

Abstract
There is evidence indicating that invariant Natural Killer T (iNKT) cells play an important role in defense against influenza A virus (IAV). However, the effect of inhibitory receptor, programmed death-1 (PD-1), and its ligands, programmed death ligand (PD-L) 1 and 2 on iNKT cells in protection against IAV remains to be elucidated. Here we investigated the effects of these co-stimulatory molecules on iNKT cells in the response to influenza. We discovered that compare to the wild type, PD-L1 deficient mice show reduced sensitivity to IAV infection as evident by reduced weight loss, decreased pulmonary inflammation and cellular infiltration. In contrast, PD-L2 deficient mice showed augmented weight loss, pulmonary inflammation and cellular infiltration compare to the wild type mice after influenza infection. Adoptive transfer of iNKT cells from wild type, PD-L1 or PD-L2 deficient mice into iNKT cell deficient mice recapitulated these findings. Interestingly, in our transfer system PD-L1−/−-derived iNKT cells produced high levels of interferon-gamma whereas PD-L2−/−-derived iNKT cells produced high amounts of interleukin-4 and 13 suggesting a role for these cytokines in sensitivity to influenza. We identified that PD-L1 negatively regulates the frequency of iNKT cell subsets in the lungs of IAV infected mice. Altogether, these results demonstrate that lack of PD-L1 expression by iNKT cells reduces the sensitivity to IAV and that the presence of PD-L2 is important for dampening the deleterious inflammatory responses after IAV infection. Our findings potentially have clinical implications for developing new therapies for influenza.
Introduction
Influenza A virus (IAV) infections represent a major public health threat, particularly in the case of children, the elderly and those with underlying diseases, all of whom are at an increased risk for disease complications and death following IAV infection [1], [2]. Seasonal outbreaks alone cause an estimated 200,000 hospitalizations and over 30,000 deaths annually in the United States [3].
Immune system plays an important role in the resolution of IAV infection. Both mucosal and systemic immunity play important roles in the elimination of infection with IAV [4], [5], [6]. Accumulating evidence in the last few years suggests an important role for conventional CD4+ and CD8+ T cells in the control and clearance of the IAV [7], [8], [9]. However, in recent years, a relatively new T cell population, invariant natural killer T (iNKT) cells, have been reported to act not only as innate lymphocytes but also as regulators of adaptive immune responses [10], [11].
iNKT cells have been suggested to play critical roles in a wide range of immune responses by acting in a pro-inflammatory or anti-inflammatory manner [12], [13]. They are a specialized subset of T lymphocytes expressing markers of the NK cell lineage and an invariant T cell receptor (TCR) [14]. In contrast to conventional T cells, iNKT cells recognize self and exogenous lipid antigens presented by the MHC class I-like molecule CD1d [15], [16]. Upon lipid recognition through their TCR, iNKT cells secrete a range of cytokines with opposing effects on immune responses, which contribute to the activation of NK, T and B cells, and dendritic cells (DCs) [17]. This functional property establishes iNKT cells as innate immune effector cells as well as regulators of adaptive immune responses. Numerous studies have shown that, upon activation, iNKT cells either suppress or enhance immune-mediated responses during inflammation, cancer, autoimmune diseases and infection [15], [18], [19], [20]. There is evidence indicating that iNKT cell responses to viral infection require interaction of iNKT cells with DCs where co-stimulatory interactions may play an important role in determining the outcome of the response.
The PD-1: PD-1 ligand co-stimulatory interaction is a recently characterized signaling pathways within the B7: CD28 superfamily. This co-stimulation consists of the PD-1 receptor and its two ligands PD-L1 (B7-H1) and PD-L2 (B7-DC). PD-L1 is expressed in a wide variety of tissues and by a number of different cell types including T cells, NK T cells and DCs [21], [22], [23], [24], and its expression is up-regulated by IFN- γ [25], [26]. The expression of PD-L2 is much more restricted and appears to be limited to a subset of bone marrow-derived cells, including DCs and macrophages [23], [27]. PD-1 is an inhibitory co-receptor that is expressed on T, iNKT and B cells after activation that delivers an inhibitory signal upon recognition of either of its ligands. Cytokines such as IFN-γ and IL-4 that are produced after T cell activation increase the expression of PD-1 ligands at mucosal surfaces, leading to attenuate the immune response [28]. Although PD-1 has been well characterized as a negative regulator of conventional CD4+ T cells, the role of PD-1 and its interaction with PD1 ligands in regulating activation and function of iNKT cells after infection with IAV has not been investigated.
In the present study, we examined the relative contribution of PD-L1 and PD-L2 to the modulation of immune responses after IAV infection. We determined that in the absence of PD-L2 animals succumb more rapidly compared to wild type animals after infection with a lethal dose of IAV virus. This is associated with an increased cellular infiltration into the lungs and enhanced pulmonary inflammation. In contrast, in PD-L1 deficient mice the severity of cellular infiltration and pulmonary inflammation is reduced. This differential regulation of the response to IAV infection is associated with alterations to cytokine production by iNKT cells. These results suggest that under normal circumstances PD-1/PD-L1 interactions are negative regulators of viral clearance, whereas PD-1/PD-L2 interactions are important for dampening deleterious inflammation. Therefore, our findings may have important clinical implications in the sense that targeting PD-1 co-stimulation in vivo may provide a novel therapeutic target for controlling and enhancing host immune responses to IAV allowing an enhanced response with reduced inflammation.
Materials and Methods
Mice
Female BALB/c ByJ mice were purchased from Jackson Laboratories. Jα18 −/− mice (backcrossed to BALB/c) were a gift from M. Taniguchi/T. Nakayama (Chiba University, Chiba, Japan) and S. Balk (Brigham and Women's Hospital, Boston, Massachusetts) [29]. PD-L1−/−, PD-L2−/− and PD-L1−/− PD-L2−/− double knockout mice backcrossed to BALB/cByJ mice for 11 generations were obtained from Arlene Sharpe (Harvard Medical School, Boston, Massachusetts) [30]. Mice were maintained and used according to institutional and National Institutes of Health guidelines in a pathogen-free facility. The Animal Care and Use Committee, University of Southern California approved all animal protocols. A loss of 25% or more of body weight was the criterion for euthanasia according to our IACUC protocol.
Influenza A infection
Six to eight week-old adult mice were anesthetized with ketamine (200 mg/kg)/xylazine (20 mg/kg) and inoculated intranasally (i.n.) with IAV (strain PR8, H1N1) in 50 μl saline. The virus was grown and harvested from 10-day embryonated chicken eggs as previously described [31]. A dose of 3000 PFU/mouse was chosen as the lethal dose. Control (mock-infected) mice were treated with i.n. allantoic fluid (AF) diluted 1∶500 in saline.
Flow cytometry analysis
In all experiments, lungs were aseptically removed, minced using sterile razor blades, and incubated in 1.6 mg/ml collagenase (CLS4, Worthington Biochemicals, Lakewood, NJ) and 30 µg/ml DNAse (Sigma-Aldrich, St. Louis, MI) at 37°C for 90 min. To achieve a single-cell suspension, lung fragments were pressed through a 70-µm pore nylon cell strainer using the flat end of a sterile 3-ml syringe plunger. Enzymatic action was terminated by washing cells twice in complete RPMI (RPMI 1640 with L-glutamine and 10% fetal bovine serum (FBS)) by centrifugation at 400× g for 5 min at 4°C. Leukocytes were isolated by centrifugation over a 30–70% Percoll gradient (GE Healthcare, Piscataway, NJ). Cells were then pre-incubated for 30 min. with normal rat serum, and washed before staining. iNKT cells were identified using various antibody combinations that included PE conjugated CD1d: PBS-57 loaded tetramer (NIH, NIAID tetramer core facility, Atlanta GA), TCRβ-allophycocyanin (APC) (clone H57-597, eBioscience, San Diego, CA) and CD4-APC-eFluor750 (clone RM4-5) (eBioscience). Cells were acquired on the FACSCanto II 8 color flow cytometer (BD Biosciences, San Jose, CA) and 10,000 events within the iNKT cell gate were collected. The data were analysed with FlowJo 8.6 software (Tree Star Inc., Ashland, OR).
Purification of iNKT cells and their sub-populations
Single cell suspensions were prepared from spleen in accordance with standard protocols. Splenocytes were labeled with PE-conjugated CD1d: PBS-57 loaded tetramer for 20 min at 4°C. Cells were then washed and magnetically labeled with anti-PE microbeads (Miltenyi Biotec, Auburn, CA). After washing iNKT cells were obtained by positive selection using an AutoMACS Pro (Miltenyi Biotec) using the Posseld program. This program is designed by the manufacturer for enrichment of rare cells using two cell enrichment columns. Purity of iNKT cells was >80% as determined by flow cytometry analysis. For detection of CD4+ and DN iNKT cells in lungs CD3-FITC and CD4-APC-eFluor750 (both eBioscience) were used. For in vitro studies, iNKT cells were negatively selected from splenocytes using a cocktail of PE conjugated mAbs against B220, CD62L, CD8α and CD11c, (BD Pharmingen, San Jose, CA) followed by incubation with anti-PE microbeads. The enriched iNKT cell splenocytes were confirmed to contain an average of >10% iNKT cells by tetramer staining and flow cytometry analysis.
Cytokine production by iNKT cells
Invariant NKT cells were positively selected from lungs of wild type, PD-L1−/− or PD-L2−/− mice five days post infection as described above. iNKT subsets were positively selected and subsequently stained with anti-CD4 antibody and sorted into CD4+ and DN subsets by FACS on a BD FACSAria III. Once positively selected iNKT cells were cultured for 48 h and the cells harvested for cytokine determination by quantitative reverse transcription polymerase chain reaction (RT-PCR) as previously described [32]. Briefly, total RNA was extracted from sorted subtypes or total iNKT cells using the RNAeasy mini kit (Qiagen, Valencia, CA) and cDNAs were generated with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA) according to the manufacturer's recommendations. Quantification of mRNA levels was carried out by quantitative real-time PCR on a CFX96 thermal cycler (Bio-Rad, Hercules, CA) with predesigned Taqman gene expression assays and reagents, as per manufacturer's instructions.
Activation of iNKT cells by dendritic cells in vitro
Negatively enriched iNKT cells were cultured in round bottom 96 well plates with 1×103 DCs positively isolated after 3 days from lungs of IAV infected wild type, PD-L1−/− or PD-L2−/− mice, as previously described [13]. CD11c+ DCs were isolated using CD11c microbeads (Miltenyi Biotec) according to the manufacturer's instructions. Supernatants were collected after 48 hours and cytokines levels were determined by ELISA (eBioscience).
Adoptive transfers
Purified iNKT cells (1×106) from PD-L1−/−, PD-L2−/− or wild type mice were adoptively transferred into Jα18−/− mice intravenously into the tail vein. Mice were challenged intranasally 24 hours after adoptive transfer with lethal viral dose of 3000 PFU in 50 µl saline.
BAL fluid analysis
After 5 days of IAV infection, the lungs from PD-L1−/−, PD-L2−/− or wild type mice were lavaged twice with 1 ml of PBS with 2% FBS and the fluid pooled as described previously [12]. The relative number of different types of leukocytes was determined from slide preparations of bronchoalveolar lavage (BAL) fluid stained with Hematoxylin and Eosin (H&E).
Lung Histology
Following 5 days post IAV infection, lungs from the sacrificed mice were perfused with 10% formalin-PBS and removed and fixed immediately in 10% neutral-buffered formalin (v/v). After overnight fixation, the lungs were processed for histology. The lung tissue was embedded in paraffin, 3 µm sections were cut and stained with H&E according to standard protocols. Sections were scanned using light microscope for inflammation. Images of these fields were captured by a Nikon eclipse TE 2000-S microscope (Nikon, USA) objective lens, total magnification 40X, with an in-line camera and assembled into multipanel figures using Adobe Photoshop software (version 7.0). Images were analyzed using the Leica Application suite (Leica Microsystems, Bannockburn, Ill).
Statistical Analysis
Data were analysed with a one-way ANOVA followed by a Tukey post-test between groups using PRISM v4 software (Graph Pad). P values of less than 0.05 were considered statistically significant with * P<0.05, ** P<0.01 and *** P<0.001. Data represents mean ± SEM.
Results
iNKT cells play an important role in the control of influenza viral infection
Previous studies have suggested an important role for iNKT cells in the protection against IAV in the C57Bl6/J background [10], [11]. Here we first tested whether iNKT cells are recruited to the lungs after IAV infection and whether these cells play a protective role in mice with BALB/c background. To this end, we first evaluated the frequency and the absolute number of iNK T cells in the lungs of BALB/c mice at day 1, 3 and 7 after infection. As shown in figure 1A and 1B, at day 3 after IAV infection there is a significantly higher number of iNKT cells in the lungs of infected mice compare to day 1, suggesting that IAV infection leads to recruitment of these cells to the lungs. At day 7 after infection, there is even higher frequency and number of iNKT cells in the lung compare to day 1 and 3.
Figure 1. iNKT cells reduce the sensitivity of BALB/c mice to IAV infection.

Jα18−/− mice and BALB/c mice (n = 10) were intranasally infected with a lethal dose (3000 PFU) of IAV. (A) Increased frequency of iNKT cells after IAV infection. Representative FACS plots of iNKT cell frequencies in the lung of BALB/c mice stained with CD1d: PBS57 loaded tetramer and TCR-β antibody, gated on viable lymphocytes with percentage of cells in the iNKT cell gate indicated next to the gate. (B) Total iNKT cell numbers in the lung were determined by flow cytometry at different days post infection (DPI). Data is presented as mean ± standard error of the mean (SEM), n = 3. * P>0.05 (C) The weight loss of Jα18−/− mice and BALB/c mice has been monitored daily after IAV infection. Jα18−/− mice show augmented weight loss compared to wild type mice. Data are representative of three independent experiments.
We next addressed the role of iNKT cells in reducing the sensitivity to IAV by infecting wild type BALB/c and Jα18−/− mice that lack iNKT cells and evaluated the loss of body weight as an index for influenza progression. As shown in figure 1C, we found that although initially both strains had the same body weight, after the second day post infection Jα18−/− mice display severer weight loss compare to wild type BALB/c mice (P<0.05). These data suggest that iNKT cells play a role in reducing the sensitivity to IAV infection also in BALB/c background and that this role of iNKT cells is not strain dependent.
PD-1 and PD-L1 expression on iNKT cells are modulated in response to IAV infection
After confirming the role of iNKT cells in reducing the sensitivity to IAV, we addressed the role of co-stimulatory molecule PD-1 and its ligands on iNKTcells. It is known that iNKT cells can express PD-1 and PD-L1 but do not express PD-L2. [13]. We sought to determine whether the expression of these molecules is modulated after IAV infection. To reach this aim, the expression of PD-1, PD-L1 and PD-L2 by iNKT cells subsets was assessed by flow cytometry 5 days after IAV infection. As shown in figure 2, expression of PD-1 is increased in CD4+ iNKT cells in IAV infected mice compare to mock treated control mice. Interestingly, double negative iNKT cells of IAV infected mice show even a greater expression of PD-1 compare to mock treated mice. Similarly, the level of PD-L1 expression by CD4+ and by double negative iNKT cells is higher in IAV infected compare to control mice. As expected, neither CD4+ nor double negative iNKT cells express PD-L2 in IAV infected or control mice. These data suggest that the protective role of iNKT cells may be mediated by co-stimulatory molecule PD-1 and its ligands.
Figure 2. Differential expansion of iNKT cell subsets to IAV infection.

Expression of PD-1, PD-L1 and PD-L2 on iNKT cells isolated from the lungs of control or 5 day post IAV infection in BALB/c mice. Dot plots are gated on iNKT cells that were identified as CD1d: PBS57 loaded tetramer+ TCR-βintermediate and show CD4 expression on the iNKT cells (Dot plots are representative of 5).
PD-L1 and PD-L2 modulate sensitivity to IAV infection in opposing directions
Next we examined the contribution of PD-1 ligands to the sensitivity to IAV infection at the functional level. To this end, we compared the severity of IAV infection in wild type BALB/c with mice lacking PD-L1 and those lacking PD-L2. Body weight loss, the composition of BAL, lung histology and virus titer in the lungs were assessed as indications of influenza severity. We found that in the absence of PD-L1, mice show significantly higher body weight as compared to wild type mice (P<0.05, Fig. 3A). In contrast, mice lacking PD-L2 show a greater weigh loss than wild type mice (P<0.05, Fig. 3A) indicating that PD-L1 contributes to sensitivity to IAV infection.
Figure 3. PD-L1 and PD-L2 affect the sensitivity and the degree of lung inflammation after infection with IAV.

PD-L2−/−, PD-L1−/− and BALB/c (n = 10) mice received a lethal dose of IAV (3000 PFU) intranasally. (A) Weight loss has been monitored by measuring body weight daily post infection (DPI) (B) BAL was performed in mice (n = 5) five days post IAV infection and analyzed for differential cellular infiltration. Bar graph shows total cell number (Total) and absolute number of eosinophils (EOS), monocytes/macrophages (MAC), lymphocytes (LYM) and neutrophils (PMN). ND: not detectable. (C) Representative image of lung sections taken 5 days post infection with 3000 PFU IAV stained with H&E (original magnification ×40, insert magnification ×100, scale bar 50 μm) from BALB/c, PD-L1−/− and PD-L2−/− mice. (D) Quantification of lung sections as shown by measurement of thickness of epithelium, left panel and number of inflammatory cells per 250 µm2, right panel. (E) Virus titration in the lungs shown as plaque forming units (PFU) per gram of lung tissue. Data are presented as mean ± SEM, * P>0.05, ** P>0.01 and *** P>0.001 as calculated by a one-way ANOVA with Tukey post hoc test and is representative of four independent experiments.
The increased weight loss of PD-L2−/− mice was associated with a significant increase in the number of macrophages and lymphocytes in BAL compared to wild type mice (P<0.05 and P<0.001, Fig. 3B). Histological analysis of lungs revealed that compare to wild type mice there is an increased number of inflammatory cells and epithelium thickness in the lungs of PD-L2−/− mice whereas, there is a decreased number of inflammatory cells and epithelium thickness in the lungs of PD-L1−/− mice (P<0.05, Fig. 3C–D). Similarly, the lungs of PD-L1−/− mice contain a significantly lower titer of IAV at day 3 and 7 post infection compare to wild type mice. IAV titer in lungs of PD-L2−/− mice is significantly higher compare to wild type mice at 3 and 7 days post infection.
Since we observed that PD-L1 and PD-L2 have opposing effects, we addressed the response to IAV infection in the absence of both of these co-stimulatory molecules. To this end, we infected PD-L1−/− PD-L2−/− double knockout or wild type BALB/c. Interestingly, we found that the there is no difference in the level of weight loss between wild type and PD-L1−/− PD-L2−/− double knockout mice (figure S1). These data suggest that PD-L1 and PD-L2 have the capacity to balance/neutralize each others effect and therefore, the absence of both results in no phenotype in IAV resistance or sensitivity.
Taken together, these results indicate that PD-L1−/− contributes to the sensitivity to infection and its severity, while PD-L2−/− plays a role in recovery and protection against IAV.
Lack of PD-L1 expression on iNKT cells reduces sensitivity to IAV infection
To determine whether the expression of PD-1 ligands on iNKT cells or development of these cells in the absence of PD-1 ligands underlies the altered sensitivity to IAV we performed a series of adoptive transfer experiments. iNKT cells were positively selected from the spleen of BALB/c, PD-L1−/− or PD-L2−/− animals and adoptively transferred into iNKT cell deficient Jα18−/− mice. As expected Jα18−/− mice that did not receive any iNKT cells had a high rate of weight loss that was reduced after the transfer of wild type iNKT cells (P<0.05, Fig. 4), confirming the essential role of these cells. In contrast mice that received PD-L2−/− iNKT cells demonstrated no significant difference in weight loss compare to animals that did not receive iNKT cells (Fig. 4). Whereas, transfer of PD-L1−/− iNKT cells resulted in enhanced protection from IAV as evident by the reduced weight loss compared to animals that received BALB/c iNKT cells and PD-L2−/− iNKT cells (P = 0.01, Fig. 4). These data clearly demonstrate that expression of PD-L1 specifically by iNKT cells contributes to influenza pathogenesis and severity. Moreover, our results suggest that although iNKT cells do not express PD-L2, their protective role against IAV requires the presence of this co-stimulatory molecule during the development of iNKT cells.
Figure 4. Lack of PD-L1 expression on iNKT cells reduces sensitivity to IAV infection.

iNKT cells (1×106) were positively isolated from spleen of BALB/c, PD-L1−/− and PD-L2−/− mice and adoptively transferred into iNKT cell deficient Jα18−/− mice. After 24 hours Jα18−/− mice received 3000 PFU influenza A intranasally. Weight loss was monitored at different time points after the infection with n = 5 per group and is representative of three independent experiments.
Cytokine profile of pulmonary iNKT cells is regulated by PD-L1 and PD-L2 co-stimulation
Our adoptive transfer studies suggest that even though iNKT cells do not express PD-L2 they are functionally affected by development in a PD-L2 deficient environment. It has been previously reported that iNKT cells from PD-L1 or PD-L2 knockout mice have altered cytokine polarization [13]. Therefore, we sought to determine how PD-L1 and PD-L2 signaling might affect the function of iNKT cells in response to IAV infection. Lung iNKT cells from IAV infected PD-L1−/− mice produced significantly higher levels of IFN-γ compared to wild type BALB/c and PD-L2−/− mice (Fig. 5). In contrast, higher levels of IL-4 were produced from IAV infected PD-L2−/− mice lung iNKT cells compared to PD-L1−/− lung iNKT cells (Fig. 5). PD-L1−/− lung iNKT cells also expressed significantly less IL-4 compared to wild type BALB/c mice after IAV infection (Fig. 5). These experiments indicate that signaling through PD-L1 and PD-L2 can skew the cytokines produced by pulmonary iNKT cells in response to IAV infection either towards more antiviral (PD-L1) or less antiviral (PD-L2) profile. The inflammatory cytokine, IL-13, was also significantly higher in PD-L2−/− mice compared PD-L1−/− mice (Fig. 5) whereas there was no significant difference in TNF-α between the groups of mice (Fig. 5).
Figure 5. PD-L1 and PD-L2 affect the cytokines produced by lung iNKT cells after IAV infection.

iNKT cells were positively selected from influenza A infected lungs on day 5 from PD-L1−/−, PD-L2−/− and wild type BALB/c mice and different cytokines were analysed by quantitative RT-PCR normalized to β-actin levels. Data presented as mean ± SEM. * P>0.05, ** P>0.01 and *** P>0.001 as calculated by a one-way ANOVA with Tukey post hoc test and is representative of five independent experiments with n of 5 per group.
PD-L1 and PD-L2 expression on dendritic cells regulates the iNKT cell after influenza infection
As demonstrated above, cytokine production by iNKT cells is altered by the absence of PD-L1 or PD-L2. As both PD-L1 and PD-L2 can be expressed by DCs [13], [33], [34] we sought to determine the role of DC expression of these co-stimulatory molecules on cytokine secretion by iNKT cells. DCs were isolated from the lungs of IAV infected PD-L1−/−, PD-L2−/− and BALB/c mice and loaded with an iNKT cell stimulating ligand, α-galactosylceramide, prior to co-culture with BALB/c iNKT cells. When using PD-L1−/− DCs production of IFN-γ was increased compared to BALB/c or PD-L2−/− DCs (Fig. 6). In contrast, PD-L2−/− DCs resulted in enhanced production of IL-4 compared to wild type and PD-L1−/− mice (Fig. 6). Therefore, PD-L1 and PD-L2 can exert effects on the cytokine profile of iNKT cells through their expression on the DCs that present antigens to and activate iNKT cells.
Figure 6. PD-L1 and PD-L2 expression on dendritic cells regulates the iNKT cell cytokine secretions after IAV infection.

Pulmonary DCs (103) were isolated from lungs of influenza A infected PD-L1−/−, PD-L2−/− and wild type BALB/c mice and loaded with 1 µg of α-galactosylceramide for 3 hours and then cultured with negatively enriched iNKT cells (1×104) from the spleen of naïve wild type mice. Supernatants were collected after 48 h and IL-4 and IFN- γ levels were measured by ELISA. Data presented as mean ± SEM. *** P>0.001 as calculated by a one-way ANOVA with Tukey post hoc test and is representative of four independent experiments with n of 5 per group.
Lack of PD-L1 leads to preferential expansion of double negative iNKT cell in IAV infected mice
Murine iNKT cells may be either CD4+CD8− (CD4+) or CD4−CD8− (DN) and it has been previously demonstrated that these different iNKT cell populations produce a different profile of cytokines [35], [36]. Therefore, we reasoned that the differences in the cytokines produced by the pulmonary iNKT cells in PD-L1−/− and PD-L2−/− compared to wild type could be due to differential composition of iNKT cell subpopulations. Hence, we determined the relative proportions of CD4+ and DN iNKT cells in the lung of IAV infected BALB/c, PD-L1−/− and PD-L2−/− at various days post infection. At five days post infection the frequency of DN iNKT was increased in the PD-L1−/− animals compared to BALB/c and PD-L2−/− mice (Fig. 7A). By day 10, the expansion of DN iNKT cells in the lung of PD-L1−/− was even greater and significantly different compared to wild type BALB/c and PD-L2−/− mice (Fig. 7A). Furthermore, in the lung of PD-L2−/− mice the frequency of CD4+ iNKT cells was significantly increased compared to wild type BALB/c mice (gray asterisk, Fig. 7A). These finding are in agreement with a recent study that demonstrated the protective role of DN iNKT cells during the resolution of IAV infection [35]. We reasoned that the protective role of DN iNKT cells was associated with enhanced IFN-γ secretion. Therefore, we analyzed IFN-γ gene expression of iNKT cell subsets following IAV infection. As shown in figure 7B both iNKT cell subpopulations from PD-L1−/− mice demonstrated enhanced IFN- γ expression compared to PD-L2−/− mice. The expression of IFN-γ by the DN iNKT cell subset from PD-L1−/− mice was significantly higher than wild type BALB/c mice (Fig. 7B) with BALB/c CD4+ iNKT cells expressing significantly more IFN-γ compared to CD4+ iNKT cells from PD-L2−/− mice (Fig. 7B). In contrast, IL-4 was increased in both iNKT cell subsets from PD-L2−/− compared to PD-L1−/− mice (Fig. 7B). These findings indicate that the role of iNKT cells in reducing the severity of IAV in absence of PD-L1 co-stimulation is associated with the increased DN iNKT cell frequency and enhanced IFN-γ production whereas in the absence of PD-L2 CD4+ iNKT cells are increased resulting in the production of more IL-4.
Figure 7. iNKT cell subpopulations are modulated in PD-L1−/− and PD-L2−/− animals after IAV infection.
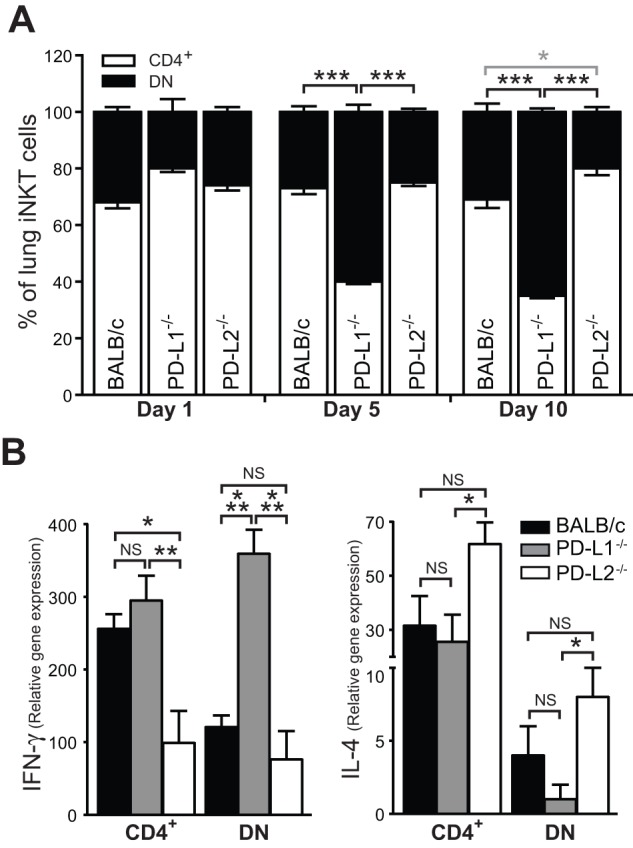
(a) Wild type BALB/c, PD-L1−/− and PD-L2−/− mice (n = 3) were infected with IAV intranasally. Lungs were collected at day 1, 5 and 10 post infection and the frequency of CD4+ and DN iNKT cells determined by flow cytometry and compared (DN: black asterisk, CD4+: gray asterisk). (b) CD4+ and DN iNKT cells subsets were sorted from IAV infected lungs on day 5 from PD-L1−/−, PD-L2−/− and wild type mice BALB/c and IL-4 and IFN- γ were analysed by quantitative RT-PCR normalized to β-actin levels (n = 8). Data presented as mean ± SEM. * P>0.05, ** P>0.01 and *** P>0.001 as calculated by a one-way ANOVA with Tukey post hoc test and is representative of five independent experiments.
Discussion
Our findings suggest an important role for the PD-1 ligands, PD-L1 and PD-L2 in regulating the immune response to infection with IAV. In the absence of PD-L1 mice are less sensitive to IAV infection as evident by reduced excessive cellular infiltration and inflammation and weight loss. In contrast, in the absence of PD-L2 pulmonary inflammation and cellular infiltration is increased and results in a more rapid weight loss. This altered response is associated with the production of different cytokines by iNKT cells. Adoptive transfer of iNKT cells from PD-L1 or PD-L2 deficient mice to animals lacking iNKT cells recapitulated the findings from the whole animal. Interestingly, we found that PD-L1 and PD-L2 have possibly balancing/neutralizing effects on each other as PD-L1−/− PD-L2−/− double knockout mice show a similar sensitivity to IAV infection as do wild type mice. We also identified a role for PD-1/PD-L interactions in controlling the expansion of iNKT cell subsets. In the absence of PD-L1 DN iNKT cells were increased whereas the opposite occurred in mice lacking PD-L2 with an increase in CD4+ iNKT cells. We have also demonstrated an important role for expression of PD-1 ligands on DCs in the control of cytokine production by iNKT cells. Altogether these results indicate that PD-1/PD-L1 interactions are vital modulators of the immune response and represent novel targets for therapeutic modification in vivo.
The role of iNKT cells in the control of viral infections is well characterized. In terms of viral immunity, iNKT cells are involved in immune surveillance and/or clearance of hepatitis B and C virus [37], [38], respiratory syncytial virus [39], herpes simplex virus 1 and 2 [40], [41], and HIV [42]. Several reports have indicated that iNKT cells are important in IAV clearance. In both murine infection models and human patients it has been demonstrated that iNKT cells are required for the generation of adaptive immune responses by reducing the suppressive effects of myeloid derived suppressor cells, which expand after infection [10]. Furthermore, activation of iNKT cells with a potent agonist, α-galactosylceramide, improves the disease course in part through enhancing innate immune responses [43]. iNKT cells have also been demonstrated to play an important role in the generation of influenza specific CD8+ T cell responses [11]. With all these findings it is not surprising that iNKT cell deficient Jα18−/− mice are more sensitive to IAV, exhibit a higher rate of weight loss and increased severity of disease. This influence of iNKT cells on multiple aspects during the course of IAV infection and clearance are likely associated with the ability of iNKT cells to produce both Th1 and Th2 cytokines very rapidly after activation [44]. Mounting evidence now suggests that two subsets of iNKT cells, based on their expression of CD4 or not (CD4+ or DN), are enriched in the production of certain cytokines [36], [45]. Therefore, it is interesting that after infection with IAV the relative proportions of iNKT cell subsets in the lungs are altered in the absence of PD-L1 or PD-L2.
In PD-L1−/− mice the frequency of DN iNKT cells, that are the major producers of IFN-γ, is increased. In contrast, in PD-L2−/− mice the proportion of CD4+ iNKT cells is increased resulting in the enhanced production of IL-4. Thus, not only does signaling through the PD-1/PD-L pathway influence the cytokines production by the iNKT cells but also controls the proportions of the iNKT cell subsets. The underlying mechanism for this finding is not clear as yet but may have important implications in attempts to polarize immune responses in a Th1 or Th2 biased manner. In addition to iNKT cells, previous reports suggest that T cells, in particular CD8+ T cells, play a major role in producing IFN-γ that assists in the clearance of IAV [46], [47]. However, recent studies provide evidence that iNKT cells are important in the generation of influenza specific CD8+ T cells responses [11]. Here, we found that iNKT cells from PD-L1−/− mice showed increased IFN-γ production after IAV infection. There is evidence indicating that IFN-γ possesses different opposing functions including pro inflammatory Th1 responses and immuneregulatory function depending on the cytokine milieu and the state of immune responses [48], [49], [50]. We also found that pulmonary iNKT cells from PD-L2−/− mice produce more IL-4 and IL-13 after influenza infection. Although not directly tested at the functional level, our findings suggest that the altered cytokine profile of iNKT cells in the absence of PD-L1 or PD-L2 may underlie the observed difference in IAV sensitivity. During pulmonary inflammation Th2 cells produce IL-4 and IL-13 [24], which interact with mast cells or eosinophils to mediate the inflammatory responses. Earlier, Moran and coworkers reported that treatment of mice with IL-4 resulted in a significant delay in viral clearance [51]. A recent report suggested that pulmonary IL-13 is responsible for the viral induced lung inflammation and airway hyperreactivity [52]. Acute viral infection leads to the production of IL-13 which has been shown to be responsible for many characteristics of asthma [52]. In our experiments we show that lack of PD-L2 is associated with increased production of IL-13 by iNKT cells which contributes to the augmented pathogenesis of IAV infection in these mice. However, on the other side, it is well established that Type I interferons are key cytokines produced by IAV-infected epithelial cells and monocytes and play an important role in clearance of virus [53], [54], [55].
The role of the molecules PD-L1 and PD-L2 in the activation and modulation of iNKT cells in influenza pathogenesis has not previously been investigated. The PD-1: PD-L pathway is best known for its ability to negatively regulate immune responses [33], [56]. Most of the evidence for this role comes from models of tolerance, cancer or chronic infections [30], [57], [58], [59], [60]. In the present study, we observed that PD-L1 expression modulates the cytokine profile of iNKT cells and consequently regulates the inflammatory phase after influenza infection. Nevertheless, our findings regarding the role of PD-1 ligands is focused on the acute phase of the influenza and the impact of these ligands on the pathogenesis of influenza during the late phase remains to be elucidated.
The PD-1 ligands exhibit distinct patterns of expression. Previous studies have shown that PD-L1 is expressed more broadly than PD-L2. PD-L1 is expressed on various hematopoietic and non-hematopoietic cells [21], [22], [33], whereas, expression of PD-L2 is predominantly on dendritic cells and macrophages [61]. It has been reported that PD-1 and PD-L1 are expressed in the lower airways of patients with IAV infection [62]. Interestingly, we found that IAV infection significantly, increases the expression of PD-1 and PD-L1 by DN respiratory iNKT cells. However PD-L2 expression by iNKT cells was minimal.
Our in vitro experiments would suggest that even though PD-L2 is not expressed on iNKT cells, development of iNKT cells in a PD-L2 deficient environment primes them towards exhibiting a Th2 like cytokine profile. This is demonstrated in the effects of adoptively transferred PD-L2−/−-derived iNKT cells on the severity of IAV infection in iNKT cell deficient mice. To further characterize what cell type could express PD-L2 and may be responsible for altering the cytokine production by iNKT cells we studied DCs. In a co-culture model naïve wild type iNKT cells activated by PD-L2−/− DCs demonstrated the same bias of cytokine production towards IL-4 and IL-13. Taken together, our findings suggest that development of iNKT cells in the absence of PD-L2 affects their phenotype towards reduced antiviral characteristics, whereas, lack of PD-L1 expression by iNKT cells or development of these cells in the absence of PD-L1 primes them towards an enhanced antiviral phenotype.
Resolution of influenza virus requires a potent anti-viral immune response on the one hand and regulating the amplitude of the elicited immune response on the other hand. The control of the balance between viral clearance and inflammation is a delicate process that is not fully understood to date. Here we have confirmed the important role of iNKT cells in the host immune response after influenza infection and have extended these findings to the influence of the PD-1/PD-L pathway on the iNKT cell response. Our data indicates that lack of PD-L1 improves the course of influenza disease, whereas the presence of PD-L2 provides signals to control the deleterious inflammation. Therefore, treatment strategies that target these pathways, including block PD-L1 or enhancing PD-L2 signaling, represent novel methods for modulating the host's response after influenza infection that allow more rapid viral clearance with minimized inflammation.
Supporting Information
PD-L1−/− PDL2−/− double knockout show a similar sensitivity to IAV as wild type BALB/c. (a) PD-L1−/− PDL2−/− double knockout and wild type BALB/c mice were infected with IAV (3000 PFU). Weight loss is monitored daily. Data presented as mean ± SEM.
(EPS)
Funding Statement
This work was funded by National Institutes of Health R01 AI066020 grant (to O.A.) and a Senior Research Training Fellowship from the American Lung Association (to V.L.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Barker WH, Mullooly JP (1980) Impact of epidemic type A influenza in a defined adult population. Am J Epidemiol 112: 798–811. [DOI] [PubMed] [Google Scholar]
- 2. Barker WH, Mullooly JP (1982) Pneumonia and influenza deaths during epidemics: implications for prevention. Arch Intern Med 142: 85–89. [PubMed] [Google Scholar]
- 3. Thompson WW, Comanor L, Shay DK (2006) Epidemiology of seasonal influenza: use of surveillance data and statistical models to estimate the burden of disease. J Infect Dis 194 Suppl 2 S82–91. [DOI] [PubMed] [Google Scholar]
- 4. Clements ML, Betts RF, Tierney EL, Murphy BR (1986) Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild-type virus. J Clin Microbiol 24: 157–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clements ML, Murphy BR (1986) Development and persistence of local and systemic antibody responses in adults given live attenuated or inactivated influenza A virus vaccine. J Clin Microbiol 23: 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murphy BR, Baron S, Chalhub EG, Uhlendorf CP, Chanock RM (1973) Temperature-sensitive mutants of influenza virus. IV. Induction of interferon in the nasopharynx by wild-type and a temperature-sensitive recombinant virus. J Infect Dis 128: 488–493. [DOI] [PubMed] [Google Scholar]
- 7. Droebner K, Haasbach E, Fuchs C, Weinzierl AO, Stevanovic S, et al. (2008) Antibodies and CD4(+) T-cells mediate cross-protection against H5N1 influenza virus infection in mice after vaccination with a low pathogenic H5N2 strain. Vaccine 26: 6965–6974. [DOI] [PubMed] [Google Scholar]
- 8. La Gruta NL, Kedzierska K, Stambas J, Doherty PC (2007) A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 85: 85–92. [DOI] [PubMed] [Google Scholar]
- 9. Lawrence CW, Ream RM, Braciale TJ (2005) Frequency, specificity, and sites of expansion of CD8+ T cells during primary pulmonary influenza virus infection. J Immunol 174: 5332–5340. [DOI] [PubMed] [Google Scholar]
- 10. De Santo C, Salio M, Masri SH, Lee LY, Dong T, et al. (2008) Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest 118: 4036–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paget C, Ivanov S, Fontaine J, Blanc F, Pichavant M, et al. (2011) Potential role of invariant NKT cells in the control of pulmonary inflammation and CD8+ T cell response during acute influenza A virus H3N2 pneumonia. J Immunol 186: 5590–5602. [DOI] [PubMed] [Google Scholar]
- 12. Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, et al. (2003) Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat Med 9: 582–588. [DOI] [PubMed] [Google Scholar]
- 13. Akbari O, Stock P, Singh AK, Lombardi V, Lee WL, et al. (2010) PD-L1 and PD-L2 modulate airway inflammation and iNKT-cell-dependent airway hyperreactivity in opposing directions. Mucosal Immunol 3: 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barral DC, Brenner MB (2007) CD1 antigen presentation: how it works. Nat Rev Immunol 7: 929–941. [DOI] [PubMed] [Google Scholar]
- 15. Bendelac A, Savage PB, Teyton L (2007) The biology of NKT cells. Annu Rev Immunol 25: 297–336. [DOI] [PubMed] [Google Scholar]
- 16. Godfrey DI, Kronenberg M (2004) Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest 114: 1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tessmer MS, Fatima A, Paget C, Trottein F, Brossay L (2009) NKT cell immune responses to viral infection. Expert Opin Ther Targets 13: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cerundolo V, Silk JD, Masri SH, Salio M (2009) Harnessing invariant NKT cells in vaccination strategies. Nat Rev Immunol 9: 28–38. [DOI] [PubMed] [Google Scholar]
- 19. Diana J, Brezar V, Beaudoin L, Dalod M, Mellor A, et al. (2011) Viral infection prevents diabetes by inducing regulatory T cells through NKT cell-plasmacytoid dendritic cell interplay. J Exp Med 208: 729–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Terabe M, Berzofsky JA (2007) NKT cells in immunoregulation of tumor immunity: a new immunoregulatory axis. Trends Immunol 28: 491–496. [DOI] [PubMed] [Google Scholar]
- 21. Dong H, Zhu G, Tamada K, Chen L (1999) B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 5: 1365–1369. [DOI] [PubMed] [Google Scholar]
- 22. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, et al. (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192: 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, et al. (2002) Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 169: 5538–5545. [DOI] [PubMed] [Google Scholar]
- 24.Kerzerho J, Maazi H, Speak AO, Szely N, Lombardi V, et al.. (2012) Programmed cell death ligand 2 regulates T(H)9 differentiation and induction of chronic airway hyperreactivity. J Allergy Clin Immunol. [DOI] [PMC free article] [PubMed]
- 25. Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, et al. (2002) Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation 9: 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rodig N, Ryan T, Allen JA, Pang H, Grabie N, et al. (2003) Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol 33: 3117–3126. [DOI] [PubMed] [Google Scholar]
- 27. Liang SC, Greenwald RJ, Latchman YE, Rosas L, Satoskar A, et al. (2006) PD-L1 and PD-L2 have distinct roles in regulating host immunity to cutaneous leishmaniasis. Eur J Immunol 36: 58–64. [DOI] [PubMed] [Google Scholar]
- 28. Maier H, Isogawa M, Freeman GJ, Chisari FV (2007) PD-1: PD-L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J Immunol 178: 2714–2720. [DOI] [PubMed] [Google Scholar]
- 29. Cui J, Shin T, Kawano T, Sato H, Kondo E, et al. (1997) Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science 278: 1623–1626. [DOI] [PubMed] [Google Scholar]
- 30. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, et al. (2006) Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med 203: 883–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Talon J, Salvatore M, O'Neill RE, Nakaya Y, Zheng H, et al. (2000) Influenza A and B viruses expressing altered NS1 proteins: A vaccine approach. Proc Natl Acad Sci U S A 97: 4309–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lombardi V, Stock P, Singh AK, Kerzerho J, Yang W, et al. (2010) A CD1d-dependent antagonist inhibits the activation of invariant NKT cells and prevents development of allergen-induced airway hyperreactivity. J Immunol 184: 2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, et al. (2004) PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A 101: 10691–10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y, Chung Y, Bishop C, Daugherty B, Chute H, et al. (2006) Regulation of T cell activation and tolerance by PDL2. Proc Natl Acad Sci U S A 103: 11695–11700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chang YJ, Kim HY, Albacker LA, Lee HH, Baumgarth N, et al. (2011) Influenza infection in suckling mice expands an NKT cell subset that protects against airway hyperreactivity. J Clin Invest 121: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coquet JM, Chakravarti S, Kyparissoudis K, McNab FW, Pitt LA, et al. (2008) Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4-NK1.1- NKT cell population. Proc Natl Acad Sci U S A 105: 11287–11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Durante-Mangoni E, Wang R, Shaulov A, He Q, Nasser I, et al. (2004) Hepatic CD1d expression in hepatitis C virus infection and recognition by resident proinflammatory CD1d-reactive T cells. J Immunol 173: 2159–2166. [DOI] [PubMed] [Google Scholar]
- 38. Kakimi K, Guidotti LG, Koezuka Y, Chisari FV (2000) Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J Exp Med 192: 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johnson TR, Hong S, Van Kaer L, Koezuka Y, Graham BS (2002) NK T cells contribute to expansion of CD8(+) T cells and amplification of antiviral immune responses to respiratory syncytial virus. J Virol 76: 4294–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ashkar AA, Rosenthal KL (2003) Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol 77: 10168–10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grubor-Bauk B, Simmons A, Mayrhofer G, Speck PG (2003) Impaired clearance of herpes simplex virus type 1 from mice lacking CD1d or NKT cells expressing the semivariant V alpha 14-J alpha 281 TCR. J Immunol 170: 1430–1434. [DOI] [PubMed] [Google Scholar]
- 42. Motsinger A, Haas DW, Stanic AK, Van Kaer L, Joyce S, et al. (2002) CD1d-restricted human natural killer T cells are highly susceptible to human immunodeficiency virus 1 infection. J Exp Med 195: 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ho LP, Denney L, Luhn K, Teoh D, Clelland C, et al. (2008) Activation of invariant NKT cells enhances the innate immune response and improves the disease course in influenza A virus infection. Eur J Immunol 38: 1913–1922. [DOI] [PubMed] [Google Scholar]
- 44. Van Dommelen SL, Degli-Esposti MA (2004) NKT cells and viral immunity. Immunol Cell Biol 82: 332–341. [DOI] [PubMed] [Google Scholar]
- 45. Liu TY, Uemura Y, Suzuki M, Narita Y, Hirata S, et al. (2008) Distinct subsets of human invariant NKT cells differentially regulate T helper responses via dendritic cells. Eur J Immunol 38: 1012–1023. [DOI] [PubMed] [Google Scholar]
- 46.Hatta Y, Hershberger K, Shinya K, Proll SC, Dubielzig RR, et al.. (2010) Viral replication rate regulates clinical outcome and CD8 T cell responses during highly pathogenic H5N1 influenza virus infection in mice. PLoS Pathog 6. [DOI] [PMC free article] [PubMed]
- 47. Hufford MM, Kim TS, Sun J, Braciale TJ (2011) Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J Exp Med 208: 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kishimoto K, Sandner S, Imitola J, Sho M, Li Y, et al. (2002) Th1 cytokines, programmed cell death, and alloreactive T cell clone size in transplant tolerance. J Clin Invest 109: 1471–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wood KJ, Sawitzki B (2006) Interferon gamma: a crucial role in the function of induced regulatory T cells in vivo. Trends Immunol 27: 183–187. [DOI] [PubMed] [Google Scholar]
- 50. Bocek P Jr, Foucras G, Paul WE (2004) Interferon gamma enhances both in vitro and in vivo priming of CD4+ T cells for IL-4 production. J Exp Med 199: 1619–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moran TM, Isobe H, Fernandez-Sesma A, Schulman JL (1996) Interleukin-4 causes delayed virus clearance in influenza virus-infected mice. J Virol 70: 5230–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, et al. (2011) Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol 12: 631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Garcia-Sastre A, Durbin RK, Zheng H, Palese P, Gertner R, et al. (1998) The role of interferon in influenza virus tissue tropism. J Virol 72: 8550–8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ronni T, Matikainen S, Sareneva T, Melen K, Pirhonen J, et al. (1997) Regulation of IFN-alpha/beta, MxA, 2′,5′-oligoadenylate synthetase, and HLA gene expression in influenza A-infected human lung epithelial cells. J Immunol 158: 2363–2374. [PubMed] [Google Scholar]
- 55. Sareneva T, Matikainen S, Kurimoto M, Julkunen I (1998) Influenza A virus-induced IFN-alpha/beta and IL-18 synergistically enhance IFN-gamma gene expression in human T cells. J Immunol 160: 6032–6038. [PubMed] [Google Scholar]
- 56. Singh AK, Stock P, Akbari O (2011) Role of PD-L1 and PD-L2 in allergic diseases and asthma. Allergy 66: 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Curran MA, Montalvo W, Yagita H, Allison JP (2010) PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 107: 4275–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dulgerian LR, Garrido VV, Stempin CC, Cerban FM (2011) Programmed death ligand 2 regulates arginase induction and modifies Trypanosoma cruzi survival in macrophages during murine experimental infection. Immunology 133: 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guleria I, Khosroshahi A, Ansari MJ, Habicht A, Azuma M, et al. (2005) A critical role for the programmed death ligand 1 in fetomaternal tolerance. J Exp Med 202: 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Paterson AM, Brown KE, Keir ME, Vanguri VK, Riella LV, et al. (2011) The programmed death-1 ligand 1: b7-1 pathway restrains diabetogenic effector T cells in vivo. J Immunol 187: 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Matsumoto K, Inoue H, Nakano T, Tsuda M, Yoshiura Y, et al. (2004) B7-DC regulates asthmatic response by an IFN-gamma-dependent mechanism. J Immunol 172: 2530–2541. [DOI] [PubMed] [Google Scholar]
- 62. Erickson JJ, Gilchuk P, Hastings AK, Tollefson SJ, Johnson M, et al. (2012) Viral acute lower respiratory infections impair CD8+ T cells through PD-1. J Clin Invest 122: 2967–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PD-L1−/− PDL2−/− double knockout show a similar sensitivity to IAV as wild type BALB/c. (a) PD-L1−/− PDL2−/− double knockout and wild type BALB/c mice were infected with IAV (3000 PFU). Weight loss is monitored daily. Data presented as mean ± SEM.
(EPS)