

Abstract
Background
Naturally occurring autoantibodies against amyloid-β (nAbs-Aβ) have been shown to exert beneficial effects on transgenic Alzheimer’s disease (AD) animals in vivo and on primary neurons in vitro. Not much is known about their effect on microglial cells. Our aim was to investigate the effect of nAbs-Aβ on amyloid-β (Aβ)-treated microglial cells in vitro with respect to cell viability, stress pathways, cytokine production and phagocytotic abilities and whether these effects can be conveyed to neurons.
Methods
Primary microglial cells isolated from Swiss Webster mouse mesencephalons on embryonic day 13.5 were pretreated with nAbs-Aβ and then treated with Aβ oligomers. After 3 hours, phagocytosis as well as western blot analysis were evaluated to measure the amount of phagocytized Aβ. Cell viability was analyzed using an MTT assay 24 hours after treatment. Pro-inflammatory cytokines in the supernatants were analyzed with ELISAs and then we treated primary neuronal cells with these conditioned microglia supernatants. Twenty-four hours later we did a MTT assay of the treated neurons. We further investigated the effect of a single nAbs-Aβ administration on Tg2576 mice in vivo.
Results
Upon co-administration of Aβ and nAbs-Aβ no change in microglia viability was observed. However, there was an increase in phosphorylated p38 protein level, an increase in the pro-inflammatory cytokines TNF-α and IL-6 and an increase in Aβ uptake by microglial cells. Treatment of primary neurons with conditioned microglia medium led to a 10% improvement in cell viability when nAbs-Aβ were co-administered compared to Aβ-treated cells alone. We were unable to detect changes in cytokine production in brain lysates of Tg2576 mice.
Conclusions
We provide evidence on the mechanism of action of nAbs-Aβ on microglia in vitro. Interestingly, our in vivo data indicate that nAbs-Aβ administration should be considered as a therapeutic strategy in AD, since there is no inflammatory reaction.
Keywords: Alzheimer’s disease, β-amyloid, Microglia, Inflammation, Immunoglobulins
Background
Alzheimer’s disease (AD) is a neurodegenerative disorder leading to the loss of cholinergic neurons in the brain, characterized by extracellular amyloid-β (Aβ) deposits and the formation of neurofibrillar tangles comprising hyperphosphorylated tau protein inside the cell. The toxicity of Aβ deposits is conveyed by oligomeric aggregates that lead to synaptic dysfunction and apoptosis of neurons [1,2].
Active immunization strategies using different forms of Aβ have been extensively tested in amyloid precursor protein (APP) transgenic mouse models. The initial animal results were promising [3,4], but since active immunization in AD patients was associated with considerable side effects [5] and some patients did not develop Aβ-antibody titers [6], passive immunization strategies came into focus. Treatment with humanized monoclonal antibodies [7] as well as passive immunization with intravenous immunoglobulins (IVIg) has been investigated in pilot clinical trials [8,9]. The rationale for its use is that IVIg contain naturally occurring autoantibodies against Aβ (nAbs-Aβ). Naturally occurring autoantibodies in the central nervous system are involved in maintaining homeostasis by removing debris and are known to prevent inflammation [10]. The beneficial effects of nAbs-Aβ have been shown both in vitro on primary neurons and neuronal cell lines as well as in vivo in transgenic mice [11,12]. The effect of IVIg on microglial cells has already been investigated by other groups. It has been shown that IVIg reduce phagocytosis in vitro via Fc receptors [13], IVIg induce tumor necrosis factor-α (TNF-α) and nitric oxide (NO) in a dose-dependent manner, whereas the greater the IgM/IgA content the higher the impact on microglial cells [14], and that IVIg enhance the secretion of matrix metalloproteinase 9, which seems to play a role in the pathogenesis of multiple sclerosis [15]. Referring to Aβ, Magga et al. found an increase in Aβ clearance when they administered IVIg in a dose-dependent manner on primary microglial cells [16]. IVIg depleted of nAbs-Aβ did not promote clearance, indicating that the enhanced clearance effect is mediated by nAbs-Aβ. In a study on the immortalized murine microglial cell line BV-2, administration of IVIg restored cells’ viability and enhanced phagocytotic ability of fibrillar Aβ [17]. In addition to these results, IVIg are limited in their availability and we therefore decided to use nAbs-Aβ in our study. Theoretically, these will be able to be cloned eventually and therefore the problem of availability can be overcome. In all experiments we used the negative fraction of IVIg, which contains all antibodies except those that were purified with the affinity chromatography, and we were not able to see the same effects as those obtained with nAbs-Aβ, coming to the conclusion that nAbs-Aβ are the active substance in IVIg responsible for the beneficial effects with regard to AD. The effect and the interaction, however, of nAbs-Aβ on microglial cells have not been investigated to date. This is especially important as microglia are purported to have a major role in the pathogenesis and propagation of AD [18]. Upon stimulation, microglial cells can react in many different ways, including phagocytosis, the secretion of immunomodulating cytokines and finally programmed cell death in order to kill pathogens or restore tissue integrity [19]. Activated microglial cells have been found surrounding plaques in histopathological sections of AD patients [20] and their role in the phagocytosis of all forms of Aβ has been investigated. Due to the immune-stimulating effect of foreign monoclonal antibodies in the human body, we were interested in the effect of nAbs-Aβ on primary microglial cells. In the following experiments we investigated the effect of nAbs-Aβ on Aβ-treated microglial cells with respect to cell viability, neuroinflammation and phagocytosis, and whether any effect on primary neurons is conveyed by treated microglial cells.
Material and methods
All chemicals were obtained from Sigma-Aldrich, St. Louis, MO, USA unless indicated otherwise.
Antibodies
Antibodies against the following proteins were used: Aβ (clone 6E10, Covance, Princeton, NJ, USA), phosphorylated (phospho) p38, horseradish peroxidase (HRP)-conjugated secondary antibodies (all Cell Signaling Technology, Danvers, MA, USA), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Novus Biologicals, Littleton, CO, USA), allophycocyanin (APC)-conjugated CD11b (eBioscience, San Diego, CA, USA).
Cells
Primary microglial cell culture
For the isolation of primary microglial cells we used a protocol based on that described by Saura et al. [21]. Microglial cells were derived from embryonic day 13.5 (E13.5) Swiss Webster mouse mesencephalons. Briefly, meninges-free mesencephalons were isolated, collected and homogenized in Leibovitz L-15 medium (PAA Laboratories, Pasching, Austria). After centrifugation cells were resuspended in Dulbecco's modified Eagle’s medium (DMEM) with L-glutamine (Lonza, Basel, Switzerland) containing 10% fetal bovine serum (FBS) (PAA Laboratories, Pasching, Austria), 100 U/mL penicillin and 100 μg/mL streptomycin (Lonza, Basel, Switzerland) and plated on polyethyleneimine (PEI)-coated plates. After 7 days cells were stimulated with granulocyte-macrophage colony-stimulating factor (GM-CSF) (Roche, Basel, Switzerland). Cells were subplated on PEI-coated plates and used for experiments on day in vitro 14 to 19.
Primary cortical neuron cell culture
Neurons were cultured from cortices of E13.5 Swiss Webster mice. Briefly, meninges-free cortices were isolated, collected and homogenized in Leibovitz L-15 medium and resuspended in Neurobasal-A Medium (Invitrogen, Grand Island, NY, USA) supplemented with B27 (Gibco, Basel, Switzerland), 100 U/mL penicillin and 100 μg/mL streptomycin and L-glutamine and plated on PEI-coated plates. Cells were used for experiments on day 6 to 8.
Animals
Twenty- to 22-month-old heterozygous adult female Tg2576 mice expressing mutant APPSWE (695(K670N,M671L) under the control of the hamster prion promoter in a hybrid C57Bl/6 × SJL background and age- and gender-matched non-transgenic wild-type control mice (WT) were used for all experiments. Tg2576 and WT mice were randomly divided into groups of five to six, independent of genotype and treatment, on a 12 hour light–dark schedule (lights on 07:00 to 19:00). They had free access to tap water, were fed ad libitum and kept under standard conditions. The sample sizes of the groups were as follows: transgenic (Tg) control n = 5, Tg nAbs-Aβ n = 5, WT n = 6. All animal procedures were approved by the office of the district president and the Institutional Animal Care and Use Committee.
Administration of nAbs-Aβ to mice
Mice were treated intraperitoneally (i.p.) with nAbs-Aβ (400 μg dissolved in 0.2 ml of physiological saline solution) or vehicle (0.2 ml physiological saline solution). The mice were sacrificed 24 hours later and samples were taken. Brains were immediately frozen in liquid nitrogen and stored at −80°C.
Preparation of oligomeric Aβ
Aβ oligomers were synthesized according to Kayed et al. [22]. Briefly, lyophilized synthetic Aβ42 peptide (PSL, Heidelberg, Germany) was initially monomerized by dissolving in hexafluoroisopropanol (HFIP) and separated into aliquots in low-binding tubes. HFIP was evaporated and aliquots were stored at −20°C until use. The peptide film was resuspended in ultrapure water with a final concentration of 232 μM. A small magnetic stirrer was added and the peptide solution was stirred for 48 hours at room temperature at 1400 rpm.
Preparation of nAbs-Aβ
nAbs-Aβ were isolated from IVIg as previously described [11]. Briefly, we used purified intravenous IgG (Octagam 5%). Ninety-six percent of protein represents normal human IgG (IgA <0.2 mg; IgM <0.1 mg). IgG subclasses are fully represented (IgG 1, 65%; IgG 2, 30%; IgG 3, 3%; IgG 4, 2%). To ensure homogenous orientation of the Aβ peptide to the affinity column, a cysteine residue was introduced at the N-terminal part of the peptide. The azlactone-activated support contains an iodoacetyl group (Ultralink; Perbio, Bonn, Germany) at the end of a hexadecyl-spacer group, which reacted with the cysteinyl-sulfhydryl group to yield a stable thioether linkage in order to reduce steric hindrance and provide maximum binding capacity of the antibodies. The bound antibodies were eluted from the column with 10 × 0.5 ml 0.1 M glycine buffer, pH 2.8. Each fraction was collected in a microreaction tube containing 35 μl 1 M Tris–HCl, pH 9. IVIg depleted of nAbs, termed flow-through (ft), was also collected and used as a negative control in experimental settings. To maintain the integrity of the antibodies, a neutral pH was adjusted immediately after elution by adding the appropriate amount of Tris-HCI or glycine buffer. nAbs-Aβ and ft were sterile filtered and stored at −20°C until use.
Cell viability assessment
Quantification of cell viability was performed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay. Microglial cells were treated with oligomeric Aβ42 (5 μM) and/or nAbs-Aβ or ft (both 0.1 μM and 1 μM) for 24 hours in DMEM without FBS. Medium was changed to medium containing 0.5 mg/mL MTT and incubated for 1 hour. Medium was removed and cells were solubilized with dimethyl sulfoxide (DMSO) (AppliChem, Darmstadt, Germany) and shaken for 30 minutes. The absorbance was measured at 570 nm on a plate reader. Neuronal cells were treated with supernatants of microglial cells. Neurobasal-A medium was removed and conditioned medium from microglial cells was added. After 24 hours, medium was replaced with DMEM containing 0.5 mg/mL MTT and incubated for 1 hour and the assay was performed as described above.
Measurement of TNF-α, IL-1β, IL-6 and interferon-γ
Primary microglial cells were pre-treated with nAbs-Aβ or ft (0.1 μM) for 30 minutes and stimulated with oligomeric Aβ42 (5 μM) for 24 hours. Supernatants were stored at −20°C until use. Mice brains were lysed using T-PER lysis buffer (Pierce, Rockford, IL, USA) containing protease inhibitors (Roche Diagnostics, Mannheim, Germany). Protein concentrations were determined using a spectrophotometer. Concentrations of TNF-α, interleukin-1β (IL-1β), IL-6 and interferon-γ (IFN-γ) were quantified using the Duoset enzyme-linked immunosorbent assay (ELISA) system (R&D, Minneapolis, MN, USA) according to the manufacturer’s protocol.
Western blot
Treated cells were washed twice with ice-cold phosphate-buffered saline (PBS) and cells were lysed using M-Per supplemented with protease and phosphatase inhibitors according to the manufacturer’s protocol. The protein concentration of the cell lysates was determined using NanoDrop. Total cell protein was separated by 4 to 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes. Membranes were blocked with 1 × Rotiblock (Carl Roth, Karlsruhe, Germany) and incubated with the appropriate primary antibody dilutions for Aβ (1:2000), phospho p38 (1:4000) or GAPDH (1:5000). Membranes were washed with Tris-buffered saline containing 5% Tween20 and incubated with secondary antibodies. Membranes were incubated with SuperSignal™ West Dura Extended Duration Substrate (Thermo Scientific, Rockford, IL, USA) and exposed to an autoradiographic film (CL-Xposure Film, Thermo Scientific, Rockford, IL, USA). Band intensity was quantified using a densitometer.
Aβ phagocytosis assay
Fluorescein isothiocyanate (FITC)-labeled Aβ42 was oligomerized as described above. Primary microglial cells were pre-incubated with nAbs-Aβ or ft (0.1 μM) in serum-free DMEM for 30 minutes at 37°C and then treated with FITC-Aβ42 (5 μM) for 3 hours. For flow cytometric analysis, microglial cells were rinsed twice with ice-cold PBS, harvested, and washed with fluorescence-activated cell sorting (FACS) buffer (PBS with 0.1% FBS). Microglial cells were stained with APC-conjugated CD11b antibody, washed and resuspended in FACS buffer containing HOECHST 33258 to sort vital cells only. Measurements were performed with a LSR II flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Analysis was performed using FlowJo software (Tree Star Inc., Ashland, OR, USA). Only vital and CD11b+ cells were used for uptake comparison. Uptake of the differently treated cells was evaluated by comparing the mean fluorescence intensity of FITC. Levels of Aβ phagocytosis were also determined by Western blot.
Statistical analysis
All results of in vitro as well as in vivo experiments are presented as the mean ± SD. We used the Student’s t test to assess the statistical significance of all experiments. For all statistical comparisons, the following definitions were used: P <0.05 (*), P <0.01 (**) or P <0.001 (***).
Results
nAbs-Aβ are not toxic to microglia
nAbs-Aβ have been shown to have beneficial effects on Aβ oligomer-induced toxicity in neuronal cells [11,12]. It has recently been reported that Aβ oligomers reduce viable microglial cells in vitro[23]. We investigated the effect of nAbs-Aβ on Aβ oligomer-induced toxicity (Figure 1). Cell viability was reduced to 50% when stimulated with Aβ42 oligomers. nAbs-Aβ had no beneficial effect on the Aβ-induced reduction in viability. nAbs-Aβ alone showed no reduction in cell viability, nor did solvent-treated cells.
Figure 1.
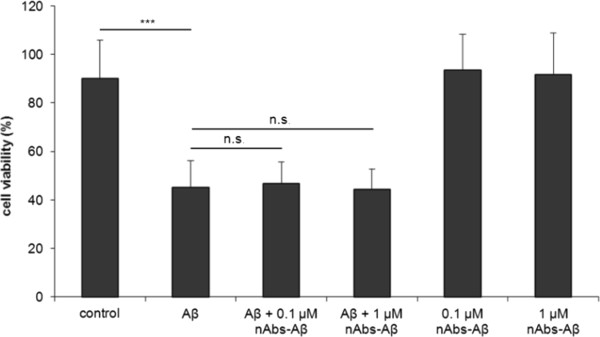
Aβ oligomers reduce cell viability, co-administered nAbs-Aβ do not attenuate cell death. Primary microglia cells were treated with Aβ42 oligomers, cell viability was measured using the MTT assay. To investigate the effect of nAbs-Aβ, microglial cells were pre-incubated for 30 minutes with different concentrations of nAbs-Aβ. Viability of untreated cells was referred to as 100% viability. Experiments were performed at least three times, independently. nAbs-Aβ, naturally occurring autoantibodies against amyloid-β; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Effect of Aβ oligomers and co-administered nAbs-Aβ on stress pathways and cytokine levels in vitro
Since Aβ-mediated cell viability was unaffected by nAbs-Aβ, we investigated changes in intracellular stress pathways upon stimulation with Aβ oligomers and in the presence of nAbs-Aβ. We observed an increase in phospho p38 protein levels upon stimulation with Aβ oligomers alone compared to untreated microglial cells (Figure 2A). nAbs-Aβ alone also induced a slight increase in phospho p38 levels. However, co-administration of Aβ oligomers and nAbs-Aβ led to a strong induction of phospho p38 with protein levels increasing 10-fold compared to untreated cells (Figure 2B).
Figure 2.

Co-administration of nAbs-Aβ and Aβ oligomers leads to a rise in phosphorylated p38 and TNF-α levels. Western blot analysis was performed to detect protein levels of phosphorylated p38. GAPDH was used as loading control (A). Phospho p38 levels were evaluated densitometrically and normalized to GAPDH (B). C + D show cytokine levels of Aβ-treated cells. Cells were treated for 24 hours and supernatants were subjected to cytokine ELISA. ELISA experiments were performed at least three times, independently. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; ELISA, enzyme-linked immunosorbent assay; nAbs-Aβ, naturally occurring autoantibodies against amyloid-β; TNF-α, tumor necrosis factor-α.
Cytokine levels were examined after 24 hours co-treatment. The results are consistent with the results obtained from the phospho p38 stress-pathway investigation: Aβ and nAbs-Aβ alone slightly increased TNF-α (Figure 2C) and IL-6 (Figure 2D) levels, whereas co-administration of both Aβ and nAbs-Aβ led to a strong and significant induction in pro-inflammatory cytokine levels. Co-administration of ft did not lead to the induction of cytokine levels (data not shown).
Influence of nAbs-Aβ on pro-inflammatory cytokines in Tg2576 transgenic animals
After observing the induction of pro-inflammatory cytokines in vitro following treatment of microglial cells with nAbs-Aβ, we next evaluated cytokine concentration in brain homogenates of wild-type animals as well as Tg2576 mice. For all analyzed cytokines we were able to detect upregulation in transgenic animals (Figure 3). IL-1β was elevated by 58% compared to wild-type animals (Figure 3A) (7.11 ± 2.05 vs. 11.23 ± 3.32, P <0.05), IFN-γ levels (Figure 3B) in Tg2576 mice brain were increased by 41% (19.78 ± 1.48 vs. 27.91 ± 8.00, P <0.05), TNF-α (Figure 3C) was increased by 24% (93.16 ± 7.97 vs. 115.62 ± 9.95, P <0.05) and IL-6 (Figure 3D) showed an increase of 24% (19.65 ± 1.48 vs. 24.45 ± 4.32, P <0.05), respectively. Treatment of Tg2576 mice with nAbs-Aβ did not lead to a significant induction or attenuation in the brain concentration of any of the aforementioned cytokines.
Figure 3.
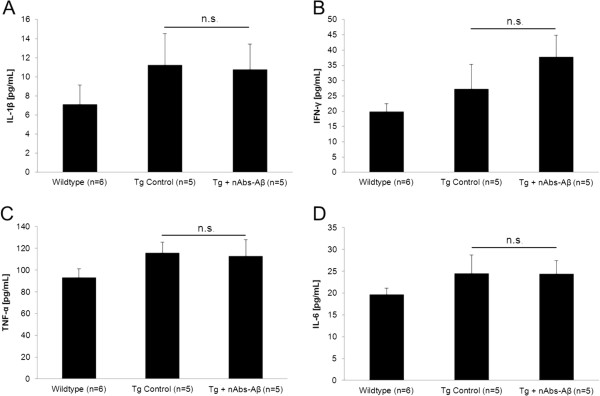
Injection of nAbs-Aβ does not change cytokine levels in Tg2576 mice brain lysates. Brain lysates of wild-type mice, Tg2576 mice and Tg2576 mice treated with nAbs-Aβ for 24 hours were subjected to cytokine ELISA. Figure 3 shows cytokine levels for IL-1β (A), INF-γ (B), TNF-α (C) and IL-6 (D). ELISA, enzyme-linked immunosorbent assay; IL, interleukin; INF, interferon; nAbs-Aβ, naturally occurring autoantibodies against amyloid-β; TNF-α, tumor necrosis factor-α.
nAbs-Aβ increase Aβ42 uptake in primary microglial cells
Aβ is taken up by microglial cells from the periphery and it has been shown that the monoclonal antibody 6E10 increases the phagocytotic ability of microglial cells [24]. To investigate the effect of co-administration of Aβ oligomers and nAbs-Aβ on the phagocytotic abilities of microglial cells in vitro, we used FITC-labeled Aβ42 and FACS analysis. Figure 4A shows a histogram of the shift in the mean fluorescence intensity of the cell population with nAbs-Aβ pre-treatment. Co-administration of nAbs-Aβ led to a doubling in the phagocytosis of Aβ42 oligomers (Figure 4B). ft did not enhance phagocytosis, pointing to a genuine effect of nAbs-Aβ.
Figure 4.

nAbs-Aβ enhance phagocytosis of Aβ oligomers in primary microglia. Histogram of the mean fluorescence intensity of FITC-Aβ and FITC-Aβ and nAbs-Aβ (0.1 μM) co-treated cells (A). (B) shows the means of three independent experiments. Values were normalized to FITC-Aβ42-treated cells. Cell lysates of Aβ42-treated cells were also subjected to western blot. To detect Aβ uptake, blots were probed with the monoclonal Aβ antibody 6E10. GAPDH was used as loading control (C). (D) shows the densitometric evaluation of three independent experiments; values are given normalized to GAPDH intensity. FITC, fluorescein isothiocyanate; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; nAbs-Aβ, naturally occurring autoantibodies against amyloid-β.
Levels of phagocytosed Aβ were also determined by western blotting (Figure 4C). The intensity of the protein bands was evaluated densitometrically (Figure 4D). As shown in Figure 4C and D the incubation of primary microglial cells with Aβ and nAbs-Aβ led to a doubling in phagocytosis. In contrast, no significant increase was observed when using ft.
Supernatants of nAbs-Aβ-co-treated microglial cells have beneficial effects on the viability of neurons compared to supernatants of Aβ-treated microglial cells
To investigate a possible indirect effect of nAbs-Aβ on primary neuronal cells, we cultured neurons in the medium of microglial cells treated with Aβ and/or nAbs-Aβ or ft for 24 hours and assessed cell viability using the MTT assay (Figure 5). Supernatants of microglial cells from three independent preparations were applied to neurons from three independent preparations. Compared to supernatants from solvent-treated microglial cells, supernatants from Aβ-treated microglial cells reduced the viability of neuronal cells to 67%. Following the co-administration of nAbs-Aβ to microglial cells, supernatants significantly reduced the number of viable neuronal cells to 77%. Pre-treatment with ft was not able to significantly restore the viability of primary neuronal cells. In addition, treatment with nAbs-Aβ alone was not harmful to neuronal cells.
Figure 5.
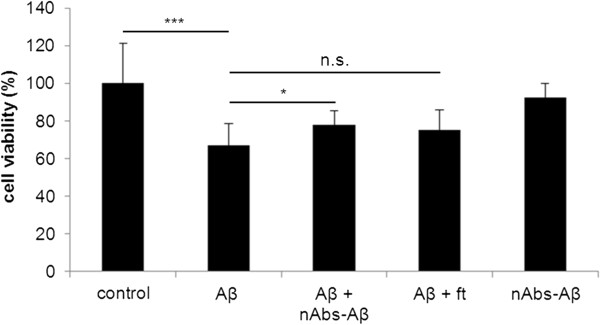
Beneficial effects of supernatants of nAbs-Aβ co-treated microglial cells on the viability of primary neurons. Supernatants of treated microglial cells were subjected to primary neurons and incubated for 24 hours. Aβ concentration was 5 μM and nAbs/ft concentration was 0.1 μM. Viability was assessed using MTT assay. Viability of control-treated cells was referred to as 100% viability. Figure 5 shows the means of at least three independent experiments. nAbs-Aβ, naturally occurring autoantibodies against amyloid-β; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Discussion
The positive effect of nAbs-Aβ on neuronal cells and the fact that IVIg may exert beneficial effects on cognition in AD patients has previously been established [9]. In addition AD patients have reduced levels of nAbs-Aβ in the CSF and plasma compared to age-matched controls, leading to the hypothesis that AD patients will have increased Aβ plaque deposits [25]. In this study we investigated the effect of nAbs-Aβ, isolated from IVIg, on Aβ-exposed microglial cells with the emphasis on cytokine production and phagocytosis of Aβ and whether changes in these physiological actions can convey beneficial effects to primary neurons.
Although the viability of neurons improves with the co-administration of nAbs-Aβ in Aβ oligomer-treated cells [11,12], we found no similar effects on the viability of microglial cells. nAbs-Aβ alone did not influence cell viability, an important property of nAbs-Aβ considering their therapeutic use. Smith et al. provided evidence for a link between increased phagocytosis of Aβ and induction of apoptosis in BV-2 microglial cells [26]. This may also hold true for the co-administration of nAbs-Aβ and could explain the difference in the effect observed for neuronal cells. Compared to the monoclonal antibody 6E10, nAbs-Aβ increased pro-inflammatory TNF-α levels in vitro, whereas no significant changes were observed for IL-6. With the additional administration of Aβ oligomers TNF-α levels and IL-6 levels rose significantly. Consistent with these results, we observed a strong increase in the phosphorylation of the stress pathway protein MAPK p38. There is contradictory evidence regarding whether pro-inflammatory cytokines are beneficial or harmful in AD; current data support the hypothesis that it is a matter of the right equilibrium [27]. Small rises in pro-inflammatory cytokines seem to have beneficial effects, whereas the induction of excessive and prolonged secretion of pro-inflammatory cytokines can lead to chronic neuroinflammation and neurodegeneration. Neuroprotective effects, especially of TNF-α, have been reported. The neuroprotective effect seems to be mediated by the regulation of peroxide formation, calcium accumulation and NF-κB activation [28]. It has been shown by several groups that Aβ overexpression induces neuroinflammation in the AD transgenic mouse models Tg2576 and B6.Cg-Tg(APPswe, PSEN1dE9)85Dbo/J [29,30]. In our present study, we also observed increased cytokine concentrations in 22-month-old Tg2576 mice compared to their wild-type littermates. In contrast to our in vitro experiments, administration of nAbs-Aβ to Tg2576 mice did not lead to an increased inflammatory reaction as measured by intracerebral cytokine levels. Instead IL-1β, TNF-α, IL-6 and IFN-γ levels were unchanged following the administration of nAbs-Aβ to Tg2576 mice. Although the brain is protected by the blood–brain barrier, it has already been shown that nAbs-Aβ can cross it [31]. In vivo studies provide an environment with astrocytes, neurons and microglial cells whereas our in vitro experiments were performed using microglial cells only. Von Bernhardi et al. observed that microglia exposed to Aβ respond with reactive morphological changes, induction of inducible nitric oxide synthase (iNOS), elevated nitric oxide production and decreased reductive metabolism. All these responses were attenuated by the presence of astrocytes [32]. Astrocytes are also involved in Aβ degradation and removal and play an important role in maintaining physiological homeostasis in the brain. However, compared to microglial cells they do not need a stimulus for phagocytosis and at the same time phagocytosis cannot be enhanced by large amounts of IVIg as it can be for microglial cells [16]. Further studies with mixed glial cell cultures are needed to investigate this specific issue.
Interestingly, our observations contrast with those of Minami and colleagues, who showed a profound induction of neuroinflammation following the administration of monoclonal antibodies to mice [33]. One explanation might be the use of a different animal model in the aforementioned study compared to the Tg2576 mouse model in our study. More likely, as a passive immunization strategy, nAbs-Aβ may preclude a neuroinflammatory reaction, unlike monoclonal antibodies in vivo. As we only treated mice for 24 hours before measuring intracerebral cytokine levels one could argue that longer treatment periods are needed to assess the influence of administering anti-Aβ antibodies on neuroinflammation. A study by Puli et al. also showed significant suppression of TNF-α levels after long-term treatment with IVIg in the APP/PS1 mouse model of AD [34]. Nevertheless, long-term studies with repeated administration of nAbs-Aβ and ft are needed to investigate this issue further and to preclude a rise in pro-inflammatory cytokines upon chronic nAbs-Aβ administration in transgenic mice. With respect to the adverse effects of active immunization observed in an early clinical study, treatment with IVIg and/or nAbs-Aβ might prove a safer approach [5]. By applying two different methods we were able to show that phagocytosis of Aβ oligomers by microglial cells doubles when nAbs-Aβ are pre-administered. This effect was not observed with the same amount of flow-through, indicating a highly specific effect of nAbs-Aβ on the uptake of Aβ from microglial cells. This might be attributed to the formation of antigen-antibody complexes between nAbs-Aβ and Aβ oligomers, which are preferentially taken up by microglial cells. Phagocytosis of antigen-antibody complexes is usually mediated via the Fcγ receptor [35]. In the experimental setup that we used, microglial cells of murine origin were exposed to human IgG. Human Fcγ receptor 1 and murine Fcγ receptor 1 share 65 to 75% homology in their extracellular domains and it is known that human Fcγ receptor 1 can bind murine IgG [36]. Unfortunately, nothing is known on the ability of murine Fcγ receptor 1 to bind to human IgG. As our nAbs-Aβ preparation contains all IgG subclasses, it might be worth testing them with regard to their influence on cytokine secretion and phagocytosis separately in the future. Very recently Smith et al. developed a mouse model where all murine Fcγ receptors have been replaced by human Fcγ receptors [37]. It would be ideal to test our human nAbs-Aβ on this mouse model to verify our results and to exclude the impact of species on the immunoglobulin-Fc receptor interaction. However, the positive effect of nAbs-Aβ on Aβ phagocytosis is consistent with results obtained for monoclonal antibodies [24]. Since the amyloid hypothesis postulates that an increase in amyloid-β leads to neurodegeneration, nAbs-Aβ support Aβ oligomer clearance. To determine whether the changes observed for microglial cells have beneficial effects on neurons, we treated primary neurons with conditioned media from microglial cell cultures. Interestingly, we were able to observe a significant improvement in neuron viability from Aβ-treated microglial cells compared to those with co-administered nAbs-Aβ. This was not the case following co-administration of flow-through. We can exclude that this effect is due to a lower amount of Aβ in nAbs-Aβ-treated cell supernatants, even though we observed an increase in Aβ uptake. It is possible that in the co-treated supernatants Aβ is bound to nAbs-Aβ or even modulated in its aggregation state and is therefore less toxic to the neuronal cells. Another explanation could be that the activated microglial cells secrete neurotrophic factors, as it has been reported for lipopolysaccharide (LPS)-treated microglial cells by Nakajima et al. [38]. It is thus likely that simultaneous stimulation of Aβ and nAbs-Aβ activates the same signaling pathways in microglial cells that lead to the production of neurotrophic factors. We measured one neurotrophic factor (brain-derived neurotrophic factor (BDNF); data not shown) but were unable to show a significant increase. Other factors such as glial cell line-derived neurotrophic factor (GDNF) may play a role in the observed effect, and further studies are currently underway.
Conclusions
In summary, we investigated the effects of nAbs-Aβ isolated from IVIg on microglial cells. Although we were not able to rescue microglial cells upon stimulation with Aβ oligomers we showed that nAbs-Aβ have positive effects on the phagocytotic ability of Aβ oligomers. The beneficial effect of microglial-conditioned media on primary neurons is promising, and should be the focus of further research. Our present data demonstrate that nAbs-Aβ are able to enhance Aβ degradation and support neuronal survival without inducing a cerebral inflammatory reaction as shown by unchanged pro-inflammatory cytokine levels in vivo. These findings support nAbs-Aβ as a potential therapy to ameliorate Aβ-induced neuronal toxicity in AD.
Competing interests
This work was partially financed by the Alzheimer Forschungsinitiative (AFI) awarded to JPB.
Authors’ contributions
MG and DM carried out the laboratory experiments. MG, DM and JPB analyzed the data, interpreted the results and wrote the paper. SR designed and carried out the animal experiments. RD discussed analyses, interpretation and presentation. In addition, he participated in the drafting of the paper. All authors have contributed to, read and approved the final manuscript.
Contributor Information
Maike Gold, Email: goldm@staff.uni-marburg.de.
David Mengel, Email: dmengel@med.uni-marburg.de.
Stephan Röskam, Email: roeskam@staff.uni-marburg.de.
Richard Dodel, Email: dodel@med.uni-marburg.de.
Jan-Philipp Bach, Email: bachjan@staff.uni-marburg.de.
Acknowledgements
We would like to thank Christine Forbach for the preparation of nAbs-Aβ. In addition we thank Prof. Dr. Michael Bacher, Department of Immunology, Philipps-University Marburg, for critical discussion during the experiments.
References
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.WNL.0000073623.84147.A8. [DOI] [PubMed] [Google Scholar]
- Schenk D. Amyloid-beta immunotherapy for Alzheimer’s disease: the end of the beginning. Nat Rev Neurosci. 2002;3:824–828. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- Blennow K, Zetterberg H, Rinne JO, Salloway S, Wei J, Black R, Grundman M, Liu E. Effect of immunotherapy with Bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer Disease. Arch Neurol. 2012;69:1002–1010. doi: 10.1001/archneurol.2012.90. [DOI] [PubMed] [Google Scholar]
- Dodel RC, Du Y, Depboylu C, Hampel H, Frolich L, Haag A, Hemmeter U, Paulsen S, Teipel SJ, Brettschneider S, Spottke A, Nölker C, Möller HJ, Wei X, Farlow M, Sommer N, Oertel WH. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2004;75:1472–1474. doi: 10.1136/jnnp.2003.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relkin NR, Szabo P, Adamiak B, Burgut T, Monthe C, Lent RW, Younkin S, Younkin L, Schiff R, Weksler ME. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2009;30:1728–1736. doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- Gold M, Pul R, Bach JP, Stangel M, Dodel R. Pathogenic and physiological autoantibodies in the central nervous system. Immunol Rev. 2012;248:68–86. doi: 10.1111/j.1600-065X.2012.01128.x. [DOI] [PubMed] [Google Scholar]
- Dodel R, Balakrishnan K, Keyvani K, Deuster O, Neff F, Andrei-Selmer LC, Roskam S, Stuer C, Al-Abed Y, Noelker C, Balzer-Geldsetzer M, Oertel W, Du Y, Bacher M. Naturally occurring autoantibodies against beta-amyloid: investigating their role in transgenic animal and in vitro models of Alzheimer’s disease. J Neurosci. 2011;31:5847–5854. doi: 10.1523/JNEUROSCI.4401-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Wei X, Dodel R, Sommer N, Hampel H, Gao F, Ma Z, Zhao L, Oertel WH, Farlow M. Human anti-beta-amyloid antibodies block beta-amyloid fibril formation and prevent beta-amyloid-induced neurotoxicity. Brain. 2003;126:1935–1939. doi: 10.1093/brain/awg191. [DOI] [PubMed] [Google Scholar]
- Stangel M, Joly E, Scolding NJ, Compston DA. Normal polyclonal immunoglobulins (‘IVIg’) inhibit microglial phagocytosis in vitro. J Neuroimmunol. 2000;106:137–144. doi: 10.1016/S0165-5728(00)00210-1. [DOI] [PubMed] [Google Scholar]
- Pul R, Nguyen D, Schmitz U, Marx P, Stangel M. Comparison of intravenous immunoglobulin preparations on microglial function in vitro: more potent immunomodulatory capacity of an IgM/IgA-enriched preparation. Clin Neuropharmacol. 2002;25:254–259. doi: 10.1097/00002826-200209000-00005. [DOI] [PubMed] [Google Scholar]
- Pul R, Kopadze T, Skripuletz T, Voss EV, Kieseier BC, Stangel M. Polyclonal immunoglobulins (IVIg) induce expression of MMP-9 in microglia. J Neuroimmunol. 2009;217:46–50. doi: 10.1016/j.jneuroim.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Magga J, Puli L, Pihlaja R, Kanninen K, Neulamaa S, Malm T, Hartig W, Grosche J, Goldsteins G, Tanila H, Koistinaho J, Koistinaho M. Human intravenous immunoglobulin provides protection against Abeta toxicity by multiple mechanisms in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2010;7:90. doi: 10.1186/1742-2094-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Istrin G, Bosis E, Solomon B. Intravenous immunoglobulin enhances the clearance of fibrillar amyloid-beta peptide. J Neurosci Res. 2006;84:434–443. doi: 10.1002/jnr.20886. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci. 2011;31:16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, Araoz C 3rd. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44:183–189. doi: 10.1002/glia.10274. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Bach JP, Mengel D, Wahle T, Kautz A, Balzer-Geldsetzer M, Al-Abed Y, Dodel R, Bacher M. The role of CNI-1493 in the function of primary microglia with respect to amyloid-beta. J Alzheimers Dis. 2011;26:69–80. doi: 10.3233/JAD-2011-110179. [DOI] [PubMed] [Google Scholar]
- Takata K, Hirata-Fukae C, Becker AG, Chishiro S, Gray AJ, Nishitomi K, Franz AH, Sakaguchi G, Kato A, Mattson MP, Laferla FM, Aisen PS, Kitamura Y, Matsuoka Y. Deglycosylated anti-amyloid beta antibodies reduce microglial phagocytosis and cytokine production while retaining the capacity to induce amyloid beta sequestration. Eur J Neurosci. 2007;26:2458–2468. doi: 10.1111/j.1460-9568.2007.05852.x. [DOI] [PubMed] [Google Scholar]
- Du Y, Dodel R, Hampel H, Buerger K, Lin S, Eastwood B, Bales K, Gao F, Moeller HJ, Oertel W, Farlow M, Paul S. Reduced levels of amyloid beta-peptide antibody in Alzheimer disease. Neurology. 2001;57:801–805. doi: 10.1212/WNL.57.5.801. [DOI] [PubMed] [Google Scholar]
- Smith AM, Gibbons HM, Dragunow M. Valproic acid enhances microglial phagocytosis of amyloid-beta(1–42) Neuroscience. 2010;169:505–515. doi: 10.1016/j.neuroscience.2010.04.041. [DOI] [PubMed] [Google Scholar]
- Wee Yong V. Inflammation in neurological disorders: a help or a hindrance? Neuroscientist. 2010;16:408–420. doi: 10.1177/1073858410371379. [DOI] [PubMed] [Google Scholar]
- Sriram K, O’Callaghan JP. Divergent roles for tumor necrosis factor-alpha in the brain. J Neuroimmune Pharmacol. 2007;2:140–153. doi: 10.1007/s11481-007-9070-6. [DOI] [PubMed] [Google Scholar]
- Abbas N, Bednar I, Mix E, Marie S, Paterson D, Ljungberg A, Morris C, Winblad B, Nordberg A, Zhu J. Up-regulation of the inflammatory cytokines IFN-gamma and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APP(SWE) transgenic mice. J Neuroimmunol. 2002;126:50–57. doi: 10.1016/S0165-5728(02)00050-4. [DOI] [PubMed] [Google Scholar]
- Wu X, Lu Y, Dong Y, Zhang G, Zhang Y, Xu Z, Culley DJ, Crosby G, Marcantonio ER, Tanzi RE, Xie Z. The inhalation anesthetic isoflurane increases levels of proinflammatory TNF-alpha, IL-6, and IL-1beta. Neurobiol Aging. 2012;33:1364–1378. doi: 10.1016/j.neurobiolaging.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher M, Depboylu C, Du Y, Noelker C, Oertel WH, Behr T, Henriksen G, Behe M, Dodel R. Peripheral and central biodistribution of (111)In-labeled anti-beta-amyloid autoantibodies in a transgenic mouse model of Alzheimer’s disease. Neurosci Lett. 2009;449:240–245. doi: 10.1016/j.neulet.2008.08.083. [DOI] [PubMed] [Google Scholar]
- von Bernhardi R, Eugenin J. Microglial reactivity to beta-amyloid is modulated by astrocytes and proinflammatory factors. Brain Res. 2004;1025:186–193. doi: 10.1016/j.brainres.2004.07.084. [DOI] [PubMed] [Google Scholar]
- Minami SS, Sidahmed E, Aid S, Shimoji M, Niikura T, Mocchetti I, Rebeck GW, Prendergast JS, Dealwis C, Wetzel R, Bosetti F, Matsuoka Y, Hoe HS, Turner RS. Therapeutic versus neuroinflammatory effects of passive immunization is dependent on Abeta/amyloid burden in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2010;7:57. doi: 10.1186/1742-2094-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puli L, Pomeshchik Y, Olas K, Malm T, Koistinaho J, Tanila H. Effects of human intravenous immunoglobulin on amyloid pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2012;9:105. doi: 10.1186/1742-2094-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessner JE, Heiken H, Tamm A, Schmidt RE. The IgG Fc receptor family. Ann Hematol. 1998;76:231–248. doi: 10.1007/s002770050396. [DOI] [PubMed] [Google Scholar]
- Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc Natl Acad Sci USA. 2012;109:6181–6186. doi: 10.1073/pnas.1203954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Honda S, Tohyama Y, Imai Y, Kohsaka S, Kurihara T. Neurotrophin secretion from cultured microglia. J Neurosci Res. 2001;65:322–331. doi: 10.1002/jnr.1157. [DOI] [PubMed] [Google Scholar]