

Abstract
Background
Congenital hyperinsulinism (CHI) is a clinically heterogeneous condition. Mutations in eight genes (ABCC8, KCNJ11, GLUD1, GCK, HADH, SLC16A1, HNF4A and HNF1A) are known to cause CHI.
Aim
To characterise the clinical and molecular aspects of a large cohort of patients with CHI.
Methodology
Three hundred patients were recruited and clinical information was collected before genotyping. ABCC8 and KCNJ11 genes were analysed in all patients. Mutations in GLUD1, HADH, GCK and HNF4A genes were sought in patients with diazoxide-responsive CHI with hyperammonaemia (GLUD1), raised 3-hydroxybutyrylcarnitine and/or consanguinity (HADH), positive family history (GCK) or when CHI was diagnosed within the first week of life (HNF4A).
Results
Mutations were identified in 136/300 patients (45.3%). Mutations in ABCC8/KCNJ11 were the commonest genetic cause identified (n=109, 36.3%). Among diazoxide-unresponsive patients (n=105), mutations in ABCC8/KCNJ11 were identified in 92 (87.6%) patients, of whom 63 patients had recessively inherited mutations while four patients had dominantly inherited mutations. A paternal mutation in the ABCC8/KCNJ11 genes was identified in 23 diazoxide-unresponsive patients, of whom six had diffuse disease. Among the diazoxide-responsive patients (n=183), mutations were identified in 41 patients (22.4%). These include mutations in ABCC8/KCNJ11 (n=15), HNF4A (n=7), GLUD1 (n=16) and HADH (n=3).
Conclusions
A genetic diagnosis was made for 45.3% of patients in this large series. Mutations in the ABCC8 gene were the commonest identifiable cause. The vast majority of patients with diazoxide-responsive CHI (77.6%) had no identifiable mutations, suggesting other genetic and/or environmental mechanisms.
Introduction
Congenital hyperinsulinism (CHI) is a clinically, genetically, morphologically and functionally heterogeneous condition (1, 2, 3). The age of presentation is variable with the severe forms typically presenting in the neonatal period and the milder forms presenting later in infancy or childhood with recurrent symptoms of hypoglycaemia (4). Symptoms generally develop after a period of fasting or when the child is unwell. However, some forms of CHI demonstrate protein and/or leucine sensitivity and symptoms are manifested or aggravated in the postprandial period following a protein-rich meal rather than a fast (5). Macrosomia is a common feature in infants, reflecting fetal hyperinsulinaemia; however, not all babies with CHI are macrosomic (6).
CHI occurs due to defects in key genes involved in regulating insulin secretion from β-cells. So far, mutations in eight different genes (ABCC8, KCNJ11, GLUD1, GCK, HADH, SLC16A1, HNF4A and HNF1A) that lead to dysregulated secretion of insulin have been described (7, 8). Recessive and dominantly acting mutations in the genes ABCC8 and KCNJ11, encoding the SUR1 and Kir6.2 subunits of the pancreatic β-cell ATP-sensitive K+ (KATP) channel respectively, are the most common known cause of CHI (9, 10, 11, 12). While the majority of patients with KATP channel mutations are medically unresponsive, a number of cases with dominant ABCC8 or KCNJ11 mutation(s) who have responded well to therapy have been reported (13). The molecular basis of recessive inactivating ABCC8 and KCNJ11 mutations involves multiple defects in channel biogenesis and turnover, in channel trafficking from the endoplasmic reticulum and Golgi apparatus to the plasma membrane and alterations in the sensitivity of the channel to both magnesium ATP and ADP (14, 15, 16). However, in dominant ABCC8/KCNJ11 mutations, channel biogenesis and subunit trafficking seem to be normal but with impaired channel activity (13).
Gain-of-function mutations in the GLUD1 gene, which encodes for the intra-mitochondrial enzyme glutamate dehydrogenase (GDH), form the molecular and biochemical basis of the hyperinsulinism and hyperammonaemia (HI/HA) syndrome and explain the link between leucine hypersensitivity and the HA (17). Heterozygous activating GCK mutations cause CHI, which in most cases is medically responsive but in some cases surgery may be required (18, 19, 20). Loss-of-function mutations in the HADH gene are associated with CHI and protein sensitivity but with normal serum ammonia levels (21, 22, 23, 24, 25). In some cases, increased levels of plasma 3-hydroxybutyrylcarnitine are observed. Mutations in the promoter region of SLC16A1 gene have been described in patients with exercise-induced HI (26, 27).
Mutations in the HNF4A gene, encoding the hepatocyte nuclear factor 4 alpha (HNF4A), are a novel cause of transient and persistent hyperinsulinaemic hypoglycaemia (HH), associated with macrosomia (6). Following a remission of the HI patients develop maturity-onset diabetes of the young type 1, characterised by autosomal dominant inheritance and impaired glucose-stimulated insulin secretion from pancreatic β-cells (28). While the majority of patients with HNF4A mutations have isolated CHI, a recent study reported a case with an HNF4A mutation causing CHI and defects in liver glycogen metabolism and renal tubular transport (8). The same study described HNF1A as another cause of CHI.
Histologically, CHI is classified into a diffuse form, where the pancreatic β-cells show increased nuclear size throughout the pancreas and a focal form characterised by the presence of a focal adenomatous hyperplasia confined to a single region of the pancreas (29). Focal adenomatous hyperplasia involves the specific loss of the maternal chromosome 11p15 region (loss of heterozygosity (LOH)) and a constitutional mutation of a paternally inherited allele of the genes ABCC8/KCNJ11 encoding the KATP channel (30). The LOH results from paternal uniparental disomy of chromosome 11p15.5-11p15.1, which unmasks the paternally inherited KATP channel mutation at 11p15.1 leading to uncontrolled secretion of insulin. Using 18F-l-DOPA-PET/CT, it is now possible to pre-operatively localise the focal lesion (31). The precise preoperative localisation and limited surgical removal of the focal domain ‘cures’ the patient. By contrast, patients with diffuse disease may require a near total pancreatectomy, which will have lifelong implications (high risk of diabetes mellitus and pancreatic exocrine insufficiency) (32).
The mainstay of medical therapy in CHI is diazoxide – a drug that binds to the intact SUR1 component of the KATP channels (4). It acts by keeping the KATP channels open, thereby preventing depolarisation of the β-cell membrane and insulin secretion. Clinically, it is useful to classify children as diazoxide responsive and diazoxide unresponsive. The aims of this study were to investigate genotype/phenotype correlations in relation to birth weight, age at presentation, responsiveness to medical therapy with diazoxide and prediction of focal disease.
Materials and methods
Patients
All patients with CHI recruited into the study were referred to Great Ormond Street Children's Hospital, which is a national and international referral centre for CHI. Three hundred patients with biochemically confirmed CHI (blood glucose concentration <3.0 mmol/l with detectable concentrations of insulin and/or C-peptide, i.v. glucose infusion rate of >8 mg/kg per min and inappropriately low concentration non-esterified fatty acids and ketone bodies) were recruited into the study. Clinical and biochemical data were collated via a standard request form (www.diabetesgenes.org), clinical letter of referral or by case note review. Responsiveness to diazoxide was assessed and defined as being able to maintain normoglycaemia (blood glucose >3.5 mmol/l) following a period of fasting, which is determined by the age of the child (6 h for a neonate, 12 h up to infancy and 18 h in a child). At the end of the fasting period, there should be an appropriate increase in the serum ketone bodies and an appropriate suppression of insulin secretion. Patients with a secondary cause of hyperinsulinaemic hypoglycaemia such as perinatal asphyxia, intra-uterine growth restriction, Rhesus isoimmunisation and syndromic forms such as Beckwith–Wiedemann syndrome were excluded from the study (33).
Birth weight SDS were calculated for each patient. The mean birth weight and age of presentation of patients with diazoxide-responsive CHI due to a KATP channel mutation(s), GLUD1 mutation, HADH mutation or an HNF4A mutation were compared using one-way ANOVA followed by the Fisher's least significant difference post-test to test for statistical significance (SPSS, version 20). The study was ethically approved by the Ethics Committee of Great Ormond Street Children's Hospital and the Institute of Child Health.
Sequence analysis strategy of genes known to cause CHI
The DNA samples were analysed for mutations in genes known to cause CHI (ABCC8, KCNJ11, HNF4A, GCK, GLUD1 and HADH genes) at the Diagnostic Molecular Genetics Laboratory at Peninsula Medical School (Exeter, UK), according to the established methods. The pathogenicity of novel variants was considered on an individual basis. Where possible, evidence to support/refute causality was sought by the following: i) co-segregation studies, ii) identification in control chromosomes (dbSNP (November 2010)/1000 genomes databases (May 2011)), iii) SIFT and PolyPhen in silico predictions and iv) in silico splicing prediction accessed through Alamut Software, version 2.1 (Interactive Biosoftware, Rouen, France).
Diazoxide-unresponsive patients
KCNJ11 and ABCC8 were sequenced concurrently in all patients. If i) no ABCC8 or KCNJ11 mutation was identified, ii) a single mutation inherited from an unaffected mother was found, or iii) a single mutation was identified in a patient with diffuse pancreatic disease, dosage analysis of the ABCC8 gene by multiplex ligation dependent probe amplification (MLPA) was undertaken. Sequencing analysis of the GCK gene was subsequently performed in all patients in whom no mutation in the ABCC8 and KCNJ11 genes had been identified.
Diazoxide-responsive patients
In patients with diazoxide-responsive CHI, mutations in GLUD1 were sought if the patient had HA or leucine sensitivity, and HADH was sequenced if there was raised plasma 3-hydroxybutyrylcarnitine or consanguinity was reported. If no mutations were identified and in the absence of the above features, KCNJ11 and ABCC8 were sequenced concurrently. MLPA analysis of the ABCC8 gene was undertaken when a single heterozygous ABCC8 mutation was identified. If no KATP channel mutation was identified, HNF4A was sequenced. Sequencing of GCK was undertaken if there was sufficient DNA and a positive family history of hypoglycaemia or delayed presentation of hypoglycaemia (>6 months). The SLC16A1 gene was not sequenced as no patients were reported to have exercise-induced HI.
If it was not possible to establish whether a patient with CHI was diazoxide responsive or not (n=12), KCNJ11 and ABCC8 followed by HNF4A sequencing was undertaken.
Results
Three hundred patients with CHI were recruited in this study. The patients were broadly classified into diazoxide-responsive (n=183) and diazoxide-unresponsive (n=105) groups. Twelve patients with CHI were not able to be grouped within these two broad categories. These included patients with transient CHI that did not require treatment with diazoxide (n=11) and one patient who was not treated with diazoxide following an anaphylactic reaction to the drug.
Analysis of ABCC8 gene mutations
Mutations in the ABCC8 gene were identified in 98 patients (Fig. 1). Of the 98 patients, 19 were compound heterozygous, 38 were homozygous and 41 were heterozygous. Dominant mutations were confirmed in 17/41 patients with a heterozygous mutation. In the remaining 24 patients, the mutation was presumed to be recessively acting. A variety of missense, frameshift, nonsense, deletion and splice site mutations, located throughout the gene, were identified. Thirty-six novel ABCC8 mutations were identified in this study. The most common mutations were the splice site mutation c.3992-9G>A and the missense mutation p.G111R, found in six patients each. The ABCC8 mutations/variants and the associated phenotype are presented in Supplementary Tables 1, 2, 3 and 4, see section on supplementary data given at the end of this article, as i) homozygous or compound heterozygous mutations, ii) heterozygous mutations and iii) variants of uncertain significance.
Figure 1.
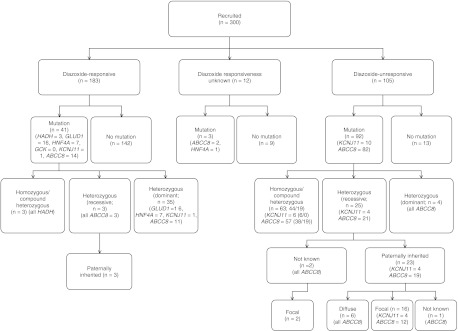
Summary of the genotypes of all patients undergoing sequencing analysis.
CHI due to homozygous or compound heterozygous recessively acting ABCC8 mutations
All patients with two ABCC8 mutations in trans that had been inherited from clinically unaffected parents were unresponsive to diazoxide therapy (n=57; Supplementary Tables 1 and 2). Of these, 38 patients underwent a near total pancreatectomy and diffuse disease was confirmed histologically. The remaining 19 patients were treated non-surgically with a combination of glucose polymer in feeds and octreotide therapy. Diazoxide was used as an adjunct in seven patients as there was clinical evidence of partial responsiveness to diazoxide with improvement in blood glucose concentrations. The mean age of presentation of these patients was 20 days (median of 1 day) and the mean birth weight was +1.67 SDS.
CHI due to heterozygous ABCC8 mutations
Forty-one patients were heterozygous for a single ABCC8 mutation (Supplementary Table 3). In all patients, gene dosage analysis by MLPA had excluded a partial or whole gene deletion on the opposite allele. This heterogeneous group of patients included 25 patients with diazoxide-unresponsive CHI and 14 patients with diazoxide-responsive CHI. A single heterozygous ABCC8 mutation was also identified in two patients where the diazoxide responsiveness is not known as they had transient CHI that resolved without trial of diazoxide therapy. This included one patient (patient 172) who was heterozygous for a novel p.G1479A mutation and one patient (patient 281) who was heterozygous for the p.R1539Q mutation known to cause mild, dominantly inherited CHI (13). The p.G1479A mutation is likely to be dominantly acting as i) the family member testing demonstrated that the mutation had arisen de novo, ii) a second mutation was not detected by sequencing or MLPA analysis and iii) a different mutation affecting the same residue has been reported previously in patients with dominantly inherited CHI (13, 34). The transient nature of the CHI in these two patients is in keeping with previous reports, which have described a variable penetrance associated with dominantly acting mutations (34).
Diazoxide-unresponsive CHI with a heterozygous ABCC8 mutation
Of the 25 with diazoxide-unresponsive CHI and a single ABCC8 mutation, four had a dominantly acting mutation (patients previously reported in (28)), 19 had a paternal mutation, and in two patients (with histologically confirmed focal disease), the inheritance was not established due to unavailability of the paternal DNA (Supplementary Table 3). All four patients with a dominantly acting mutation had diffuse disease while 12 of the 19 patients with a recessively acting paternal mutation had histologically confirmed focal disease that was managed by resection of the focal lesion/limited pancreatectomy. Six of the remaining patients had diffuse disease with a mutation inherited from an unaffected father. Two of these (patients 97 and 270) are heterozygous for the same novel mutation (p.A1263T). One other patient with a paternally inherited mutation was treated non-surgically with glucose polymer in feeds and octreotide and it was not determined whether he had a focal lesion or not.
Diazoxide-responsive CHI with a heterozygous ABCC8 mutation
Fourteen patients with diazoxide-responsive CHI had a single heterozygous ABCC8 mutation (Supplementary Table 3). The patients in this category have varying durations of HH, ranging from transient disease of 5-month duration (patient 10) to persistent disease requiring continued diazoxide therapy at 10.7 years of age (patient 194). Similar to the other groups of patients with an ABCC8 mutation, the mean age of presentation in this group was early at 29.2 days (median 1 day) and the mean birth weight was 1.53 SDS. This category includes 11 patients (four related and seven unrelated) with seven different dominantly inherited mutations. The clinical phenotype of these patients and the functional characterisation of the mutations have been described (34). Apart from the patients with dominantly inherited mutations, there are three patients where a single mutation was inherited from an asymptomatic parent.
ABCC8 variants of uncertain clinical significance
In seven patients, a heterozygous ABCC8 novel variant of uncertain clinical significance was identified (Supplementary Table 4). All patients were responsive to diazoxide and four of the patients had inherited the variant from an unaffected parent (three maternal and one paternal). In three patients, the parental origin of the variant could not be determined as samples were not available for testing.
Analysis of KCNJ11 gene mutations
Eleven different KCNJ11 mutations were identified, six of which are novel. The genotype and phenotype of these patients are summarised in Supplementary Table 5, see section on supplementary data given at the end of this article. The mean age of presentation of these patients was 5.5 days (median 1 day) and the mean birth weight was +2.02 SDS. Six patients had diazoxide-unresponsive disease associated with a homozygous mutation. Four of these patients underwent a near total pancreatectomy while two were treated non-surgically with a combination of glucose polymer in feeds and octreotide therapy. Diffuse disease was confirmed histologically in the four patients who underwent a near total pancreatectomy and in a further patient via a pancreatic tail biopsy. The histological diagnosis was not established in one patient. Four patients with diazoxide-unresponsive disease and a paternally inherited KCNJ11 mutation had focal disease, confirmed on histological examination following a limited/sub-total pancreatectomy. In one of these patients (patient 148), who has been reported previously, a giant focal lesion was observed (35).
Diazoxide-responsive disease
One patient with diazoxide-responsive disease was heterozygous for a KCNJ11 mutation (Supplementary Table 5). This patient had inherited the mutation from their affected father. The father had a history of hyperinsulinaemic hypoglycaemia in the neonate, thus confirming dominant inheritance of this mutation.
KCNJ11 variants of uncertain clinical significance
In two patients with transient CHI, a novel heterozygous KCNJ11 variant of uncertain clinical significance was identified (Supplementary Table 6, see section on supplementary data given at the end of this article). One patient had inherited the mutation from an unaffected mother. The inheritance of the mutation in the second patient could not be determined as the mutation was not present in the maternal DNA and a sample from the father was not available for testing.
Diazoxide-responsive CHI without an ABCC8 or KCNJ11 mutation
The largest category of patients with CHI in this cohort included patients with diazoxide-responsive CHI. Of the 183 patients with diazoxide-responsive disease, a genetic diagnosis was possible in 41 patients (22.4%; Fig. 1).
Twenty patients with diazoxide-responsive CHI (19 patients with HA and one with leucine sensitivity without HA) had sequencing analysis of GLUD1 gene and mutations were identified in 16 patients. The genotype–phenotype correlations of these patients have been reported (36). Seven of the diazoxide-responsive patients had HNF4A mutations. The phenotypic characteristics of these patients have been previously reported (37). No patients with a GCK mutation were identified in this cohort.
Clinical characteristics according to genetic aetiology
The clinical characteristics of patients with transient and/or diazoxide-responsive CHI were compared according to genetic aetiology. Patients with a HNF4A mutation presented earlier and were born heavier than patients with a GLUD1 mutation (1 day vs 157 days, P=0.004, and +2.36 birth weight SDS vs −0.10 birth weight SDS, P=0.001 respectively). In comparison with patients with a HADH mutation, patients with a HNF4A mutation were heavier (birth weight SDS +2.36 (HNF4A) vs −1.08 (HADH), P=0.002). No difference in the age of presentation was observed (1 day vs 125 days, P=0.109). In addition, no differences in the mean age at presentation were observed between patients with a HNF4A or KATP channel mutation(s) with diazoxide-responsive CHI (the range varied from 1 day to 1 year for patients with a KATP channel mutation). The difference in the birth weights between these two groups were also not significant (+2.36 vs +1.96 SDS, P=0.551).
Within the other groups of patients, the age of presentation and birth weight of patients with a GLUD1 or HADH mutation were similar (157 vs 125 days and birth weight SDS −0.10 vs – 1.08, P=0.63 and P=0.32 respectively). The mean birth weight and age of presentation of the patients with a GLUD1 mutation was significantly different from patients with a KATP channel mutation (P=0.003 and P=0.001 respectively). Patients with a HADH mutation were also significantly smaller at birth than patients with a KATP channel mutation (P=0.003). The mean age of presentation of patients with a HADH mutation was 125 days in comparison with 27.4 days in patients with KATP channel mutations; however, the difference was not statistically significant (P=0.15).
Discussion
In this study, a genetic diagnosis for CHI was made for 45.3% of patients that were studied. In the category of children who were unresponsive to therapy with diazoxide, mutations in the ABCC8 gene were the commonest identifiable cause. All patients with two ABCC8 mutations in trans that had been inherited from clinically unaffected parents were unresponsive to diazoxide therapy. Out of the 105 patients who were unresponsive to diazoxide treatment, mutations in ABCC8/KCNJ11 were found in 92. This suggests that from the clinical point of view, the vast majority of patients who do not respond to therapy with diazoxide are likely to harbour mutations in the ABCC8/KCNJ11 genes.
By contrast, the phenotype of those patients who inherited a heterozygous ABCC8 mutation was distinct from those with two ABCC8 mutations. The heterozygous ABCC8 mutation carriers were either responsive or unresponsive to therapy with diazoxide. Those that responded to diazoxide mostly had a dominant form of CHI. Thirteen patients (from nine families) were heterozygous for eight different dominantly inherited mutations occurring in the NBD2 area of the SUR1 protein. NBD1 appears to be the principal site for ATP binding, whereas NBD2 binds MgADP (38). These binding domains cooperate with each other in mediating the nucleotide regulation of the pore function (39). MgADP and diazoxide activate KATP channels in the presence of inhibitory concentrations of ATP, and both processes require Mg2+ (40, 41, 42). Mutations in NBD2 can abolish channel activation by diazoxide or MgADP (42). One of these mutations (p.G1479A) is novel. The phenotype associated with these dominantly inherited mutations confirms the reported phenotype associated with dominant mutations in the NBD2 area (13, 43); however, dominant CHI mutations also occur outside the NBD2 (44).
The observations suggest that the clinical presentation of patients with dominant inactivating ABCC8 missense mutations is variable, ranging from mild medically responsive to severe early-onset CHI requiring a near total pancreatectomy. The clinical presentation of patients at the time of diagnosis cannot distinguish between recessive and dominantly acting ABCC8 mutations. A genetic diagnosis is important as finding a de novo dominant mutation in a patient with diazoxide-unresponsive CHI confers a lower recurrence risk for future siblings (i.e. the risk of germ-line mosaicism) compared with a recessive mutation, but a higher risk of CHI (50%) for the next generation.
This study also demonstrated that a paternal ABCC8/KCNJ11 mutation in association with diazoxide-unresponsive disease is not synonymous with a focal lesion. Of the 23 patients with diazoxide-unresponsive disease and an identifiable paternal mutation (4, KCNJ11 and 19, ABCC8), 16 had confirmed focal disease, six had diffuse disease and the histological diagnosis was not confirmed in one. The mechanism of diffuse disease in patients with a heterozygous paternal ABCC8/KCNJ11 mutation is not understood, but similar observations have been documented in a previous study (45). However, in the previous study, differentiation of diffuse and focal were based on radiological rather than histological grounds, and this might have resulted in the excess of paternal-only cases. It is possible that the sequencing and dosage analysis was unable to detect a second ABCC8 mutation. It is also possible that there is a second hit within the pancreas, for e.g. loss of a part of the maternal 11p15 affecting the entire pancreas (leading to a large focal lesion) or a second pancreatic specific mutation. Another possibility is that some of these mutations are dominantly acting and the lack of co-segregation with disease in the family reflects incomplete penetrance. Genetic analysis of the pancreatic samples is warranted to test this hypothesis.
Three patients (patients 10, 329 and 129) with diazoxide-responsive CHI were heterozygous for a single ABCC8 mutation (Supplementary Table 3). These mutations (c.3992-9G>A and p.D1472N) are known to act recessively, and it is possible that a second germ-line, or somatic mutation within the pancreas, has not been identified by Sanger sequencing or dosage analysis. However, the phenotype of these patients does not fit in with recessively inherited CHI that is generally diazoxide unresponsive (as is also seen in this series). This series identified a missense mutation p.G111R in six unrelated patients with severe, diazoxide-unresponsive disease, of Indian background. Two ABCC8 mutations, c.3992-9G>A and p.F1388del, are associated with CHI in the Ashkenazi Jewish population (11) and the p.V187D mutation has been associated with the Finnish population (46). The recognition of the p.G111R mutation exclusively in patients from an Indian ethnic background suggests a founder effect.
It was striking that those patients with HNF4A mutations presented earlier with hypoglycaemia and had a higher birth weight when compared with patients with other genetic aetiologies. However, as an overlap between birth weights and age at presentation was observed between patients with HNF4A and KATP channel mutations, it is not possible to distinguish between the two groups on the basis of these features. Although a family history of diabetes is more likely to suggest a HNF4A mutation, it is also not a distinguishing feature. Hence, given the higher prevalence of KATP channel mutations and the high rate of diabetes phenocopies in the population, we recommend to sequence KCNJ11 and ABCC8 initially in all patients with diazoxide-responsive CHI, followed by HNF4A if HI is diagnosed within the first week of life.
In contrast to patients with a HNF4A or KATP channel mutation, patients with a GLUD1/HADH mutation were diagnosed later and were of normal birth weight. It was not possible to distinguish patients with a GLUD1 mutation from those with a HADH mutation based on these two features. Patients with GLUD1 or HADH mutation share further similarities in terms of being leucine sensitive. In the vast majority of patients, a serum ammonia concentration would help in distinguishing between the two aetiologies (raised in the majority of patients with a GLUD1 mutation). The other distinguishing feature would be a history of consanguinity as all HADH mutations reported so far have been recessively inherited while GLUD1 mutations are dominantly inherited. In the category of patients who were responsive to diazoxide, no identifiable mutations in the known genes were found in nearly 80% of patients. This suggests that there may well be other novel genetic/environmental mechanisms involved in regulating insulin secretion. Unravelling the genetic basis of CHI in these patients might provide an opportunity to gain further insights into pancreatic β-cell physiology.
In conclusion, a genetic diagnosis was made for 45.3% of patients in this large series. Mutations in the ABCC8 gene were the commonest identifiable cause. The vast majority of patients with diazoxide-responsive CHI had no identifiable mutations, suggesting other genetic and/or environmental mechanisms.
Supplementary data
This is linked to the online version of the paper at http://dx.doi.org/10.1530/EJE-12-0673.
Acknowledgements
The authors would like to thank Annet Damhuis, Karen Stals, Michael Day and Andrew Parrish for their technical assistance and Dr Jayne Houghton for her contribution to the genetic studies.
Footnotes
*(R R Kapoor and S E Flanagan contributed equally to this work)
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by the Medical Research Council (grant number 1DMA), Wellcome Trust (081188/A/06/Z) and Diabetes UK.
References
- de Lonlay P, Fournet JC, Touati G, Groos MS, Martin D, Sevin C, Delagne V, Mayaud C, Chigot V, Sempoux C, et al. Heterogeneity of persistent hyperinsulinaemic hypoglycaemia. A series of 175 cases. European Journal of Pediatrics. 2002;161:37–48. doi: 10.1007/s004310100847. [DOI] [PubMed] [Google Scholar]
- Eds C Stanley & DD de Leon. In Monogenic Hyperinsulinemic Hypoglycemia Disorders (Frontiers in Diabetes). Philadelphia: Karger, 2012.
- Monogenic disorders of insulin secretion: congenital hyperinsulinism and neonatal diabetes, March 15–16, 2012, Faculty Synopses. Pediatric Diabetes 13 344–368, 2012 ( 10.1111/j.1399-5448.2012.00887.x).
- Aynsley-Green A, Hussain K, Hall J, Saudubray JM, Nihoul-Fekete C, De Lonlay-Debeney P, Brunelle F, Otonkoski T, Thornton P, Lindley KJ. Practical management of hyperinsulinism in infancy. Archives of Disease in Childhood. Fetal and Neonatal Edition. 2000;82:F98–F107. doi: 10.1136/fn.82.2.F98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu BY, Kelly A, Thornton PS, Greenberg CR, Dilling LA, Stanley CA. Protein-sensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndrome. Journal of Pediatrics. 2001;138:383–389. doi: 10.1067/mpd.2001.111818. [DOI] [PubMed] [Google Scholar]
- Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, Ellard S, Ferrer J, Hattersley AT. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Medicine. 2007;4:e118. doi: 10.1371/journal.pmed.0040118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James C, Kapoor RR, Ismail D, Hussain K. The genetic basis of congenital hyperinsulinism. Journal of Medical Genetics. 2009;46:289–299. doi: 10.1136/jmg.2008.064337. [DOI] [PubMed] [Google Scholar]
- Stanescu DE, Hughes N, Kaplan B, Stanley CA, De Leon DD. Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. Journal of Clinical Endocrinology and Metabolism. 2012;97:E2026–E2030. doi: 10.1210/jc.2012-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, Aguilar-Bryan L, Gagel RF, Bryan J. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science. 1995;268:426–429. doi: 10.1126/science.7716548. [DOI] [PubMed] [Google Scholar]
- Thomas P, Ye Y, Lightner E. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Human Molecular Genetics. 1996;5:1809–1812. doi: 10.1093/hmg/5.11.1809. [DOI] [PubMed] [Google Scholar]
- Nestorowicz A, Wilson BA, Schoor KP, Inoue H, Glaser B, Landau H, Stanley CA, Thornton PS, Clement JP, Bryan J, et al. Mutations in the sulonylurea receptor gene are associated with familial hyperinsulinism in Ashkenazi Jews. Human Molecular Genetics. 1996;5:1813–1822. doi: 10.1093/hmg/5.11.1813. [DOI] [PubMed] [Google Scholar]
- Dunne MJ, Kane C, Shepherd RM, Sanchez JA, James RF, Johnson PR, Aynsley-Green A, Lu S, Clement JP, Lindley KJ, et al. Familial persistent hyperinsulinemic hypoglycemia of infancy and mutations in the sulfonylurea receptor. New England Journal of Medicine. 1997;336:703–706. doi: 10.1056/NEJM199703063361005. [DOI] [PubMed] [Google Scholar]
- Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, Ganguly A, Shyng SL, Stanley CA. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. Journal of Clinical Investigation. 2008;118:2877–2886. doi: 10.1172/JCI35414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane C, Shepherd RM, Squires PE, Johnson PR, James RF, Milla PJ, Aynsley-Green A, Lindley KJ, Dunne MJ. Loss of functional KATP channels in pancreatic β-cells causes persistent hyperinsulinemic hypoglycemia of infancy. Nature Medicine. 1996;2:1344–1347. doi: 10.1038/nm1296-1344. [DOI] [PubMed] [Google Scholar]
- Matsuo M, Trapp S, Tanizawa Y, Kioka N, Amachi T, Oka Y, Ashcroft FM, Ueda K. Functional analysis of a mutant sulfonylurea receptor, SUR1-R1420C, that is responsible for persistent hyperinsulinemic hypoglycemia of infancy. Journal of Biological Chemistry. 2000;275:41184–41191. doi: 10.1074/jbc.M006503200. [DOI] [PubMed] [Google Scholar]
- Cartier EA, Conti LR, Vandenberg CA, Shyng SL. Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. PNAS. 2001;98:2882–2887. doi: 10.1073/pnas.051499698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley CA. Hyperinsulinism/hyperammonemia syndrome: insights into the regulatory role of glutamate dehydrogenase in ammonia metabolism. Molecular Genetics and Metabolism. 2004;81(Suppl 1):S45–S51. doi: 10.1016/j.ymgme.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Cuesta-Munoz AL, Huopio H, Otonkoski T, Gomez-Zumaquero JM, Nanto-Salonen K, Rahier J, Lopez-Enriquez S, Garcia-Gimeno MA, Sanz P, Soriguer FC, et al. Severe persistent hyperinsulinemic hypoglycemia due to a de novo glucokinase mutation. Diabetes. 2004;53:2164–2168. doi: 10.2337/diabetes.53.8.2164. [DOI] [PubMed] [Google Scholar]
- Christesen HB, Tribble ND, Molven A, Siddiqui J, Sandal T, Brusgaard K, Ellard S, Njolstad PR, Alm J, Brock Jacobsen B, et al. Activating glucokinase (GCK) mutations as a cause of medically responsive congenital hyperinsulinism: prevalence in children and characterisation of a novel GCK mutation. European Journal of Endocrinology. 2008;159:27–34. doi: 10.1530/EJE-08-0203. [DOI] [PubMed] [Google Scholar]
- Sayed S, Langdon DR, Odili S, Chen P, Buettger C, Schiffman AB, Suchi M, Taub R, Grimsby J, Matschinsky FM, et al. Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes. 2009;58:1419–1427. doi: 10.2337/db08-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton PT, Eaton S, Aynsley-Green A, Edginton M, Hussain K, Krywawych S, Datta V, Malingre HE, Berger R, van den Berg IE. Hyperinsulinism in short-chain l-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of β-oxidation in insulin secretion. Journal of Clinical Investigation. 2001;108:457–465. doi: 10.1172/JCI11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molven A, Matre GE, Duran M, Wanders RJ, Rishaug U, Njolstad PR, Jellum E, Sovik O. Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes. 2004;53:221–227. doi: 10.2337/diabetes.53.1.221. [DOI] [PubMed] [Google Scholar]
- Hussain K, Clayton PT, Krywawych S, Chatziandreou I, Mills P, Ginbey DW, Geboers AJ, Berger R, van den Berg IE, Eaton S. Hyperinsulinism of infancy associated with a novel splice site mutation in the SCHAD gene. Journal of Pediatrics. 2005;146:706–708. doi: 10.1016/j.jpeds.2005.01.032. [DOI] [PubMed] [Google Scholar]
- Kapoor RR, James C, Flanagan SE, Ellard S, Eaton S, Hussain K. 3-Hydroxyacyl-coenzyme A dehydrogenase deficiency and hyperinsulinemic hypoglycemia: characterization of a novel mutation and severe dietary protein sensitivity. Journal of Clinical Endocrinology and Metabolism. 2009;94:2221–2225. doi: 10.1210/jc.2009-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Chen P, Palladino A, Narayan S, Russell LK, Sayed S, Xiong G, Chen J, Stokes D, Butt YM, et al. Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. Journal of Biological Chemistry. 2010;285:31806–31818. doi: 10.1074/jbc.M110.123638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otonkoski T, Kaminen N, Ustinov J, Lapatto R, Meissner T, Mayatepek E, Kere J, Sipila I. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes. 2003;52:199–204. doi: 10.2337/diabetes.52.1.199. [DOI] [PubMed] [Google Scholar]
- Otonkoski T, Jiao H, Kaminen-Ahola N, Tapia-Paez I, Ullah MS, Parton LE, Schuit F, Quintens R, Sipila I, Mayatepek E, et al. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic β cells. American Journal of Human Genetics. 2007;81:467–474. doi: 10.1086/520960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor RR, Locke J, Colclough K, Wales J, Conn JJ, Hattersley AT, Ellard S, Hussain K. Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes. 2008;57:1659–1663. doi: 10.2337/db07-1657. [DOI] [PubMed] [Google Scholar]
- Rahier J, Guiot Y, Sempoux C. Persistent hyperinsulinaemic hypoglycaemia of infancy: a heterogeneous syndrome unrelated to nesidioblastosis. Archives of Disease in Childhood. Fetal and Neonatal Edition. 2000;82:F108–F112. doi: 10.1136/fn.82.2.F108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkarre V, Fournet JC, de Lonlay P, Gross-Morand MS, Devillers M, Rahier J, Brunelle F, Robert JJ, Nihoul-Fekete C, Saudubray JM, et al. Paternal mutation of the sulfonylurea receptor (SUR1) gene and maternal loss of 11p15 imprinted genes lead to persistent hyperinsulinism in focal adenomatous hyperplasia. Journal of Clinical Investigation. 1998;102:1286–1291. doi: 10.1172/JCI4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otonkoski T, Nanto-Salonen K, Seppanen M, Veijola R, Huopio H, Hussain K, Tapanainen P, Eskola O, Parkkola R, Ekstrom K, et al. Noninvasive diagnosis of focal hyperinsulinism of infancy with [18F]-DOPA positron emission tomography. Diabetes. 2006;55:13–18. doi: 10.2337/diabetes.55.01.06.db05-1128. [DOI] [PubMed] [Google Scholar]
- Fekete CN, de Lonlay P, Jaubert F, Rahier J, Brunelle F, Saudubray JM. The surgical management of congenital hyperinsulinemic hypoglycemia in infancy. Journal of Pediatric Surgery. 2004;39:267–269. doi: 10.1016/j.jpedsurg.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Kapoor RR, Flanagan SE, James C, Shield J, Ellard S, Hussain K. Hyperinsulinaemic hypoglycaemia. Archives of Disease in Childhood. 2009;94:450–457. doi: 10.1136/adc.2008.148171. [DOI] [PubMed] [Google Scholar]
- Kapoor RR, Flanagan SE, James CT, McKiernan J, Thomas AM, Harmer SC, Shield JP, Tinker A, Ellard S, Hussain K. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia. 2011;54:2575–2583. doi: 10.1007/s00125-011-2207-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura K, Flanagan SE, Zadek B, Lethby M, Zubcevic L, Girard CA, Petz O, Mannikko R, Kapoor RR, Hussain K, et al. Adjacent mutations in the gating loop of Kir6.2 produce neonatal diabetes and hyperinsulinism. EMBO Molecular Medicine. 2009;1:166–177. doi: 10.1002/emmm.200900018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor RR, Flanagan SE, Fulton P, Chakrapani A, Chadefaux B, Ben-Omran T, Banerjee I, Shield JP, Ellard S, Hussain K. Hyperinsulinism–hyperammonaemia syndrome: novel mutations in the GLUD1 gene and genotype–phenotype correlations. European Journal of Endocrinology. 2009;161:731–735. doi: 10.1530/EJE-09-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan SE, Kapoor RR, Mali G, Cody D, Murphy N, Schwahn B, Siahanidou T, Banerjee I, Akcay T, Rubio-Cabezas O, et al. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. European Journal of Endocrinology. 2010;162:987–992. doi: 10.1530/EJE-09-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti LR, Radeke CM, Shyng SL, Vandenberg CA. Transmembrane topology of the sulfonylurea receptor SUR1. Journal of Biological Chemistry. 2001;276:41270–41278. doi: 10.1074/jbc.M106555200. [DOI] [PubMed] [Google Scholar]
- Ueda K, Matsuo M, Tanabe K, Morita K, Kioka N, Amachi T. Comparative aspects of the function and mechanism of SUR1 and MDR1 proteins. Biochimica et Biophysica Acta. 1999;1461:305–313. doi: 10.1016/S0005-2736(99)00157-1. [DOI] [PubMed] [Google Scholar]
- Dunne MJ, Petersen OH. Intracellular ADP activates K+ channels that are inhibited by ATP in an insulin-secreting cell line. FEBS Letters. 1986;208:59–62. doi: 10.1016/0014-5793(86)81532-0. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Shyng S, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. Journal of General Physiology. 1997;110:643–654. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko HL, Rahier J, Vauhkonen I, Kere J, Laakso M, Ashcroft F, et al. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. Journal of Clinical Investigation. 2000;106:897–906. doi: 10.1172/JCI9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macmullen CM, Zhou Q, Snider KE, Tewson PH, Becker SA, Aziz AR, Ganguly A, Shyng SL, Stanley CA. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the β-cell sulfonylurea receptor SUR1. Diabetes. 2011;60:1797–1804. doi: 10.2337/db10-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellanne-Chantelot C, Saint-Martin C, Ribeiro MJ, Vaury C, Verkarre V, Arnoux JB, Valayannopoulos V, Gobrecht S, Sempoux C, Rahier J, et al. ABCC8 and KCNJ11 molecular spectrum of 109 patients with diazoxide-unresponsive congenital hyperinsulinism. Journal of Medical Genetics. 2010;47:752–759. doi: 10.1136/jmg.2009.075416. [DOI] [PubMed] [Google Scholar]
- Otonkoski T, Ammala C, Huopio H, Cote GJ, Chapman J, Cosgrove K, Ashfield R, Huang E, Komulainen J, Ashcroft FM, et al. A point mutation inactivating the sulfonylurea receptor causes the severe form of persistent hyperinsulinemic hypoglycemia of infancy in Finland. Diabetes. 1999;48:408–415. doi: 10.2337/diabetes.48.2.408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.