

Abstract
Mice lacking the 4th-group paralog Hoxd4 display malformations of the anterior vertebral column, but are viable and fertile. Here, we report that zebrafish embryos having decreased function of the orthologous hoxd4a gene manifest striking perturbations in vasculogenesis, angiogenesis and primitive and definitive hematopoiesis. These defects are preceded by reduced expression of the hemangioblast markers scl1, lmo2 and fli1 within the posterior lateral plate mesoderm (PLM) at 13 hours post fertilization (hpf). Epistasis analysis revealed that hoxd4a acts upstream of meis1.1 but downstream of cdx4 as early as the shield stage in ventral-most mesoderm fated to give rise to hemangioblasts, leading us to propose that loss of hoxd4a function disrupts hemangioblast specification. These findings place hoxd4a high in a genetic hierarchy directing hemangioblast formation downstream of cdx1/cdx4 and upstream of meis1.1. An additional consequence of impaired hoxd4a and meis1.1 expression is the deregulation of multiple Hox genes implicated in vasculogenesis and hematopoiesis which may further contribute to the defects described here. Our results add to evidence implicating key roles for Hox genes in their initial phase of expression early in gastrulation.
Introduction
The processes of vasculogenesis, angiogenesis and hematopoiesis establish the circulatory system and blood lineages of the embryo [1]–[4]. In zebrafish, the precursors of the vascular and hematopoietic systems derive from unipotential progenitors as well as a common progenitor known as the hemangioblast [5]. The hemangioblast is identifiable by the expression of the scl gene within the PLM [1], [3]. The first blood vessels are laid down through the process of vasculogenesis whereby angioblasts under the influence of vascular endothelial growth factor (VEGF) migrate and then aggregate as endothelial cells to form a vascular cord [2]. Subsidiary blood vessels are then formed by sprouting from existing vessels in a process termed angiogenesis [2].
In zebrafish, markers of vasculogenesis are apparent at 12 hpf with the formation of scl1-expressing cells in the anterior lateral plate mesoderm (ALM) and scl- and fli1-expressing angioblasts in the PLM [2], [3]. Between 18 and 22 hpf, angioblasts migrate to the midline, merge and lumenize to form the primary blood vessel [2]. Recent evidence suggests that the second major axial vessel, the posterior cardinal vein (PCV) is produced from the ventral side of the primary vessel through a process of ventral sprouting between 21 and 24 hpf; it expresses ephb4a and is distinguished from the dorsal aorta (DA) which expresses the arterial marker efnb2a [2]. From 20 hpf, new vessels sprout from the preexisting DA and at 32 hpf from the PCV [2]. From the DA, the primary intersegmental vessels (ISV) sprout dorsally, followed by the secondary ISV from PCV [2].
As in other vertebrates, blood development in zebrafish initiates with primitive hematopoiesis and is then followed by definitive hematopoiesis which establishes the adult blood lineages [1], [3]. In mammals and birds, the extra-embryonic yolk sac is the primary site of hematopoiesis, while in zebrafish markers of primitive hematopoiesis are apparent at 10 to 11 hpf (2 somite stage) in the ALM and PLM as revealed by the expression of hemangioblast markers scl1 and lmo2 [1], [3]. The first signs of commitment to primitive hematopoiesis are given by the expression of gata1 in a subset of scl1 positive cells of the PLM [1]. The myeloid progenitors and some endothelial cells are produced in the ALM. By 14 hpf (10 somite stage), precursors in the PLM commence migration to the midline and there form the intermediate cell mass (ICM), the major site of primitive hematopoiesis in the posterior of the embryo [1], [3]. Circulation initiates at 24 hpf following which a transient site of erythromyelopoiesis, the posterior blood island (PBI), functions over the next 12 hours [4].
The second and definitive wave of hematopoiesis begins in the ventral wall of the DA at 24 hpf [1], [3]. The ventral DA is considered to be the counterpart of the aorta-gonad-mesonephros (AGM) region of amniotes. HSCs arise in the AGM beginning at 24 hpf and are identifiable by expression of runx1 and cmyb. Starting at 48 hpf, the HSCs then seed what will become the hematopoietic stroma of the caudal hematopoietic tissue (CHT) at 3 dpf, followed by expansion and differentiation [4], [6]. The CHT, which assumes the role of the fetal liver in mammals, is responsible for definitive hematopoiesis until the kidney marrow becomes the final hematopoietic site beginning at 4 days post-fertilization (dpf). The kidney marrow and the thymus become the lifelong primary hematopoietic sites in larval and adult life [1], [3].
Several genes have been implicated in the control of successive steps in the formation of blood and the vasculature. In zebrafish, the earliest acting gene to be identified to date is cloche [7] which may be synonymous with the acyltransferase-encoding gene lycat [8]. Also very high in the genetic hierarchy governing vasculogenesis and hematopoiesis are cdx1 and cdx4 [9], [10]. The severe hematopoietic defects seen in zebrafish cdx4 single mutants and cdx1/cdx4 doubly deficient embryos are accompanied by the down-regulation of the hemangioblast and hematopoietic stem cell (HSC) marker scl1 and a number of Hox genes, and normal hematopoiesis can be rescued by the forced expression of hoxa9a [10].
Hox genes have also been implicated in hematopoietic and vasculogenic processes in mammals [11]–[13]. Their protein products are characterized by a conserved DNA-binding homeodomain and bind DNA cooperatively with members of the TALE homeoprotein family of cofactors [14] such as MEIS (Myeloid Ecotropic Integration Site), PBX (Pre-B-Cell Leukemia Homeobox), and PREP/PKNOX (Pbx Knotted Homeobox). There are 39 Hox genes in human and mouse organized into four clusters [15]. Extensive evidence implicates both Hox gene products and their cofactors in hematopoietic function [14], [16]–[21]. The ablation of any of several murine Hox genes, including Hoxb4, leads to defects in blood lineages. Hoxb4 and indeed all remaining Hox genes of paralog group 4 (Hoxa4, Hoxc4, Hoxd4) display potent HSC-promoting functions [13], [22]. Similar activities are shared by the murine HOX cofactors PREP1, MEIS1 and PBX1 as revealed by hematopoietic and cardiac deficiencies in corresponding mutant animals [14], [21], [23]–[27]. The natural role for Hox and Meis1 genes in normal hematopoiesis is further reflected in their well-documented contributions to human and murine leukemias [13], [28]–[31].
In teleosts, a genome duplication event generated 8 Hox clusters, with zebrafish retaining 7 clusters and 47 genes following evolutionary loss [32]. In zebrafish, hoxb6b, hoxb7a, and hoxa9a are known to regulate primitive hematopoiesis and are required for hematopoietic stem cell formation [9], [10], while deficiencies in pbx and meis function strongly compromise primitive and definitive hematopoiesis as well as the vasculature [18]–[20] pointing to a conservation of function across vertebrates.
Mice lacking Hoxd4 function show abnormalities of the anterior-most vertebrae, the atlas and axis, but are viable and fertile [33], [34]. We explored the extent to which function was conserved in zebrafish by assessing the phenotype of hoxd4a loss-of-function morphants. We demonstrate a surprising role for zebrafish hoxd4a in vasculogenesis, angiogenesis and primitive and definitive hematopoiesis. Defects resulting from hoxd4a deficiency can be rescued by capped mRNA for hoxd4a, meis1.1, scl, and fli1 but not by cdx4. Impaired meis1.1 function following hoxd4a knockdown leads to widespread Hox gene deregulation including the reduced expression of Hox genes previously implicated in hematopoiesis, vasculogenesis and angiogenesis. The cdx4, hoxd4a and meis1.1 genes are spatially and temporally co-expressed in shield-stage embryos within the ventral-most presumptive mesoderm, the site from which fate mapping studies have shown the hemangioblast to arise [5], [35], [36]. Moreover, shield-stage hoxd4a morphants display decreased meis1.1 expression in ventral-most presumptive mesoderm, and this meis1.1 expression is rescued by prior injection of capped hoxd4a mRNA. Thus, our data indicate that hoxd4a functions at the earliest times and near the top of a regulatory hierarchy directing hemangioblast formation, acting downstream or in parallel to cdx genes, but upstream or parallel to meis1.1. Another consequence of impaired hoxd4a and meis1.1 function is the subsequent deregulation of multiple Hox genes previously implicated in vasculogenic and hematopoietic processes. We conclude that hoxd4a has acquired (or retained) a much more important role in hematopoiesis and vasculogenesis than observed for its murine ortholog Hoxd4. Additionally, our results add to a growing body of evidence defining functions for Hox genes during their early phase of expression at gastrulation, well before they are deployed along the antero-posterior axis proper.
Methods
Ethics Statement
All animal work conformed to the Institutional Animal Care and Use Committee (IACUC) guidelines at Nanyang Technological University and was reviewed and approved under protocol number ARF SBS/NIE-A 0144 AZ.
Zebrafish
Wild-type AB and transgenic (fli1:EGFP) [37] and Tg(gata1:dsRed) [38] lines of zebrafish were maintained as described [39]. Zebrafish embryos were staged as detailed previously [40].
Antisense Morpholino and mRNA Microinjection
Antisense morpholino oligonucleotides (MOs) were obtained from Gene Tools Inc and injected into zebrafish embryos at 1–4 cell stage. Splice MOs targeting the 5′ splice site intron-exon junction (splice acceptor) are designated as follows MO1∶5′-GTT CAC TGT GAA GGA CAA AAT CAC A-3′ and exon-intron junction (splice donor), MO2∶5′- GCA AAG AGA GTG GAT CTT ACC CGT A-3′. MOs were diluted in Danieau’s buffer (0.4 mM MgSO4, 0.6 mM CaCl2, 0.7 mM KCl, 58 mM NaCl, and 5 mM Hepes, pH 7.6). Optimal doses for each MO were tested based on phenotypic effects, and each experiment was performed in parallel with a non-specific MO (standard control MO supplied by GeneTools Inc) injected at the same concentration.
Full-length cDNA for hoxd4a was generated by PCR using the primers shown in Table S1 in File S1 and cloned into pCR®II-TOPO®. The mMESSAGE mMACHINE Kit (Ambion) was used to synthesize capped mRNA. All mRNAs used for rescue experiments were assessed over a range of concentrations and the following amounts were chosen as the minimum sufficient to induce rescue: scl1 (100 pg), fli1a (30 pg), meis1.1 (100 pg) and hoxd4a (50 pg). All injections were done at the 1-cell stage.
qRT-PCR
Total RNA was extracted from embryos at 26–28 hpf using the PureLink™ Micro-to-Midi™ Total RNA Purification System (Invitrogen). 1 µg total RNA was treated with 1 U DNase I (Fermentas) at 37°C for 15 min and used for reverse transcription with SuperScript® III First-Strand Synthesis (Invitrogen). Quantitative reverse transcriptase PCR (qRT-PCR) was performed using SYBR GreenER™ qPCR SuperMix (Invitrogen) on a BioRad iCycler iQ5. The data (in biological triplicates) were normalized against zebrafish β-actin. The sequences of the oligonucleotides used for qRT-PCR are given in Table S2 in File S1.
Whole Mount in situ Hybridization and Imaging
Whole mount in situ hybridization (WISH) was performed as previously described [41]. For nkx2.5, a PCR fragment was amplified from 26–28 hpf embryonic cDNA using the primers shown in Table S1 in File S1. The reverse primer incorporated a T7 promoter allowing the PCR product to be used directly for probe production. DIG- labeled antisense RNA probes were transcribed from linearized template using T3, T7 or SP6 RNA polymerase (Roche). Probes for hoxb6b and hoxb7a were derived by RT-PCR-mediated amplification from RNA from 26–28 hpf embryos using primers described in Wan et al [42]. Embryos were incubated with anti-DIG antibody (Roche) and probes were detected using NBT/BCIP (nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate, toluidine salt) from Roche. Images were obtained on a Zeiss lumar V.12 stereo microscope with an Axio Cam MRc (Zeiss) and Axio Vision software. Some embryos were then dissected away from the yolk and flat-mounted prior to photography.
Alkaline Phosphatase Staining of Blood Vessels
Embryos at 72 hpf were fixed with 4% paraformaldehyde in PBS (phosphate buffered saline) at room temperature for 30 min, followed by treatment with pre-cooled acetone for 30 min at −20°C. After rinsing with PBS twice (5 min each), the embryos were equilibrated with NTMT buffer (100 mM Tris pH 9.5, 50 mM MgCl2, 100 mM NaCl, 0.1% Tween 20) for three times (each for 15 min) at room temperature. For alkaline phosphatase staining, the embryos were incubated in NBT/BCIP (Roche) solution for 30 min.
O-dianisidine Staining
Staining with o-dianisidine was carried out as described [43]. Control and MO-injected embryos at 48 and 72 hpf were manually dechorionated and fixed with 4% paraformaldehyde overnight. Fixed embryos were washed three times in PBS and then incubated in the staining buffer (0.6 mg/ml o-dianisidine, 10 mM sodium acetate (pH 5.2), 0.65% hydrogen peroxide, and 40% ethanol) for 15 min in the dark. Stained embryos were cleared and stored in benzyl benzoate/benzyl alcohol (2∶1, vol/vol).
Flow Cytometry
Control- and morpholino-injected Tg(gata1:dsRed) embryos (150 embryos each) were dechorionated manually at 48 hpf, rinsed for 15 min in calcium-free Ringer’s solution and passed five times through a 200 µl pipette tip to remove the yolk. The embryos were dissociated in 0.25% trypsin and 1 mM EDTA for 60 minutes at 28.5°C, during which the sample was passed six times through a 200 µl pipette tip every 10 minutes in order to obtain a single cell suspension. The dissociated cell suspension was centrifuged at 1000 g for 9 min at 4°C, the supernatant discarded, and the cells resuspended in ice cold 0.9× PBS plus 5% fetal bovine serum and passed by gravity through a 40 µm nylon mesh filter. Flow cytometry was performed on a BD LSII instrument (BD Biosciences). We used wild type zebrafish embryos as a negative control.
Results
Expression Pattern of hoxd4a
The expression of hoxd4a during zebrafish development was examined by WISH. Maternal transcripts were seen at the 1 cell stage, and zygotic transcripts were readily detected from 3 hpf to 48 hpf ( Fig. 1A–G,I,L ) and in a majority of cells until at least 75% epiboly (8–9 hpf) ( Fig. 1D ). An anterior expression border in neurectoderm is visible by 10 hpf (bud stage; data not shown) with further resolution of the border between rhombomeres 6 and 7 (r6/7) by 12 hpf ( Fig. 1E ). As observed previously [44], [45], at 26–28 hpf hoxd4a is expressed in the hindbrain with an anterior border at r6/7, in neural crest migrating to the future branchial arches, and in the pectoral fin fields ( Fig. 1F ). We also observed hoxd4a transcripts in the PBI ( Fig. 1I ). At no point were hoxd4a transcripts detected in the PLM or ICM of control embryos. By 48 hpf, hoxd4a expression was apparent in the AGM and in patches in the area of the caudal vein plexus ( Fig. 1L ).
Figure 1. Expression pattern of hoxd4a and phenotype of hoxd4a morphants.

(A–D) Detection of hoxd4a transcripts in early zebrafish embryos. Animal pole is to the top. (A) 1 cell. (B) 3 hpf. (C) 50% epiboly. (D) 75% epiboly. (E) Dorsal view of flat-mounted embryo at 12 hpf (5–6 somites) doubly stained for expression of hoxd4a and krox20a. The arrow indicates the anterior expression border of hoxd4a, while asterisks denote expression of krox20a in r3 and r5. Anterior is to the left. The rostral-most portion of the embryo is not captured in the image. (F) Dorsal view of a flat-mounted embryo at 26–28 hpf (>26 somites) showing hoxd4a expression in hindbrain, branchial arches (white arrowhead, left side only) and pectoral fin field (black arrowhead, right side only). The white arrow marks the hoxd4a anterior expression border in the hindbrain at the boundary between r6 and r7. Asterisks denote krox20a expression in r3 and r5. (G–H) Lateral views of 26–28 hpf embryos showing hoxd4a expression in the central nervous system (arrows) of control embryos (G) and reduced expression in hoxd4a morphants (H). (I–K) hoxd4a expression in the caudal half at 26–28 hpf as shown in lateral views, anterior to the left. hoxd4a expression seen in the PBI of control embryos (I, white arrow) is greatly reduced in hoxd4a morphants (J). Expression is rescued following co-injection with capped hoxd4a mRNA (K). (L–N) Results of In situ hybridization for hoxd4a at 48 hpf showing expression in the AGM and caudal vein plexus (site of future CHT) of control injected embryos (L, black arrows), greatly reduced expression in hoxd4a morphants (M), and rescued expression in embryos simultaneously injected with capped mRNA for hoxd4a (N). (O–T) Hemoglobin within RBCs revealed by o-dianisidine staining. Ventral views at 48 hpf (O,P) and 72 hpf (Q,R) show greatly reduced levels of hemoglobin in the ducts of Cuvier in hoxd4a morphants (P and R) vs controls (O and Q). Co-injection with capped mRNA for hoxd4a results in rescued RBC production (S). (T) O-dianisidine staining of the caudal half of a control larva (upper panel) and hoxd4a morphant (middle panel) showing overall reduction in hemoglobin levels at 72 hpf in morphants, and rescue by co-injection of capped hoxd4a mRNA (lower panel). (U,V) Lateral views of trunk regions of Tg(gata1:dsRed) embryos at 26 hpf, anterior to the left. The expression of dsRed within proerythroblasts is readily detected in the ICM (arrow) and PBI (arrowhead) of control-injected embryos (U) but not hoxd4a morphants (V). Scale bars = 100 µm. Ratios indicate the number of embryos showing the presented phenotype. (W) qRT-PCR shows an overall 3-fold reduction of hoxd4a expression in morphants at 26–28 hpf. Error bars = standard error. p = 0.02. (X) Quantitation by flow cytometry of dsRed-positive cells in Tg(gata1:dsRed) embryos at 48 hpf showing that morphants display an 88% reduction in RBCs relative to control-injected embryos. Error bars = standard deviation. p<0.0002.
hoxd4a Morphants Exhibit a Defect in Primitive Hematopoiesis
To understand the function of hoxd4a during zebrafish embryogenesis, we used two splice-blocking antisense MOs against either the hoxd4a splice donor or splice acceptor flanking the single intron interrupting the coding region. Both MOs provoked highly similar phenotypes and the results obtained with the splice-acceptor blocker are reported below. Injection of hoxd4a MO (8 ng) at the 1–4 cell stage resulted in severely deformed embryos that died by 7 dpf. However, at a reduced dosage of MO (4 ng), embryos exhibited grossly normal morphology up to 30 hpf but with a developmental delay of approximately 9 h. In all experiments reported below, we compared morphants and control embryos at equivalent developmental stages. In embryos of at least 26 hpf, we routinely assessed the length between the eye and the otic vesicle as suggested previously [40], as well as the size of the eye field. In some cases, we also counted somites.
Following injection of the anti-hoxd4a MO, in situ hybridization revealed a marked reduction of hoxd4a transcripts at 26–28 hpf (>26 somites) ( Fig. 1G–H and I–J ) confirmed by qRT-PCR ( Fig. 1W ). In particular, hoxd4a expression in the PBI ( Fig. 1I ) was lost in 26 hpf morphants ( Fig. 1J ). Transcripts for hoxd4a likewise failed to appear in the AGM and caudal vein plexus in 48 hpf morphants ( Fig. 1L–M ). Co-injection of capped hoxd4a mRNA rescued expression in all tissues ( Fig. 1K,N ). By contrast, the expression of pax2.1 (a marker for intermediate mesoderm, Fig. S1A–C vs B–D in File S1), nkx2.5 (a marker for cardiac mesoderm, Fig. S1E–F in File S1) and myod (a marker of paraxial mesoderm, Fig. S1G–H in File S1) was unaltered in the morphants, demonstrating that there was no gross defect in the overall patterning of the mesoderm.
The onset of blood circulation in zebrafish is at 25 to 26 hpf. In control-injected embryos at 48 hpf, red blood cells (RBCs) streamed normally over the yolk, through the ducts of Cuvier toward the heart, and through the dorsal aorta, posterior cardinal vein and caudal plexus. By contrast, in hoxd4a morphants the number of circulating RBCs was considerably reduced (n = 96/100) (Videos S1, S2, S3), but was strongly rescued by co-injection with capped mRNA for hoxd4a (Video S2). Reduced RBC production at 48 hph ( Fig. 1O–P ) and 72 hpf ( Fig. 1Q–T ) was confirmed by staining for hemoglobin with o-dianisidine. RBC production was strongly rescued by co-injection with capped hoxd4a mRNA ( Fig. 1S,T ). O-dianisidine staining also revealed pooling of blood in the head and trunk in some embryos (Fig. S1I–J in File S1; Video S3) suggestive of vascular defects. A lack of RBCs was also apparent by observation of the heart (Fig. S1K–L in File S1). Apart from anemia, pericardial edema and areas of edema over the yolk sac were also observed from 36 hpf (Fig. S1M–O in File S1). In addition, the heart rate was also mildly but significantly slower in morphants at 26–28 hpf and 48 hpf (Fig. S1P in File S1), and may have affected definitive hematopoiesis (see Discussion).
As an independent assessment of the extent of erythropoiesis in hoxd4a morphants, we made use of the Tg(gata1:dsRed) transgenic line [38] in which the gata1 promoter drives expression of the Discosoma red fluorescent protein (dsRed) in erythrocytic lineages during primitive and definitive hematopoiesis. While a strong signal was observed in the ICM and PBI of control embryos at 26 hpf, little could be detected in hoxd4a morphants ( Fig. 1U,V ) suggesting a deficit of erythrocytic progenitors during primitive hematopoiesis. To quantify the apparent reduction of erythrocyte numbers, we performed flow cytometry on cells isolated from Tg(gata1:dsRed) controls and morphants at 48 hpf. The results show that morphants have only 12% as many dsRed-positive cells as controls ( Fig. 1X ). Together, these findings support a marked failure of primitive hematopoiesis following hoxd4a knockdown.
To assess the effect of reduced hoxd4a function on the genetic programme directing erythroid development, expression of the erythroid lineage-specific markers gata1 and β embryonic globin 1 (hbbe1) was analyzed by WISH. Both gata1 and hbbe1 transcripts were diminished in the posterior PLM at 13 hpf (∼ 8 somites) ( Fig. 2A–B vs C–D) and the ICM and PBI at 26–28 hpf (Fig. S2A–B vs C–D in File S1), establishing a defect in primitive hematopoiesis. Co-injection of capped hoxd4a mRNA along with the hoxd4a MO significantly rescued the expression of both erythroid markers ( Fig. 2E–F ; Fig. S2E–F in File S1), specifically implicating hoxd4a in the phenotype.
Figure 2. hoxd4a knockdown disrupts primitive hematopoiesis and is highly specific.

In situ hybridization revealing expression at 13 hpf of erythroid lineage markers gata1 (A,C,E) and β embryonic globin 1 (hbbe1) (B,D,F). (A,B) Normal expression of gata1 and hbbe1 in the LPM. (C,D) Expression of gata1 and hbbe1 is strongly down-regulated in hoxd4a morphants, but rescued by co-injection of capped mRNA for hoxd4a (E,F). All embryos have been flat-mounted and are shown in dorsal view. Anterior is to the left. Ratios in the bottom left corner of all panels indicate the number of embryos showing the presented phenotype. ctrl, embryos injected with a non-specific morpholino. MO, embryos injected with the anti-hoxd4a morpholino. hoxd4a mRNA, embryos simultaneously injected with the anti-hoxd4a MO plus capped mRNA for hoxd4a. Scale bars equal 100 µm. All images are at the same magnification.
Loss of hoxd4a Impairs Definitive Hematopoiesis
Between 26 to 30 hpf, definitive hematopoiesis originates at the ventral wall of the DA, with expression of runx1 and cmyb marking the HSCs [1], [3]. The expression of runx1 and cmyb in hoxd4a morphants at 26–28 hpf ( Fig. 3A–B vs C-D) and 48 hpf ( Fig. 3E–F vs G-H) was severely down-regulated. The expression levels of markers of primitive and definitive hematopoiesis were quantitated by qRT-PCR and showed a strong reduction in hoxd4a morphants and significant rescue upon co-injection with capped hoxd4a mRNA ( Fig. 3I ). Thus, the knockdown of hoxd4a results in the impairment of both primitive and definitive hematopoietic lineages.
Figure 3. hoxd4a expression is required for transient and definitive hematopoiesis.

In situ hybridization on 28 hpf (A–D) and 48 hpf (E–H) embryos showing expression of runx1 (A,C,E,G) and cmyb (B,D,F,H) in presumptive HSCs arising in the PBI (arrows in A and B) and AGM (arrows in E and F). Expression of both genes was severely reduced in hoxd4a morphants (C,D and G,H). Ratios in the bottom right corner of images indicate the fraction of embryos showing the presented phenotype. ctrl, embryos injected with a non-specific morpholino. MO, embryos injected with the anti-hoxd4a morpholino. hoxd4a mRNA, embryos simultaneously injected with the anti-hoxd4a MO plus capped mRNA for hoxd4a. Scale bars equal 100 µm. All images are at the same magnification. (I) qRT-PCR confirms the strong depletion of hematopoietic gene expression in hoxd4a morphants at 26–28 hpf, and restored expression following co-injection with capped mRNA for hoxd4a. Samples were normalized to β-actin. Error bars indicate standard error. By comparison to controls and rescuants, the gene expression levels of all morphants were statistically different to p≤0.02 except for gata1 control vs morphant (p = 0.04) and hbbe1 rescuant vs morphant (p = 0.09).
Knockdown of hoxd4a Disrupts Endothelial Development
In addition to reduced numbers of blood cells, hoxd4a morphants appeared to lack a normal vasculature. At 48 hpf, blood cells circulated normally in the axial vessels and tail region in control embryos, whereas in hoxd4a morphants, little or no blood flow could be observed or was highly irregular with RBCs appearing to become blocked in their path through the ISVs. Blood flow was strongly rescued by co-injection with capped hoxd4a mRNA (Video S2).
The state of the vasculature in hoxd4a morphants was investigated through the use of the fli1:EGFP transgenic line which expresses enhanced GFP under the control of the fli1 locus in endothelial cells [37]. While control embryos at 72 hpf exhibited normal vascular phenotypes with the formation of primary ISV joining the dorsal longitudinal anastomotic vessel (DLAV) ( Fig. 4A–B ), their morphant counterparts displayed severely impaired sprouting of ISV precursors and greatly weakened GFP signal from the region of the presumptive DA ( Fig. 4C–D ). In approximately a fifth of morphant embryos, the caudal vein plexus was replaced by an amorphous mass of endothelial tissue ( Fig. 4E–F ), while anterior bifurcation of the presumptive aortic vessel could not be observed in others (data not shown).
Figure 4. Loss of hoxd4a function impairs development of the vasculature.

(A–D) Fluorescent images of the trunk and tail regions of Tg(fli1:EGFP) embryos at 48 hpf. The panels present merged bright field and fluorescent images (A,C) or fluorescent images only (B,D) The normal pattern of the vasculature (A,B) is severely disrupted in hoxd4a morphants (C,D) Dorsal extremities of ISV sprouts that fail to contact the DLAV are marked by white dots (D). The caudal vein plexus of control embryos (E, arrowheads) is replaced by a disorganized mass of endothelial tissue in hoxd4a morphants (F, arrowheads). (G–I) Alkaline phosphatase staining at 72 hpf revealing the vasculature in (G) control-injected larvae, (H) hoxd4a morphants, and (I) rescued larvae co-injected with capped mRNA for hoxd4a. Dorsal aorta (DA), posterior cardinal vein (PCV), inter-segmental vessels (ISV), caudal artery (CA), dorsal longitudinal anastomotic vessel (DLAV), caudal vein (CV) and vertebral artery (VTA). All images show lateral views, with anterior to the left and dorsal on top. Scale bars equal 100 µm.
The effects of hoxd4a knockdown on vasculogenesis and angiogenesis were also assessed by exploiting the high endogenous levels of alkaline phosphatase characteristic of endothelial cells. In control embryos, intense signals revealed well-formed ISV, subintestinal vessels (SIVs) and vertebral artery (VTA), whereas such vessels were indiscernible or highly reduced in hoxd4a morphants ( Fig. 4G–H ). Co-injection with hoxd4a mRNA yielded a marked rescue of ISV, SIV and VTA ( Fig. 4I ). The pooling of blood that we observed might be attributable to hemorrhaging resulting from these defects in vascular development (Fig. S1I–J in File S1; Video S3). Similar hemorrhaging has been observed in fli1 morphants [46]. Together, our results confirm profound defects in vasculo- and angiogenic processes in hoxd4a morphants.
To evaluate the effect of hoxd4a knockdown on genetic programs directing vasculogenesis and angiogenesis, we performed in situ hybridization for the endothelial markers fli1 and flk1. Both genes were significantly reduced in the LPM of hoxd4a morphants at 13 hpf ( Fig. 5A–B vs C–D), and in the major trunk vessels and ISVs at 26 hpf (Fig. S3A–B vs C–D in File S1), implying that hoxd4a morphants are defective in axial vasculature. Co-injection of capped mRNA for hoxd4a, rescued both fli1 and flk1 expression, establishing a specific role for hoxd4a in the altered gene expression pattern ( Fig. 5E–F ; Fig. S3E–F in File S1).
Figure 5. Knockdown of hoxd4a disrupts the endothelial programme in zebrafish embryos.

In situ hybridization at 13 hpf revealing expression of pan-endothelial markers fli1 and flk1 in control-injected embryos (A,B), hoxd4a morphants (C,D), and rescued embryos co-injected with anti-hoxd4a MO and capped mRNA for hoxd4a (E,F). (G,I) Expression of the marker of arterial identity efnb2a in controls (G) and hoxd4a morphants (I). (H,J) Expression of the endothelial inducer vegf in controls (H) and hoxd4a morphants (J). (K,L) Expression of the venous marker ephb4a in controls (K) and hoxd4a morphants (L). Ratios indicate the fraction of embryos showing the presented phenotype. All images show dorsal views with anterior to the left except H, J, K and L which are lateral views with anterior to the left. Scale bars equal 100 µm. (M) qRT-PCR results showing depletion at 26–28 hpf of angioblast and vascular gene expression in hoxd4a morphants and rescue by co-injection of capped mRNA for hoxd4a. Samples were normalized to β-actin. Error bars indicate standard error. By comparison to controls and rescuants, the gene expression levels of all morphants were statistically different to p≤0.02 except for lmo2 control vs morphant (p = 0.04) and lmo2 rescuant vs morphant (p = 0.05).
Early in development, a common primary blood vessel forms at the midline, partly under the influence of VEGF secreted by ventral somitic tissue. The DA and PCV derive from this common primary vessel through a process of ventral sprouting dependent on efnb2a and ephb4a [2]. We observed an amorphous mass of tissue in the region of the caudal vein plexus at 48 hpf ( Fig. 4F ), a site of hoxd4a expression ( Fig. 1H ) and a failure of anterior aortic bifurcation (data not shown), suggesting defects in primary vessel formation. We therefore assessed the expression of vegf, the arterial marker efnb2a and the venous marker ephb4a. At 13 hpf, the expression of efnb2a and vegf was significantly reduced in the morphants, implicating defective artery specification ( Fig. 5G–H vs I–J). Likewise, the expression of the vein marker ephb4a was reduced at 13 hpf ( Fig. 5K–L ). The expression of efnb2a remained attenuated at 26–28 hpf, but that of ephb4a had recovered (Fig. S3 G–J in File S1). Results for fli1, flk1, vegf and efnb2a were quantified by qRT-PCR on RNA extracted from 26–28 hpf embryos. The results confirm the decreased expression of these genes in hoxd4a morphants, and the ability of co-injected mRNA for hoxd4a to rescue their expression ( Fig. 5M ).
A Role for hoxd4a in Hemangioblast Formation through Regulation of meis1.1
The hemangioblast is the common precursor to the blood and endothelial lineages and is defined by early expression of such genes as fli1, scl1 and lmo2 in both the ALM and PLM, and gata5 in the ALM only [1], [3], [46], [47]. The severe defects in both blood and endothelial lineages in hoxd4a morphants suggested that the hemangioblast itself may be compromised. To assess this idea, the expression of hemangioblast markers scl1 and lmo2 was analyzed by in situ hybridization. Significant down-regulation of both genes was observed in morphants at 13 hpf ( Fig. 6A–B vs C–D). The impaired expression of scl and lmo2 persisted at 26 hpf (Fig. S4A–B vs C–D in File S1), suggesting that their later independent functions in definitive hematopoiesis were also compromised. Co-injection of capped hoxd4a mRNA rescued scl and lmo2 expression at 13 hpf ( Fig. 6E–F ) and 26 hpf (Fig. S4E–F in File S1).
Figure 6. hoxd4a is required for hemangioblast formation.

(A,B) Normal expression at 13 hpf of posterior hemangioblast markers scl1 (A) and lmo2 (B) in the PLM. (C,D) Expression of these markers is greatly reduced in hoxd4a morphants, but is rescued by co-injection of capped mRNA for hoxd4a (E,F). Ratios indicate the fraction of embryos showing the presented phenotype. Anterior is to the left. A, C and E are dorsal views of flat-mounted specimens while B, D and F are lateral views. Scale bars equal 100 µm. A is at a lower magnification than C and E.
To establish the hierarchy between these key regulators, the hoxd4a MO was co-injected with capped mRNA for either scl or fli1. The expression of scl and lmo2 was rescued in both cases ( Fig. 7A–B vs C-D; Fig. S5A–B vs C–D in File S1), placing the action of hoxd4a above these early acting effectors of hemangioblast specification [3], [46].
Figure 7. scl1 and lmo2 act downstream of hoxd4a to direct formation of the hemangioblast.
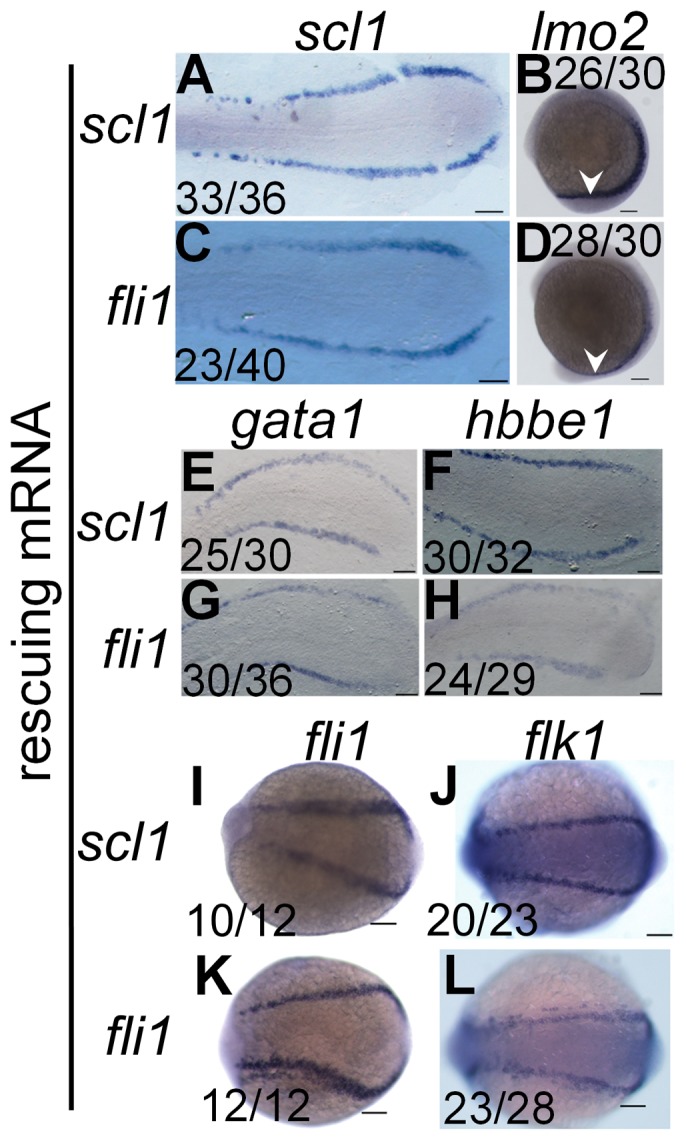
All images are of hoxd4a morphants at 13 hpf. In situ hybridization was performed to detect expression of scl1 and lmo2 (A–D), gata1 and hbbe1 (E–H) and fli1 and flk1 (I–L). To test for rescue of gene expression, embryos were co-injected with capped mRNA for either scl1 or fli1 as indicated on the left. Ratios indicate the fraction of embryos showing the presented phenotype. Scale bars equal 100 µm.
Consistent with the above findings, expression of gata1 and hbbe1 at 13 and 26 hpf was rescued by co-injection of scl and fli1 mRNA ( Fig. 7E–H ; Fig. S5E–H in File S1), though rescue by fli1 at 13 hpf was incomplete. Expression of the vasculogenic markers fli1 and flk1 was likewise rescued by scl and fli1 mRNA at 13 hpf ( Fig. 7I–L ) and 26 hpf (Fig. S5I–L in File S1). Together, these results reveal that hoxd4a occupies a very high position in the transcriptional hierarchy governing hemangioblast formation or function.
A number of studies have demonstrated major regulatory roles for Hox transcription factors in hematopoietic cell fate decisions [11]–[13], [22]. HOX proteins interact with other DNA-binding cofactors including TALE homeoproteins of the MEIS and PBX families, and Meis homologs have been implicated in the earliest stages of hematopoiesis and vasculogenesis in mice and zebrafish [18]–[20]. We therefore examined the expression of meis1.1 in hoxd4a morphants.
At 13 hpf, meis1.1 expression was significantly reduced in the PLM of hoxd4a morphants (Fig. S6E–F in File S1), while at 26–28 hpf meis1.1 expression was reduced throughout the embryo ( Fig. 8A–B and below) and could be rescued with capped hoxd4a transcripts ( Fig. 8C ). These results strongly suggested that one of the primary causes of hematopoietic and vasculogenic defects in hoxd4a morphants may be the loss of meis1.1 function. To test this notion, we attempted to rescue the expression of the genetic markers of hemangioblast formation, primitive hematopoiesis and vasculogenesis (scl, gata1 and fli1) in hoxd4a morphants by co-injection with mRNA for meis1.1. All three markers were strongly rescued at 13 hpf ( Fig. 8D–L ). Likewise, restoration of meis1.1 rescued the normal development of the vasculature including the DA, ISV, DALV and SIV as revealed in fli1:EGFP transgenics ( Fig. 8M ) and following visualization of the vasculature with alkaline phosphatase ( Fig. 8N ). The circulation is also largely restored in these embryos, especially robust in the DA, though less evident in the return flow through the caudal vein plexus and PCV (Video S4).
Figure 8. meis1.1 is down regulated in hoxd4a morphants and meis1.1 mRNA rescues hematopoietic and vasculogenic gene expression in hoxd4a morphants.

Expression of meis1.1 in control (A), morphant (B) and hoxd4a-rescuant (C) embryos at 26–28 hpf. (D–I) At 13 hpf, normal expression of gata1, scl1 and fli1 (D–F) is reduced in hoxd4a morphants (G–I), but rescued upon co-injection with capped mRNA for meis1.1 (J–L). (M,N) Rescue of vascular patterning in hoxd4a morphants by co-injection with capped mRNA for meis1.1 as visualized in Tg(fli1:EGFP) transgenics at 48 hpf (M) and by alkaline phosphatase staining at 72 hpf (N). Scale bars equal 100 µm.
Along with cloche [7], the cdx1 and cdx4 genes are among the earliest effectors of the hemangioblast lineage [9], [10]. Doubly deficient cdx1/cdx4 embryos lack midline vasculature and show a complete failure of hematopoiesis [9]. In hoxd4a morphants, cdx4 expression is unaltered at 13 hpf (Fig. S6A–B in File S1) and 26 hpf (Fig. S7Q–R in File S1). Moreover, injection of capped cdx4 mRNA fails to rescue defects of hematopoiesis or the vasculature in hoxd4a morphants (data not shown). We conclude that hoxd4a acts downstream or parallel to cdx1/cdx4 but upstream or parallel to meis1.1.
We then asked whether cdx4, hoxd4a and meis1.1 are ever temporally and spatially co-expressed so as to directly implement this hierarchy in a cell-autonomous fashion. Prior fate-mapping studies have established that precursors to the PLM hemangioblasts arise from the ventral-most presumptive mesoderm of shield-stage embryos [5], [35], [36], a tissue in which hoxd4a is expressed ( Fig. 1C ). In situ hybridization on shield-stage embryos showed strong overlapping expression of cdx4, hoxd4a and meis1.1 in ventral-most mesoderm ( Fig. 9A,C,F ). Importantly, knockdown of hoxd4a provoked a reduction in hoxd4a and meis1.1 expression in this same tissue ( Fig. 9D,G ). By contrast, cdx4 expression was unaffected ( Fig. 9B ). Importantly, co-injection of hoxd4a mRNA restored hoxd4a transcript levels and rescued the expression of meis1.1 ( Fig. 9E,H ). qRT-PCR on mRNA from controls, hoxd4a morphants and rescuants at the shield-stage validated results from in situ hybridization ( Fig. 9I ). The co-expression of hoxd4a with cdx4 and meis1.1 in tissue fated to give rise to PLM hemangioblasts, and the control of meis1.1 expression by hoxd4a in this same tissue, strongly suggests that the failure to form PLM hemangioblasts in hoxd4a morphants is due to defects initiated at the shield stage.
Figure 9. Expression of meis1.1, but not cdx4, is impaired in hoxd4a morphants at the shield stage.

Expression of cdx4 (A,B), hoxd4a (C–E) and meis1.1 (F–H) in control (A,C,F), hoxd4a morphants (B,D,G) and rescuants injected with hoxd4a mRNA (E,H) observed at the shield stage. Asterisks denote the ventral-most mesoderm fated to give rise to hemangioblast in addition to unipotential hematopoietic and angiogenic progenitors. Ratios indicate the fraction of embryos showing the presented phenotype. Scale bars equal 100 µm. (I) qRT-PCR was used to quantitate relative mRNA levels in controls, morphants and rescuants as indicated. Error bars present the standard error. Decreased expression of hoxd4a and meis1.1 in hoxd4a morphants is significantly lower than both controls and rescuants to p≤0.02.
Members of the MEIS family regulate Hox gene expression in both invertebrates and vertebrates [14], [48], [49]. Both gene families have likewise been implicated in hematopoietic and angiogenic processes [13], [18]–[20], [22]. We therefore wished to determine whether knockdown of hoxd4a and subsequent reduction in meis1.1 function might have provoked Hox gene deregulation. We assessed the expression of multiple Hox genes following hoxd4a knockdown at 26–28 hpf by quantitative RT-PCR ( Fig. 10A ). More than half of the Hox genes tested showed significant down-regulation, including hoxb4a, hoxb6b, hoxb7a and hoxa9a, all of which have been strongly implicated in hematopoietic processes. Likewise, hoxd3a, whose ortholog has been shown to promote angiogenesis in the mouse [50], [51], was also down-regulated. Consistent with results from in situ hybridization, cdx4 expression was unaffected. In situ hybridization on a number of these Hox genes confirmed decreased expression at 13 and 26–28 hpf (Fig. S7 in File S1). These results validate the suggestion that the early loss of meis1.1 function leads to the deregulation of several, though not all, Hox genes, thereby providing a fuller explanation for how the initial impairment of a single Hox gene, hoxd4a, can lead to massive defects in hematopoiesis, vasculogenesis, and angiogenesis.
Figure 10. cdx4, meis1.1 and Hox gene expression in hoxd4a morphants, and a genetic pathway for specification of hemangioblasts and unipotential stem cells.

(A) qRT-PCR showing decreased expression of meis1.1 and many, but not all, hox genes in hoxd4a morphants at 26–28 hpf. By contrast, cdx4 levels are unchanged. Samples were normalized to β-actin. Error bars indicate standard error. All pairs marked with an asterisk meet statistical significance (p≤0.02). (B) Based on the results presented here and those in the literature, we propose a pathway in which hoxd4a and meis1.1 occupy sequential steps downstream of cloche and cdx1/4 in a genetic programme leading to the specification of hemangioblasts and unipotential angiogenic and hematopoietic stem cells. The effects of hoxd4a knockdown may be magnified through positive cross-regulatory interactions with meis1.1. The observed effects on the expression of multiple Hox genes could be due to the direct action of hoxd4a and meis1.1. Non-exclusively, cdx1 and cdx4 may act in conjunction with hoxd4a and meis1.1 in a feed-forward type of mechanism to regulate one or more of these same Hox genes with widespread consequences for vasculogenesis, angiogenesis and hematopoiesis at all levels.
Discussion
This study places hoxd4a near the top of a regulatory cascade directing hematopoiesis, vasculogenesis and angiogenesis in zebrafish embryos ( Fig. 10B ). This unexpected role of hoxd4a is at least partly due to its positive regulation of meis1.1, and is likely to have both direct and indirect consequences. Most directly, decreased hoxd4a function leads to reduced expression of meis1.1 in the ventral-most presumptive mesoderm of the shield-stage embryo, a tissue fated to produce the hemangioblast in addition to unipotential erythrocytic and endothelial progenitors [5], [35], [36]. A few hours later, hemangioblast markers are severely reduced in the PLM, leading us to propose that hoxd4a and meis1.1 loss of function at the shield stage interferes with the specification of hemangioblasts and unipotential progenitors. Less directly, hoxd4a knockdown leads to massive deregulation of Hox gene expression, including a number of Hox genes previously implicated in these processes such as hoxb4a, hoxb6b, hoxb7a and hoxa9a [9], [10].
In support of this model, injection of capped mRNA for either hoxd4a or meis1.1 rescues essentially all aspects of the knockdown phenotype. It is likely that much of the effect of the loss of meis1.1 expression follows from the impaired function of MEIS-PBX and MEIS-PBX-HOX transcriptional complexes [14], [20], [52], though functions of MEIS that are independent of HOX or PBX should also be affected.
Three reports describe the effects of meis1.1 loss of function on hematopoiesis and vasculogenesis in zebrafish embryos [18]–[20]. Only Ouwehand and co-workers [18] observed, as we do, defects in both processes. Suzuki and co-workers [19] observed defects in the vasculature, heart edema and weakened heartbeat resulting from decreased meis1.1 function, and implicated a downstream impairment in flk1 and vegf expression, consistent with our findings. Waskiewicz and co-workers [20] found that loss of meis1.1 function caused severe defects in erythropoiesis, but did not observe vasculature defects nor detect alterations in either flk1 or Hox expression. Together, these results suggest that the complement of defects we observe in this study are likely to exceed what can be explained by a simple loss of meis1.1 function. A full account of the phenotypes reported here will likely have to consider the roles of meis1.1, hoxd4a itself, and the downstream effects on the expression of multiple Hox genes.
The phenotype we report here is in some ways closer to that reported for cdx1 plus cdx4 double loss-of-function embryos, including severe hematopoietic (both primitive and definitive) and vasculogenic defects, and impaired expression of multiple Hox genes [9], [10]. By contrast, Hox gene expression is not affected in meis1.1 or meis3 morphants [20], [53]. However, we do not observe significant changes in cdx4 expression in hoxd4a morphants, and the hoxd4a morphant phenotype cannot be rescued by the injection of capped mRNA for cdx4. It may be that hoxd4a is an immediate downstream effector of cdx1/4 function. Alternatively, hoxd4a may act in parallel with cdx1 and cdx4 such that their function, but not expression, is compromised. Such a mechanism is implied by recent studies showing that products of murine CDX1 and HOXD4 form functional heterodimers [54]. A direct role for cdx1 and cdx4 in definitive hematopoiesis could not be established with certainty due to major effects in the development of the dorsal aorta in doubly deficient embryos [9]. By contrast, hoxd4a morphants do form distinctly separate DA and PCV, and these vessels are open to circulation. Nonetheless, we did observe impaired expression of the arterial marker efnb2a, suggesting that the DA may not be appropriately specified in hoxd4a morphants, an event that could lead to defective HSC formation. The mildly reduced heart rate of morphants may contribute to decreased efnb2a expression and impaired specification of HSCs due to sub-optimal levels of nitrous oxide (NO) signaling [55]. This raises the possibility that the observed impairment of definitive hematopoiesis is a secondary event, and we are therefore cautious in assigning a direct role for hoxd4a and meis1.1 in this process. Nonetheless, depressed NO signaling cannot explain all of the defects noted here, since the reduced flk1 expression that we observe at 13 and 26 hpf is not an expected consequence of decreased heart rate [56], [57]. In addition, hoxd4a morphants show reduced efnb2a expression in tissues that should not be affected by NO levels consistent with a direct role for hoxd4a or meis1.1 and similar observations in meis1.1 morphants [18], [19].
Some of the phenotypic defects observed here can be explained by effects on relatively early embryonic events, such as impaired formation and function of hemangioblast precursors, as indicated by the loss of hemangioblast markers scl, lmo2 and fli1, and the ability of the capped mRNAs for these genes to rescue development. However, other aspects of the phenotype may represent later and considerably more downstream functions. For example, decreased expression of efnb2a points to a defect in arterial specification downstream of hemangioblast formation and function, as has been noted in meis1.1 morphants [18], [19]. Likewise, much of the vasculogenic and angiogenic defects could be explained by the impairment of the downstream roles of fli1, vegf and flk1 in these processes. The vegf gene is also known to play a role in the initiation of hematopoiesis [4], and its downregulation is therefore likely to contribute to many aspects of the phenotype reported here.
Expression of gata1 commits hematopoietic precursors to erythropoiesis during primitive hematopoiesis in the ICM. Later in development, runx1 and cmyb are required for myeloid development during definitive hematopoiesis in the equivalent of the AGM [1], [3]. Following hoxd4a knockdown, decreased expression of scl and lmo2 is likely to contribute to a failure in the induction of gata1 and defective primitive hematopoiesis as evidenced by a loss of expression of hbbe1; but loss of scl and lmo2 function would also account for the observed defect in runx1 and cmyb expression and the subsequent failure of definitive hematopoiesis. Interestingly, we observe the expression of hoxd4a at 26–28 hpf in the PBI and at 48 hpf in the AGM and caudal vein plexus. The caudal vein plexus is the site of the future CHT [4], an early site of definitive hematopoiesis, functionally equivalent to the fetal liver of mice [4], [6]. Thus, hoxd4a might play a direct role in primitive and definitive hematopoiesis apart from its indirect role through meis1.1 earlier in development. Both early and later functions of meis1.1 in hematopoiesis and vasculogenesis are also likely, given very early effects on the expression of hemangioblast markers and later expression of meis1.1 in the ICM as described by others [19], [58]. Multiple sites of action along a pathway leading from hemangioblast specification to vasculogenesis, angiogenesis and primitive and definitive hematopoiesis could also help to explain why epistasis experiments give apparently conflicting results. For example, one report places meis1.1 downstream of scl for the induction of primitive hematopoiesis [18], whereas our own work places meis1.1 upstream of scl at an earlier time point during hemangioblast specification.
Hox gene expression in vertebrates is implemented in two phases, an early phase in presumptive mesoderm around gastrulation and a late phase in the body proper [59]–[65]. During gastrulation in Xenopus, timed interactions between the organizer and non-organizer mesoderm induce Hox expression and pattern the AP axis [59], while impaired Hox gene expression can alter the timing of ingression through the primitive streak in the gastrulating chick [65]. Although the widespread deregulation of late-phase Hox gene expression seen after hoxd4a knockdown could be attributed to defects in gastrulation, two observations argue against this. First, not all the Hox genes we tested were affected, an observation inconsistent with a global gastrulation defect. Second, our demonstration that the expression of markers of paraxial mesoderm (myod), cardiac mesoderm (nkx2.5) and intermediate mesoderm (pax2.1) was unaffected does not support a major impairment of gastrulation movements. Nonetheless, our findings emphasize the importance of Hox gene function in the early peri-gastrulation phase [61]–[65].
Mice lacking Hoxd4 function are viable and fertile, and display defects of the anterior vertebral skeleton [33], [34]. Although an extensive analysis of vasculogenesis and hematopoiesis has not been undertaken in Hoxd4 null mice, their viability suggests that there is no severe impairment of these processes. By contrast, mutation of the paralogous Hoxb4 gene does lead to hematopoietic defects and some loss of viability, though these mice are still able to complete embryogenesis and many survive to adulthood [17], [34], [66]–[68]. In striking contrast to the mouse, we show here that loss of hoxd4a function has severely deleterious consequences for hematopoiesis, vasculogenesis and angiogenesis. We suggest that this may be due to a dependency of meis1.1 expression on hoxd4a function that was either acquired by teleosts (or simply acquired by zebrafish) or lost in mammals. It may also be that while Hoxb4 has the predominant role in these processes in mammals, evolution has selected for hoxd4a to take on these functions in teleosts. This situation may be partly analogous to that of the midkine orthologs in mice and zebrafish; the two teleost midkine genes are strongly expressed in the adult brain, whereas the mouse ortholog is not [69], implying the acquisition or loss of function in the course of evolution.
Supporting Information
Supporting information. Contains supplementary Figures S1 to S7 and supplementary Tables S1 and S2.
(PDF)
Patterning and morphology in hoxd4a morphants. (A–H) Knockdown of hoxd4a does not perturb overall patterning of the mesoderm. (A–D) Expression of pax2.1 shows that intermediate mesoderm forms and matures normally in hoxd4a morphants at 13 hpf (B) and 26–28 hpf (D) vs controls (A,C). (E,F) nkx2.5 expression in the precardiac lateral plate mesoderm at 13 hpf is normal in control (E) and morphant embryos (F). (G,H) myod expression in paraxial mesoderm is normal in control (G) and morphant embryos (H). Images in C, D, G and H are lateral views with anterior to the left. A, B, E and F show dorsal views with anterior to the top (A,B) or left (E,F). (I to O) Lateral views of control and hoxd4a-MO-injected larvae at 72 hpf. (I,J) Staining of hemoglobin with o-dianisidine reveals areas of hemorrhage such as in the head (I, arrowheads) and trunk (J, arrowheads) in some hoxd4a morphants. (K,L) Control larvae (K) but not hoxd4a morphants (L) show abundant RBCs passing through the heart (arrowheads). (M–O) Unlike control larvae (M), hoxd4a morphants display pericardial edema and edema over the adjacent yolk (N,O, arrowheads). Scale bars equal 100 µm. (P) The heart rate in morphants at 26–28 and 48 hpf was mildly reduced, but in a statistically significant manner as determined by unpaired Student’s t test (p<0.0001). Error bars give standard deviation.
(TIF)
Reduced expression of markers of primitive hematopoiesis gata1 and β embryonic globin ( hbbe1 ) in hoxd4a morphants. WISH in control and morphant embryos at 26–28 hpf showing the expression of gata1 (A,C,E) and hbbe1 (B,D,F) in the ICM and PBI (white arrowheads). Normal expression of gata1 and hbbe1 (A,B) is severely reduced in hoxd4a morphants (C,D) and rescued by co-injection with capped mRNA for hoxd4a (E,F). All images are lateral views with anterior to the left. ctrl, embryos injected with a non-specific morpholino. MO, embryos injected with the anti-hoxd4a morpholino. hoxd4a mRNA, embryos simultaneously injected with the anti-hoxd4a MO plus capped mRNA for hoxd4a. Scale bars equal 100 µm. All images are at the same magnification.
(TIF)
Reduced expression of markers of angiogenesis and venous specification in hoxd4a morphants. WISH in control and morphant embryos at 26–28 hpf showing the expression of fli1 (A,C,E) and flk1 (B,D,F). Normal expression of fli1 and flk1 (A,B) is severely reduced in hoxd4a morphants (C,D) and rescued by co-injection with capped mRNA for hoxd4a (E,F). White or black dots denote the tips of dorsally sprouting ISVs. Relative to controls (G,H), the expression of the arterial marker efnb2a is reduced in morphants at 26–28 hpf (I), while the venous marker ephb4a in morphants has recovered (J). Scale bars equal 100 µm.
(TIF)
Reduced expression of scl and lmo2 in hoxd4a morphants at 26–28 hpf. (A–J) Expression analysis of scl (A,C,E) and lmo2 (B,D,F) at 26–28 hpf. Normal expression of scl and lmo2 (A,B) is severely reduced in hoxd4a morphants (C,D) and rescued by co-injection with capped mRNA for hoxd4a (E,F) All images present lateral views with anterior to the left and dorsal on top. Scale bars equal 100 µm. All images are at the same magnification.
(TIF)
scl1 and fli1 act downstream of hoxd4a to direct formation of the hemangioblast. All images are of hoxd4a morphants at 26–28 hpf previously injected with capped mRNAs for either scl1 or fli1 as indicated on the left. WISH was performed to detect expression of scl1 and lmo2 (A–D), gata1 and hbbe1 (E–H) and fli1 and flk1 (I–L). Scale bars equal 100 µm. All images are at the same magnification.
(TIF)
Knockdown of hoxd4a results in decreased expression of meis1.1 but not cdx4 at 13 hpf (∼8 somites). (A–F) Expression of cdx4 (A,B), hoxd4a (C,D) and meis1.1 (E,F) in control (A,C,E) and hoxd4a morphants (B,D,F) at the shield stage. The white arrowheads in C and D denote the hoxd4a anterior expression boundary in the hindbrain. Scale bars equal 100 µm. All images are at the same magnification.
(TIF)
The expression of multiple hox genes is reduced at 26–28 hpf in hoxd4a morphants. Images are dorsal views (A–H) and lateral views (I–P) of embryos taken through in situ hybridization for the indicated hox genes. Relative to control embryos (A,C,E,G,I,K,M,O), hox gene expression is reduced in hoxd4a morphants (B,D,F,H,J,L,N,P). All embryos were simultaneously probed for krox20a expression in r3 and r5 as in Figure 1C. (Q–R) cdx4 expression is unchanged in control (Q) and hoxd4a morphants (R) at 26–28 hpf. Scale bars equal 100 µm. All images are at the same magnification.
(TIF)
Primers used for cDNA cloning.
(TIF)
Primers for quantitative RT-PCR.
(TIF)
Anterior blood flow in control and hoxd4a morphant embryos. Lateral view focusing on the anterior half of a 48 hpf embryo including the region of the heart, future branchial arches and yolk of control embryos and hoxd4a morphants. In particular, note robust streaming of blood cells through the ducts of Cuvier over the yolk in the control, but an almost complete absence of circulation in the hoxd4a morphant.
(7Z)
Trunk blood flow in control, hoxd4a morphant and rescued embryos. Lateral view of the trunk at 48 hpf in a control, hoxd4a morphant and rescuant previously injected with capped mRNA for hoxd4a. Circulation is vigorous in control and rescuant embryos, with abundant RBCs flowing caudally along the DA, streaming dorsally through the ISVs, caudally through the DLAV, and rostrally through the caudal vein and PCV. By contrast, the number of blood cells is greatly reduced in morphants and blood cells are unable to transit the truncated ISVs.
(7Z)
Tail blood flow in control and hoxd4a morphant. Lateral view of the tail region in control and hoxd4a morphant embryos at 48 hpf. Control embryos display vigorous blood flow through the DA, ISVs, DLAV and caudal vein plexus. By contrast, morphants show a highly reduced blood cell count with individual blood cells moving slowly caudally through the DA and returning sporadically and haltingly through the caudal vein plexus. Blood cells do not transit from the DA to the DLAV along the ISVs, unlike control embryos. The reddish appearance of the tissue in the caudal vein plexus (white arrows) appears to be due to the accumulation of RBCs.
(7Z)
Rescue by meis1.1 mRNA of trunk blood flow in hoxd4a morphant embryos. Video first showing vigorous circulation through the blood vessels of a control embryo at 48 hpf, followed by the absence of blood and weak circulation in a hoxd4a morphant. The last clip demonstrates significant rescue of blood cell count and vasculature in hoxd4a morphants rescued by co-injection of meis1.1 mRNA, including the ability of blood cells to traverse from the DA to the DLAV along intact ISVs.
(7Z)
Acknowledgments
We would like to thank the following for plasmids: P. Ingham (cmyb, myod), B.C. Low (gata1, hbbe1), R. Patient (lmo2, gata5, fli1), G. Pei (cdx4), V. Prince (hoxb4a, hoxd4a), K. Sampath (pax2,1, scl1), T. Suzuki (efnb2a, ephb4a, vegf, meis1.1), C. Thisse and B. Thisse (hoxd3a), A. Waskiewicz (meis1.1) and Z. Wen (runx1). We thank S.X. Chee for cloning the full length hoxd4a cDNA, and V. Korzh for the Tg(dsRed:gata1) transgenic line.
Funding Statement
This project was supported by an Academic Research Fund Tier-2 grant (T207B3107) from the Ministry of Education, Singapore, to CW, and a grant (10/1/22/19/663) from the Biomedical Research Council (BMRC) of the Agency for Science, Technology and Research (A*STAR), Singapore, to MF. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Paik EJ, Zon LI (2010) Hematopoietic development in the zebrafish. Int J Dev Biol 54: 1127–1137. [DOI] [PubMed] [Google Scholar]
- 2. Ellertsdottir E, Lenard A, Blum Y, Krudewig A, Herwig L, et al. (2010) Vascular morphogenesis in the zebrafish embryo. Dev Biol 341: 56–65. [DOI] [PubMed] [Google Scholar]
- 3. Ciau-Uitz A, Liu F, Patient R (2010) Genetic control of hematopoietic development in Xenopus and zebrafish. Int J Dev Biol 54: 1139–1149. [DOI] [PubMed] [Google Scholar]
- 4. Chen AT, Zon LI (2009) Zebrafish blood stem cells. J Cell Biochem 108: 35–42. [DOI] [PubMed] [Google Scholar]
- 5. Vogeli KM, Jin SW, Martin GR, Stainier DY (2006) A common progenitor for haematopoietic and endothelial lineages in the zebrafish gastrula. Nature 443: 337–339. [DOI] [PubMed] [Google Scholar]
- 6. Kissa K, Murayama E, Zapata A, Cortes A, Perret E, et al. (2008) Live imaging of emerging hematopoietic stem cells and early thymus colonization. Blood 111: 1147–1156. [DOI] [PubMed] [Google Scholar]
- 7. Stainier DY, Weinstein BM, Detrich HW 3rd, Zon LI, Fishman MC (1995) Cloche, an early acting zebrafish gene, is required by both the endothelial and hematopoietic lineages. Development 121: 3141–3150. [DOI] [PubMed] [Google Scholar]
- 8. Xiong JW, Yu Q, Zhang J, Mably JD (2008) An acyltransferase controls the generation of hematopoietic and endothelial lineages in zebrafish. Circulation Research 102: 1057–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davidson AJ, Zon LI (2006) The caudal-related homeobox genes cdx1a and cdx4 act redundantly to regulate hox gene expression and the formation of putative hematopoietic stem cells during zebrafish embryogenesis. Dev Biol 292: 506–518. [DOI] [PubMed] [Google Scholar]
- 10. Davidson AJ, Ernst P, Wang Y, Dekens MP, Kingsley PD, et al. (2003) cdx4 mutants fail to specify blood progenitors and can be rescued by multiple hox genes. Nature 425: 300–306. [DOI] [PubMed] [Google Scholar]
- 11. Eklund E (2011) The role of Hox proteins in leukemogenesis: insights into key regulatory events in hematopoiesis. Critical Reviews in Oncogenesis 16: 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Argiropoulos B, Humphries RK (2007) Hox genes in hematopoiesis and leukemogenesis. Oncogene 26: 6766–6776. [DOI] [PubMed] [Google Scholar]
- 13. Abramovich C, Pineault N, Ohta H, Humphries RK (2005) Hox genes: from leukemia to hematopoietic stem cell expansion. Annals of the New York Academy of Sciences 1044: 109–116. [DOI] [PubMed] [Google Scholar]
- 14. Moens CB, Selleri L (2006) Hox cofactors in vertebrate development. Dev Biol 291: 193–206. [DOI] [PubMed] [Google Scholar]
- 15. Kmita M, Duboule D (2003) Organizing axes in time and space; 25 years of colinear tinkering. Science 301: 331–333. [DOI] [PubMed] [Google Scholar]
- 16. Ko KH, Lam QL, Zhang M, Wong CK, Lo CK, et al. (2007) Hoxb3 deficiency impairs B lymphopoiesis in mouse bone marrow. Exp Hematol 35: 465–475. [DOI] [PubMed] [Google Scholar]
- 17. Magnusson M, Brun AC, Lawrence HJ, Karlsson S (2007) Hoxa9/hoxb3/hoxb4 compound null mice display severe hematopoietic defects. Exp Hematol 35: 1421–1428. [DOI] [PubMed] [Google Scholar]
- 18. Cvejic A, Serbanovic-Canic J, Stemple DL, Ouwehand WH (2011) The role of meis1 in primitive and definitive hematopoiesis during zebrafish development. Haematologica 96: 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Minehata K, Kawahara A, Suzuki T (2008) meis1 regulates the development of endothelial cells in zebrafish. Biochem Biophys Res Commun 374: 647–652. [DOI] [PubMed] [Google Scholar]
- 20. Pillay LM, Forrester AM, Erickson T, Berman JN, Waskiewicz AJ (2010) The Hox cofactors Meis1 and Pbx act upstream of gata1 to regulate primitive hematopoiesis. Dev Biol 340: 306–317. [DOI] [PubMed] [Google Scholar]
- 21. Argiropoulos B, Yung E, Humphries RK (2007) Unraveling the crucial roles of Meis1 in leukemogenesis and normal hematopoiesis. Genes & Development 21: 2845–2849. [DOI] [PubMed] [Google Scholar]
- 22. Iacovino M, Hernandez C, Xu Z, Bajwa G, Prather M, et al. (2009) A conserved role for Hox paralog group 4 in regulation of hematopoietic progenitors. Stem Cells Dev 18: 783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Azcoitia V, Aracil M, Martinez AC, Torres M (2005) The homeodomain protein Meis1 is essential for definitive hematopoiesis and vascular patterning in the mouse embryo. Dev Biol 280: 307–320. [DOI] [PubMed] [Google Scholar]
- 24. Di Rosa P, Villaescusa JC, Longobardi E, Iotti G, Ferretti E, et al. (2007) The homeodomain transcription factor Prep1 (pKnox1) is required for hematopoietic stem and progenitor cell activity. Dev Biol 311: 324–334. [DOI] [PubMed] [Google Scholar]
- 25. DiMartino JF, Selleri L, Traver D, Firpo MT, Rhee J, et al. (2001) The Hox cofactor and proto-oncogene Pbx1 is required for maintenance of definitive hematopoiesis in the fetal liver. Blood 98: 618–626. [DOI] [PubMed] [Google Scholar]
- 26. Hisa T, Spence SE, Rachel RA, Fujita M, Nakamura T, et al. (2004) Hematopoietic, angiogenic and eye defects in Meis1 mutant animals. Embo J 23: 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stankunas K, Shang C, Twu KY, Kao SC, Jenkins NA, et al. (2008) Pbx/Meis deficiencies demonstrate multigenetic origins of congenital heart disease. Circ Res 103: 702–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mamo A, Krosl J, Kroon E, Bijl J, Thompson A, et al. (2006) Molecular dissection of Meis1 reveals 2 domains required for leukemia induction and a key role for Hoxa gene activation. Blood 108: 622–629. [DOI] [PubMed] [Google Scholar]
- 29. Wang GG, Pasillas MP, Kamps MP (2006) Persistent transactivation by meis1 replaces hox function in myeloid leukemogenesis models: evidence for co-occupancy of meis1-pbx and hox-pbx complexes on promoters of leukemia-associated genes. Mol Cell Biol 26: 3902–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML (2007) Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev 21: 2762–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Z, Iwasaki M, Ficara F, Lin C, Matheny C, et al. (2010) GSK-3 promotes conditional association of CREB and its coactivators with MEIS1 to facilitate HOX-mediated transcription and oncogenesis. Cancer cell 17: 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoegg S, Boore JL, Kuehl JV, Meyer A (2007) Comparative phylogenomic analyses of teleost fish Hox gene clusters: lessons from the cichlid fish Astatotilapia burtoni. BMC Genomics 8: 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Horan GS, Kovacs EN, Behringer RR, Featherstone MS (1995) Mutations in paralogous Hox genes result in overlapping homeotic transformations of the axial skeleton: evidence for unique and redundant function. Dev Biol 169: 359–372. [DOI] [PubMed] [Google Scholar]
- 34. Horan GS, Ramirez-Solis R, Featherstone MS, Wolgemuth DJ, Bradley A, et al. (1995) Compound mutants for the paralogous hoxa-4, hoxb-4, and hoxd-4 genes show more complete homeotic transformations and a dose-dependent increase in the number of vertebrae transformed. Genes Dev 9: 1667–1677. [DOI] [PubMed] [Google Scholar]
- 35. Warga RM, Kane DA, Ho RK (2009) Fate mapping embryonic blood in zebrafish: multi- and unipotential lineages are segregated at gastrulation. Dev Cell 16: 744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kimmel CB, Warga RM, Schilling TF (1990) Origin and organization of the zebrafish fate map. Development 108: 581–594. [DOI] [PubMed] [Google Scholar]
- 37. Lawson ND, Weinstein BM (2002) In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol 248: 307–318. [DOI] [PubMed] [Google Scholar]
- 38. Traver D, Paw BH, Poss KD, Penberthy WT, Lin S, et al. (2003) Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nature Immunology 4: 1238–1246. [DOI] [PubMed] [Google Scholar]
- 39.Westerfield M (2000) The zebrafish book: A guide for the laboratory use of zebrafish (Danio rerio). Eugene: University of Oregon Press.
- 40. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310. [DOI] [PubMed] [Google Scholar]
- 41. Thisse C, Thisse B, Schilling TF, Postlethwait JH (1993) Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development 119: 1203–1215. [DOI] [PubMed] [Google Scholar]
- 42. Wan X, Hu B, Liu JX, Feng X, Xiao W (2011) Zebrafish mll gene is essential for hematopoiesis. J Biol Chem 286: 33345–33357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Detrich HW 3rd, Kieran MW, Chan FY, Barone LM, Yee K, et al (1995) Intraembryonic hematopoietic cell migration during vertebrate development. Proc Natl Acad Sci U S A 92: 10713–10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nolte C, Rastegar M, Amores A, Bouchard M, Grote D, et al. (2006) Stereospecificity and PAX6 function direct Hoxd4 neural enhancer activity along the antero-posterior axis. Dev Biol 299: 582–593. [DOI] [PubMed] [Google Scholar]
- 45. Prince VE, Joly L, Ekker M, Ho RK (1998) Zebrafish hox genes: genomic organization and modified colinear expression patterns in the trunk. Development 125: 407–420. [DOI] [PubMed] [Google Scholar]
- 46. Liu F, Walmsley M, Rodaway A, Patient R (2008) Fli1 acts at the top of the transcriptional network driving blood and endothelial development. Curr Biol 18: 1234–1240. [DOI] [PubMed] [Google Scholar]
- 47. Peterkin T, Gibson A, Patient R (2009) Common genetic control of haemangioblast and cardiac development in zebrafish. Development 136: 1465–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mann RS, Affolter M (1998) Hox proteins meet more partners. Curr Opin Genet Dev 8: 423–429. [DOI] [PubMed] [Google Scholar]
- 49. Mann RS, Chan SK (1996) Extra specificity from extradenticle: the partnership between HOX and PBX/EXD homeodomain proteins. Trends Genet 12: 258–262. [DOI] [PubMed] [Google Scholar]
- 50. Bahrami SB, Veiseh M, Dunn AA, Boudreau NJ (2011) Temporal changes in Hox gene expression accompany endothelial cell differentiation of embryonic stem cells. Cell adhesion & migration 5: 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Charboneau A, East L, Mulholland N, Rohde M, Boudreau N (2005) Pbx1 is required for Hox D3-mediated angiogenesis. Angiogenesis 8: 289–296. [DOI] [PubMed] [Google Scholar]
- 52. Shanmugam K, Green NC, Rambaldi I, Saragovi HU, Featherstone MS (1999) PBX and MEIS as non-DNA-binding proteins in trimeric complexes with HOX proteins. Mol Cell Biol 19: 7577–7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. diIorio P, Alexa K, Choe SK, Etheridge L, Sagerstrom CG (2007) TALE-family homeodomain proteins regulate endodermal sonic hedgehog expression and pattern the anterior endoderm. Dev Biol 304: 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lafontaine CA, Grainger S, Hess BL, Beland M, Lohnes D (2012) Cdx1 Interacts Physically with a Subset of Hox Proteins. Biochemistry. [DOI] [PubMed]
- 55. North TE, Goessling W, Peeters M, Li P, Ceol C, et al. (2009) Hematopoietic stem cell development is dependent on blood flow. Cell 137: 736–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Isogai S, Lawson ND, Torrealday S, Horiguchi M, Weinstein BM (2003) Angiogenic network formation in the developing vertebrate trunk. Development 130: 5281–5290. [DOI] [PubMed] [Google Scholar]
- 57. North TE, de Bruijn MF, Stacy T, Talebian L, Lind E, et al. (2002) Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity 16: 661–672. [DOI] [PubMed] [Google Scholar]
- 58. Waskiewicz AJ, Rikhof HA, Hernandez RE, Moens CB (2001) Zebrafish Meis functions to stabilize Pbx proteins and regulate hindbrain patterning. Development 128: 4139–4151. [DOI] [PubMed] [Google Scholar]
- 59. Wacker SA, Jansen HJ, McNulty CL, Houtzager E, Durston AJ (2004) Timed interactions between the Hox expressing non-organiser mesoderm and the Spemann organiser generate positional information during vertebrate gastrulation. Dev Biol 268: 207–219. [DOI] [PubMed] [Google Scholar]
- 60. Wacker SA, McNulty CL, Durston AJ (2004) The initiation of Hox gene expression in Xenopus laevis is controlled by Brachyury and BMP-4. Dev Biol 266: 123–137. [DOI] [PubMed] [Google Scholar]
- 61. Gaunt SJ, Strachan L (1994) Forward spreading in the establishment of a vertebrate Hox expression boundary: the expression domain separates nito anterior and posterior zones, and the spread occurs across implanted glass barriers. Dev Dyn 199: 229–240. [DOI] [PubMed] [Google Scholar]
- 62. Deschamps J, van den Akker E, Forlani S, De Graaff W, Oosterveen T, et al. (1999) Initiation, establishment and maintenance of Hox gene expression patterns in the mouse. Int J Dev Biol 43: 635–650. [PubMed] [Google Scholar]
- 63. Deschamps J, Wijgerde M (1993) Two phases in the establishment of HOX expression domains. Developmental Biology 156: 473–480. [DOI] [PubMed] [Google Scholar]
- 64. Durston AJ, Jansen HJ, Wacker SA (2010) Review: Time-space translation regulates trunk axial patterning in the early vertebrate embryo. Genomics 95: 250–255. [DOI] [PubMed] [Google Scholar]
- 65. Iimura T, Pourquie O (2006) Collinear activation of Hoxb genes during gastrulation is linked to mesoderm cell ingression. Nature 442: 568–571. [DOI] [PubMed] [Google Scholar]
- 66. Bjornsson JM, Larsson N, Brun AC, Magnusson M, Andersson E, et al. (2003) Reduced proliferative capacity of hematopoietic stem cells deficient in Hoxb3 and Hoxb4. Mol Cell Biol 23: 3872–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brun AC, Bjornsson JM, Magnusson M, Larsson N, Leveen P, et al. (2004) Hoxb4-deficient mice undergo normal hematopoietic development but exhibit a mild proliferation defect in hematopoietic stem cells. Blood 103: 4126–4133. [DOI] [PubMed] [Google Scholar]
- 68. Ramírez-Solis R, Zheng H, Whiting J, Krumlauf R, Bradley A (1993) Hoxb-4 (Hox-2.6) mutant mice show homeotic trasformation of a cervical vertebra and defects in the closure of the sternal rudiments. Cell 73: 279–294. [DOI] [PubMed] [Google Scholar]
- 69. Winkler C, Schafer M, Duschl J, Schartl M, Volff JN (2003) Functional divergence of two zebrafish midkine growth factors following fish-specific gene duplication. Genome Res 13: 1067–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information. Contains supplementary Figures S1 to S7 and supplementary Tables S1 and S2.
(PDF)
Patterning and morphology in hoxd4a morphants. (A–H) Knockdown of hoxd4a does not perturb overall patterning of the mesoderm. (A–D) Expression of pax2.1 shows that intermediate mesoderm forms and matures normally in hoxd4a morphants at 13 hpf (B) and 26–28 hpf (D) vs controls (A,C). (E,F) nkx2.5 expression in the precardiac lateral plate mesoderm at 13 hpf is normal in control (E) and morphant embryos (F). (G,H) myod expression in paraxial mesoderm is normal in control (G) and morphant embryos (H). Images in C, D, G and H are lateral views with anterior to the left. A, B, E and F show dorsal views with anterior to the top (A,B) or left (E,F). (I to O) Lateral views of control and hoxd4a-MO-injected larvae at 72 hpf. (I,J) Staining of hemoglobin with o-dianisidine reveals areas of hemorrhage such as in the head (I, arrowheads) and trunk (J, arrowheads) in some hoxd4a morphants. (K,L) Control larvae (K) but not hoxd4a morphants (L) show abundant RBCs passing through the heart (arrowheads). (M–O) Unlike control larvae (M), hoxd4a morphants display pericardial edema and edema over the adjacent yolk (N,O, arrowheads). Scale bars equal 100 µm. (P) The heart rate in morphants at 26–28 and 48 hpf was mildly reduced, but in a statistically significant manner as determined by unpaired Student’s t test (p<0.0001). Error bars give standard deviation.
(TIF)
Reduced expression of markers of primitive hematopoiesis gata1 and β embryonic globin ( hbbe1 ) in hoxd4a morphants. WISH in control and morphant embryos at 26–28 hpf showing the expression of gata1 (A,C,E) and hbbe1 (B,D,F) in the ICM and PBI (white arrowheads). Normal expression of gata1 and hbbe1 (A,B) is severely reduced in hoxd4a morphants (C,D) and rescued by co-injection with capped mRNA for hoxd4a (E,F). All images are lateral views with anterior to the left. ctrl, embryos injected with a non-specific morpholino. MO, embryos injected with the anti-hoxd4a morpholino. hoxd4a mRNA, embryos simultaneously injected with the anti-hoxd4a MO plus capped mRNA for hoxd4a. Scale bars equal 100 µm. All images are at the same magnification.
(TIF)
Reduced expression of markers of angiogenesis and venous specification in hoxd4a morphants. WISH in control and morphant embryos at 26–28 hpf showing the expression of fli1 (A,C,E) and flk1 (B,D,F). Normal expression of fli1 and flk1 (A,B) is severely reduced in hoxd4a morphants (C,D) and rescued by co-injection with capped mRNA for hoxd4a (E,F). White or black dots denote the tips of dorsally sprouting ISVs. Relative to controls (G,H), the expression of the arterial marker efnb2a is reduced in morphants at 26–28 hpf (I), while the venous marker ephb4a in morphants has recovered (J). Scale bars equal 100 µm.
(TIF)
Reduced expression of scl and lmo2 in hoxd4a morphants at 26–28 hpf. (A–J) Expression analysis of scl (A,C,E) and lmo2 (B,D,F) at 26–28 hpf. Normal expression of scl and lmo2 (A,B) is severely reduced in hoxd4a morphants (C,D) and rescued by co-injection with capped mRNA for hoxd4a (E,F) All images present lateral views with anterior to the left and dorsal on top. Scale bars equal 100 µm. All images are at the same magnification.
(TIF)
scl1 and fli1 act downstream of hoxd4a to direct formation of the hemangioblast. All images are of hoxd4a morphants at 26–28 hpf previously injected with capped mRNAs for either scl1 or fli1 as indicated on the left. WISH was performed to detect expression of scl1 and lmo2 (A–D), gata1 and hbbe1 (E–H) and fli1 and flk1 (I–L). Scale bars equal 100 µm. All images are at the same magnification.
(TIF)
Knockdown of hoxd4a results in decreased expression of meis1.1 but not cdx4 at 13 hpf (∼8 somites). (A–F) Expression of cdx4 (A,B), hoxd4a (C,D) and meis1.1 (E,F) in control (A,C,E) and hoxd4a morphants (B,D,F) at the shield stage. The white arrowheads in C and D denote the hoxd4a anterior expression boundary in the hindbrain. Scale bars equal 100 µm. All images are at the same magnification.
(TIF)
The expression of multiple hox genes is reduced at 26–28 hpf in hoxd4a morphants. Images are dorsal views (A–H) and lateral views (I–P) of embryos taken through in situ hybridization for the indicated hox genes. Relative to control embryos (A,C,E,G,I,K,M,O), hox gene expression is reduced in hoxd4a morphants (B,D,F,H,J,L,N,P). All embryos were simultaneously probed for krox20a expression in r3 and r5 as in Figure 1C. (Q–R) cdx4 expression is unchanged in control (Q) and hoxd4a morphants (R) at 26–28 hpf. Scale bars equal 100 µm. All images are at the same magnification.
(TIF)
Primers used for cDNA cloning.
(TIF)
Primers for quantitative RT-PCR.
(TIF)
Anterior blood flow in control and hoxd4a morphant embryos. Lateral view focusing on the anterior half of a 48 hpf embryo including the region of the heart, future branchial arches and yolk of control embryos and hoxd4a morphants. In particular, note robust streaming of blood cells through the ducts of Cuvier over the yolk in the control, but an almost complete absence of circulation in the hoxd4a morphant.
(7Z)
Trunk blood flow in control, hoxd4a morphant and rescued embryos. Lateral view of the trunk at 48 hpf in a control, hoxd4a morphant and rescuant previously injected with capped mRNA for hoxd4a. Circulation is vigorous in control and rescuant embryos, with abundant RBCs flowing caudally along the DA, streaming dorsally through the ISVs, caudally through the DLAV, and rostrally through the caudal vein and PCV. By contrast, the number of blood cells is greatly reduced in morphants and blood cells are unable to transit the truncated ISVs.
(7Z)
Tail blood flow in control and hoxd4a morphant. Lateral view of the tail region in control and hoxd4a morphant embryos at 48 hpf. Control embryos display vigorous blood flow through the DA, ISVs, DLAV and caudal vein plexus. By contrast, morphants show a highly reduced blood cell count with individual blood cells moving slowly caudally through the DA and returning sporadically and haltingly through the caudal vein plexus. Blood cells do not transit from the DA to the DLAV along the ISVs, unlike control embryos. The reddish appearance of the tissue in the caudal vein plexus (white arrows) appears to be due to the accumulation of RBCs.
(7Z)
Rescue by meis1.1 mRNA of trunk blood flow in hoxd4a morphant embryos. Video first showing vigorous circulation through the blood vessels of a control embryo at 48 hpf, followed by the absence of blood and weak circulation in a hoxd4a morphant. The last clip demonstrates significant rescue of blood cell count and vasculature in hoxd4a morphants rescued by co-injection of meis1.1 mRNA, including the ability of blood cells to traverse from the DA to the DLAV along intact ISVs.
(7Z)