [Google Scholar]
- 4.Mikawa T. Cardiac lineages. In: Harvey RP, Rosenthal N, editors. Heart Development. San Diego, CA: Academic Press; 1999. pp. 19–33. [Google Scholar]
- 5.Zhou B, Ma Q, Kong SW, Hu Y, Campbell PH, McGowan FX, Ackerman KG, Wu B, Zhou B, Tevosian SG, Pu WT. Fog2 is critical for cardiac function and maintenance of coronary vasculature in the adult mouse heart. J Clin Invest. 2009;119:1462–1476. doi: 10.1172/JCI38723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crispino JD, Lodish MB, Thurberg BL, Litovsky SH, Collins T, Molkentin JD, Orkin SH. Proper coronary vascular development and heart morphogenesis depend on interaction of GATA-4 with FOG cofactors. Genes Dev. 2001;15:839–844. doi: 10.1101/gad.875201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner N, Wagner KD, Theres H, Englert C, Schedl A, Scholz H. Coronary vessel development requires activation of the TrkB neurotrophin receptor by the Wilms’ tumor transcription factor Wt1. Genes Dev. 2005;19:2631–2642. doi: 10.1101/gad.346405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin FJ, Qin J, Tang K, Tsai SY, Tsai MJ. Coup d’Etat: an orphan takes control. Endocr Rev. 2011;32:404–421. doi: 10.1210/er.2010-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pereira FA, Qiu Y, Zhou G, Tsai MJ, Tsai SY. The orphan nuclear receptor COUP-TFII is required for angiogenesis and heart development. Genes Dev. 1999;13:1037–1049. doi: 10.1101/gad.13.8.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.You LR, Takamoto N, Yu CT, Tanaka T, Kodama T, Demayo FJ, Tsai SY, Tsai MJ. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proc Natl Acad Sci USA. 2005;102:16351–16356. doi: 10.1073/pnas.0507832102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.You LR, Lin FJ, Lee CT, DeMayo FJ, Tsai MJ, Tsai SY. Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature. 2005;435:98–104. doi: 10.1038/nature03511. [DOI] [PubMed] [Google Scholar]
- 12.Pereira FA, Qiu Y, Tsai MJ, Tsai SY. Chicken ovalbumin upstream promoter transcription factor (COUP-TF): expression during mouse embryogenesis. J Steroid Biochem Mol Biol. 1995;53:503–508. doi: 10.1016/0960-0760(95)00097-j. [DOI] [PubMed] [Google Scholar]
- 13.Merki E, Zamora M, Raya A, Kawakami Y, Wang J, Zhang X, Burch J, Kubalak SW, Kaliman P, Izpisua Belmonte JC, Chien KR, Ruiz-Lozano P. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc Natl Acad Sci USA. 2005;102:18455–18460. doi: 10.1073/pnas.0504343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Luca C, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, Ludwig T, Liu SM, Chua SC., Jr Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest. 2005;115:3484–3493. doi: 10.1172/JCI24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin FJ, Chen X, Qin J, Hong YK, Tsai MJ, Tsai SY. Direct transcriptional regulation of neuropilin-2 by COUP-TFII modulates multiple steps in murine lymphatic vessel development. J Clin Invest. 2010;120:1694–1707. doi: 10.1172/JCI40101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, Graef IA, Crabtree GR. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–663. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Takamoto N, You LR, Moses K, Chiang C, Zimmer WE, Schwartz RJ, DeMayo FJ, Tsai MJ, Tsai SY. COUP-TFII is essential for radial and anteroposterior patterning of the stomach. Development. 2005;132:2179–2189. doi: 10.1242/dev.01808. [DOI] [PubMed] [Google Scholar]
- 18.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 19.Bolós V, Grego-Bessa J, de la Pompa JL. Notch signaling in development and cancer. Endocr Rev. 2007;28:339–363. doi: 10.1210/er.2006-0046. [DOI] [PubMed] [Google Scholar]
- 20.Niessen K, Karsan A. Notch signaling in cardiac development. Circ Res. 2008;102:1169–1181. doi: 10.1161/CIRCRESAHA.108.174318. [DOI] [PubMed] [Google Scholar]
- 21.Timmerman LA, Grego-Bessa J, Raya A, Bertrán E, Pérez-Pomares JM, Díez J, Aranda S, Palomo S, McCormick F, Izpisúa-Belmonte JC, de la Pompa JL. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Qin J, Cheng CM, Tsai MJ, Tsai SY. COUP-TFII Is a Major Regulator of Cell Cycle and Notch Signaling Pathways. Mol Endocrinol. 2012;26:1268–1277. doi: 10.1210/me.2011-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 24.Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 25.Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 26.Männer J, Pérez-Pomares JM, Macías D, Muñoz-Chápuli R. The origin, formation and developmental significance of the epicardium: a review. Cells Tissues Organs (Print) 2001;169:89–103. doi: 10.1159/000047867. [DOI] [PubMed] [Google Scholar]
- 27.Watt AJ, Battle MA, Li J, Duncan SA. GATA4 is essential for formation of the proepicardium and regulates cardiogenesis. Proc Natl Acad Sci USA. 2004;101:12573–12578. doi: 10.1073/pnas.0400752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellgren AM, Smith CL, Olsen GS, Eskiocak B, Zhou B, Kazi MN, Ruiz FR, Pu WT, Tallquist MD. Platelet-derived growth factor receptor beta signaling is required for efficient epicardial cell migration and development of two distinct coronary vascular smooth muscle cell populations. Circ Res. 2008;103:1393–1401. doi: 10.1161/CIRCRESAHA.108.176768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.del Monte G, Casanova JC, Guadix JA, MacGrogan D, Burch JB, Pérez-Pomares JM, de la Pompa JL. Differential Notch signaling in the epicardium is required for cardiac inflow development and coronary vessel morphogenesis. Circ Res. 2011;108:824–836. doi: 10.1161/CIRCRESAHA.110.229062. [DOI] [PubMed] [Google Scholar]
- 30.de la Pompa JL, Epstein JA. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev Cell. 2012;22:244–254. doi: 10.1016/j.devcel.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luna-Zurita L, Prados B, Grego-Bessa J, Luxán G, del Monte G, Benguría A, Adams RH, Pérez-Pomares JM, de la Pompa JL. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest. 2010;120:3493–3507. doi: 10.1172/JCI42666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe Y, Kokubo H, Miyagawa-Tomita S, Endo M, Igarashi K, Aisaki K, Kanno J, Saga Y. Activation of Notch1 signaling in cardiogenic mesoderm induces abnormal heart morphogenesis in mouse. Development. 2006;133:1625–1634. doi: 10.1242/dev.02344. [DOI] [PubMed] [Google Scholar]
- 33.Grieskamp T, Rudat C, Lüdtke TH, Norden J, Kispert A. Notch signaling regulates smooth muscle differentiation of epicardium-derived cells. Circ Res. 2011;108:813–823. doi: 10.1161/CIRCRESAHA.110.228809. [DOI] [PubMed] [Google Scholar]
- 34.Qin J, Tsai MJ, Tsai SY. Essential roles of COUP-TFII in Leydig cell differentiation and male fertility. PLoS ONE. 2008;3:e3285. doi: 10.1371/journal.pone.0003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sucov HM, Gu Y, Thomas S, Li P, Pashmforoush M. Epicardial control of myocardial proliferation and morphogenesis. Pediatr Cardiol. 2009;30:617–625. doi: 10.1007/s00246-009-9391-8. [DOI] [PubMed] [Google Scholar]
- 36.Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J, Ornitz DM. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, Jiang D, von Gise A, Ikeda S, Chien KR, Pu WT. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. 2008;454:109–113. doi: 10.1038/nature07060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai CL, Martin JC, Sun Y, Cui L, Wang L, Ouyang K, Yang L, Bu L, Liang X, Zhang X, Stallcup WB, Denton CP, McCulloch A, Chen J, Evans SM. A myocardial lineage derives from Tbx18 epicardial cells. Nature. 2008;454:104–108. doi: 10.1038/nature06969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pérez-Pomares JM, Macías D, García-Garrido L, Muñoz-Chápuli R. The origin of the subepicardial mesenchyme in the avian embryo: an immunohistochemical and quail-chick chimera study. Dev Biol. 1998;200:57–68. doi: 10.1006/dbio.1998.8949. [DOI] [PubMed] [Google Scholar]
- 40.Dettman RW, Denetclaw W, Jr, Ordahl CP, Bristow J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev Biol. 1998;193:169–181. doi: 10.1006/dbio.1997.8801. [DOI] [PubMed] [Google Scholar]
- 41.Tümer Z, Harboe TL, Blennow E, Kalscheuer VM, Tommerup N, Brøndum-Nielsen K. Molecular cytogenetic characterization of ring chromosome 15 in three unrelated patients. Am J Med Genet A. 2004;130A:340–344. doi: 10.1002/ajmg.a.30035. [DOI] [PubMed] [Google Scholar]
- 42.Peoples R, Milatovich A, Francke U. Hemizygosity at the insulin-like growth factor I receptor (IGF1R) locus and growth failure in the ring chromosome 15 syndrome. Cytogenet Cell Genet. 1995;70:228–234. doi: 10.1159/000134040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.