

Abstract
Background
Alzheimer’s Disease (AD) is a progressive neurodegenerative disease, especially affecting the hippocampus. Impairment of cognitive and memory functions is associated with amyloid β-peptide-induced oxidative stress and alterations in lipid metabolism. In this scenario, the dual role of peroxisomes in producing and removing ROS, and their function in fatty acids β-oxidation, may be critical. This work aims to investigating the possible involvement of peroxisomes in AD onset and progression, as studied in a transgenic mouse model, harboring the human Swedish familial AD mutation. We therefore characterized the peroxisomal population in the hippocampus, focusing on early, advanced, and late stages of the disease (3, 6, 9, 12, 18 months of age). Several peroxisome-related markers in transgenic and wild-type hippocampal formation were comparatively studied, by a combined molecular/immunohistochemical/ultrastructural approach.
Results
Our results demonstrate early and significant peroxisomal modifications in AD mice, compared to wild-type. Indeed, the peroxisomal membrane protein of 70 kDa and acyl-CoA oxidase 1 are induced at 3 months, possibly reflecting the need for efficient fatty acid β-oxidation, as a compensatory response to mitochondrial dysfunction. The concomitant presence of oxidative damage markers and the altered expression of antioxidant enzymes argue for early oxidative stress in AD. During physiological and pathological brain aging, important changes in the expression of peroxisome-related proteins, also correlating with ongoing gliosis, occur in the hippocampus. These age- and genotype-based alterations, strongly dependent on the specific marker considered, indicate metabolic and/or numerical remodeling of peroxisomal population.
Conclusions
Overall, our data support functional and biogenetic relationships linking peroxisomes to mitochondria and suggest peroxisomal proteins as biomarkers/therapeutic targets in pre-symptomatic AD.
Keywords: Peroxisome, Brain aging, Alzheimer’s disease, Neurodegeneration, Oxidative stress, Lipid metabolism, Catalase, Superoxide dismutase, Glutathione peroxidase, Acyl-CoA beta-oxidation
Background
Alzheimer’s disease (AD) is the most common form of dementia, characterized by progressive neurodegeneration, particularly affecting the hippocampal formation. Impairment of cognitive and memory functions is associated with amyloid β-peptide (Aβ) accumulation, increased oxidative stress, lipid metabolism alteration, and inflammation [1-5].
The central role of peroxisomes in reactive oxygen species (ROS) metabolism has emerged since their discovery [6]. In fact, peroxisomes participate in both ROS generation and removal, under physiological or pathological conditions. Moreover, peroxisomes are involved in a wide range of catabolic and anabolic functions, including β-oxidation of very long chain fatty acids (VLCFAs) [7,8], biosynthesis of polyunsaturated fatty acids and plasmalogens [9,10], and calcium homeostasis [11,12]. Emergent studies have revealed that peroxisomes can also function as intracellular signaling compartments and organizing platforms that orchestrate important developmental decisions from inside the cell [13]. These dynamic and versatile organelles respond to physiological and pathological changes in cellular environment by adapting their morphology, number and enzyme content accordingly [14]. Peroxisomal dysfunction has been shown to be associated with cellular aging as well as with age-related degenerative diseases [13]. The potential role of peroxisomes in human AD, especially in relationship with lipid metabolism, has recently been suggested [15]. Indeed, a decrease in plasmalogens and, conversely, an increase in VLCFAs, were described in cases with advanced Braak stages [16]. Moreover, in an in vitro model of advanced AD a decrease of peroxisomes in hippocampal neurons was reported, while induction of peroxisomal proliferation attenuated Aβ-dependent toxicity [17]. We previously demonstrated that peroxisomes are involved in early stages of AD, as studied either in vivo, in a transgenic mouse model [18], or in vitro, on Aβ treated cortical neurons [19].
The aim of the present work was to investigate the role of peroxisomes during the progression of AD and in normal aging. To this purpose, we utilized the Tg2576 (Tg) mouse model, compared to its wild-type (WT) counterpart. Differently from other mouse models, this strain displays a slowly progressive AD pathology, offering the opportunity to study even subtle age-dependent alterations [20-23]. By combined molecular and morphological approaches, we examined the expression of peroxisome-related proteins in the hippocampal formation at early and advanced AD stages (3, 6, 9, 12, and 18 months). We focused on the CA1 hippocampal region, as pyramidal cells in this field are the most profoundly affected cell type in human AD [24]. Specifically, the expression and immunolocalization patterns of peroxisomal membrane protein of 70 kDa (PMP70), peroxin 14p (Pex14p), catalase (CAT), acyl-CoA oxidase 1 (AOX), and 3-ketoacyl-CoA thiolase (THL) were studied, to get an insight into the biogenesis and functioning of peroxisomes in the diseased brain. Given the involvement of peroxisomes in the maintenance of the redox status, antioxidant enzymes other than CAT, namely selenium-dependent glutathione peroxidase (GPX1), Cu,Zn-superoxide dismutase (SOD1), and Mn-superoxide dismutase (SOD2) were also investigated in Tg animals and in their WT littermates. It is worth recalling that these proteins, though mainly localized to different cell compartments, were also found in peroxisomes [25]. At selected stages, markers for oxidative damage to lipids and nucleic acids - acrolein and 8-hydroxy(deoxy)guanosine (8-OH(d)G), respectively - were also studied.
Notably, the size and functions of peroxisomal population are regulated by a class of ligand-activated transcription factors (peroxisome proliferator-activated receptors, PPARs) [26]. Among these, we studied PPARα which is directly involved in peroxisomal induction [27], plays a neuroprotective role in age-related inflammation [28] and enhances memory consolidation [29]. It is worth-mentioning in this context that PPARα specific agonists have been demonstrated to exert a neuroprotective action against Aβ-mediated toxicity in vitro[17]. Finally, we examined the expression of PPARγ coactivator-1γ (PGC-1α), in view of its synergism with PPARα and its implication in peroxisomal remodeling and biogenesis [30,31].
Results
The distribution of peroxisomes in Tg and WT hippocampal formation from 3-, 6-, 9-, 12-, and 18-month-old mice was investigated by analyzing the expression of peroxisomal membrane (PMP70, Pex14p) and matrix (CAT, AOX, THL) proteins. Markers of oxidative damage to nucleic acids (8-OH(d)G) and to lipids (acrolein), as well as antioxidant enzymes (SOD1, SOD2, GPX1), were also studied. Further, to get an insight into the transcriptional regulation of these genes, PPARα and PGC-1α expression was examined. At selected stages, the presence of PMP70 and PPARα in neuronal and/or astroglial cells was studied by immunofluorescence and their ultrastructural localization was investigated by immunoelectron microscopy.
Peroxisomal membrane proteins: Pex14p and PMP70
Pex14p is a membrane-anchored peroxin, involved in the peroxisomal import of matrix proteins [32-34], and thus considered a dependable marker for peroxisomal number [35]. Pex14p hippocampal levels, as assessed by densitometric analysis of WB (Figure 1a), show no genotype-dependent differences throughout the examined period, except for 18 months of age, when remarkably higher protein concentration in Tg, compared to WT, is detected. Rather, age-related decreases are observed in both normal and diseased animals, namely between 3 and 6 months in the former, and between 6 and 9 months in the latter. This pattern is consistent with Pex14p immunohistochemical staining of pyramidal neurons in CA1 hippocampal field (Figure 1b).
Figure 1.

Pex14p protein levels and distribution in the hippocampus of WT and Tg mice. (a) Densitometric values of Pex14p WB, performed on hippocampal protein extracts of 3-, 6-, 9-, 12-, and 18-month-old WT and Tg mice. Data are expressed as mean ± SD. *P <0.05; **P <0.01; ***P <0.001. (b) Pex14p immunohistochemical localization in CA1 hippocampal field of 3-, 6-, 9-, and 18-month-old WT and Tg mice. or, stratum oriens; pyr, stratum pyramidale; rad, stratum radiatum. Scale bars, 25 μm.
PMP70, a major component of mammalian peroxisomal membranes, is also considered as a good marker for the overall size of peroxisomal population [36]. This ATP-binding cassette transporter, also known as ABCD3, is suggested to be responsible for the metabolic transport of long and branched-chain fatty acyl-CoAs [37]. Our WB data (Figure 2a) show a significant PMP70 induction in 3-month-old Tg hippocampus compared to control. A decrease at 6 months of age ensues in both genotypes, being especially dramatic in the diseased mice. Neither age- nor genotype-related variations are detected at 9 and 12 months, while a peak of PMP70 protein levels is observed at 18 months, particularly in the pathological genotype.
Figure 2.

PMP70 protein levels and distribution in the hippocampus of WT and Tg mice. (a) Densitometric values of PMP70 WB, obtained analyzing the hippocampal protein extracts of 3-, 6-, 9-, 12-, and 18-month-old WT and Tg mice. Data are expressed as mean ± SD. *P <0.05; **P <0.01; ***P <0.001. (b) Immunohistochemical distribution of PMP70 in CA1 hippocampal field of 3-, 6-, 9-, and 18-month-old WT and Tg mice. or, stratum oriens; pyr, stratum pyramidale; rad, stratum radiatum. Scale bars, 25 μm. (c) PMP70 pre-embedding immunoelectron microscopy of 3-month-old CA1 pyramidal neurons of WT and Tg animals. In both genotypes positive peroxisomes (arrows) are observed and they appear more numerous in the cytoplasm of Tg neurons. N, neuronal nucleus; m, mitochondrion. Scale bars, 1 μm.
PMP70 immunohistochemical results on the CA1 hippocampal field are in agreement with molecular data (Figure 2b). Indeed, 3-month-old Tg pyramidal cell layer displays stronger immunoreactivity than its WT counterpart, while, at 6 months, decreased immunostaining is observed in both genotypes. At 9–12 months of age, immunostaining levels remain stable, and they remarkably increase in 18-month-old hippocampus, especially in the somata of Tg pyramidal neurons. PMP70 pre-embedding immunoelectron microscopy allowed us to identify positive peroxisomes in 3-month-old CA1 pyramidal neurons, in both WT and Tg animals (Figure 2c). The immunoreaction product appears confined to the membrane, as expected. Consistent with molecular and immunohistochemical data, ultrastructural analysis shows numerous PMP70 immunoreactive peroxisomes in hippocampal cells of young Tg mice.
The strong PMP70 immunoreactivity observed at 18 months suggests possible contribution by astrogliosis to peroxisome numerical increase. To address this issue, we performed GFAP immunohistochemistry and immunoblotting (Figures 3a and b), as well as double immunofluorescence of PMP70 in combination with GFAP (Figure 3c). As expected, astrogliosis is present in the aging hippocampus, particularly in the Tg animals, and also associates with senile plaques. Confocal images of 18-month-old WT and Tg CA1 fields show bright PMP70 immunofluorescence in GFAP– and GFAP+ cells, in both genotypes. PMP70+/GFAP+ cells are especially numerous in Tg, demonstrating that peroxisomal increase in the AD senescent hippocampus is importantly contributed by astroglial cells.
Figure 3.

GFAP protein levels and distribution in the hippocampus of WT and Tg mice. (a) GFAP immunohistochemical localization in CA1 hippocampal field of 9-, 12-, and 18-month-old WT and Tg mice. or, stratum oriens; pyr, stratum pyramidale; rad, stratum radiatum. Scale bars, 25 μm. (b) Densitometric analysis of GFAP WB performed on hippocampal protein extracts of 3-, 6-, 9-, 12-, and 18-month-old mice. Values are expressed as mean ± SD. **P < 0.01; ***P < 0.001. (c) Double immunofluorescence of PMP70 (green) in combination with GFAP (red) in the CA1 hippocampal field of 18-month-old WT and Tg brain. Several PMP70+/GFAP+ cells (yellow) are especially numerous in the pathological genotype. Scale bars, 25 μm.
Peroxisomal fatty acid β-oxidation enzymes
Even though the important role of peroxisomal β-oxidation in brain development and functioning is well recognized [8], no information on this metabolic pathway in AD is presently available. We therefore analyzed the expression of two major peroxisomal β-oxidation enzymes, namely AOX and THL at the onset and during the progression of AD. The former is the rate-limiting enzyme catalyzing the first step of the cycle, while THL is the last enzyme of the pathway. In 3-month-old Tg hippocampus, AOX expression is significantly higher than in its WT counterpart (Figure 4a). Starting from 6 months, protein levels significantly decrease and remain relatively low throughout the progression of disease. This pattern is supported by AOX immunohistochemical analysis, showing intense immunoreactivity in CA1 pyramidal cell somata of 3-month-old Tg hippocampus, not observed at subsequent stages (Figure 4b). The expression pattern of THL displays no genotype- or age-linked variations, except for 18 months, when hippocampal levels are strongly decreased in both genotypes, suggesting age-dependent impairment of peroxisomal β-oxidation (Figure 4c).
Figure 4.

Peroxisomal fatty acid β-oxidation enzymes in the hippocampus of WT and Tg mice. (a, c) Densitometric values of AOX and THL WB performed on hippocampal protein extracts of 3-, 6-, 9-, 12-, and 18-month-old WT and Tg brains. Data are expressed as mean ± SD. *P <0.05; **P <0.01; ***P <0.001. (b) AOX immunolocalization in CA1 hippocampal field of 3-, 6-, and 18-month-old WT and Tg brains. or, stratum oriens; pyr, stratum pyramidale; rad, stratum radiatum. Scale bars, 25 μm.
ROS-scavenging enzymes: CAT, GPX1, SOD1 and SOD2
Peroxisomes play a pivotal role in the protection against ROS, mainly through their CAT activity, but also thanks to their content in SOD1, SOD2, and GPX1 [25,38,39]. High CAT levels are found in 3-month-old hippocampus irrespective of the genotype, while at 6 months significantly higher values are observed in Tg, as compared to WT (Figure 5a). At 9 months, CAT expression in the Tg decreases to WT levels, which are maintained at later ages in both genotypes. These data are confirmed by immunohistochemical results, CA1 pyramidal neurons being especially CAT positive in 6-month-old Tg hippocampus (Figure 5b).
Figure 5.

H2O2-scavenging enzymes in WT and Tg hippocampus. (a, c) Densitometric values of CAT and GPX1 WB performed on hippocampal protein extracts of 3-, 6-, 9-, 12-, and 18-month-old WT and Tg brains. Values are expressed as mean ± SD. *P <0.05; **P <0.01; ***P <0.001. (b, d) CAT and GPX1 immunolocalization in CA1 hippocampal field of 3-, 6-, 9- and 18-month-old WT and Tg brains. or, stratum oriens; pyr, stratum pyramidale; rad, stratum radiatum. Scale bars, 25 μm.
The other major H2O2–scavenging enzyme, GPX1, shows no significant variations during normal aging, while differences in its expression levels are seen in the pathological genotype, during AD progression. Indeed, at 3 months, GPX1 expression in Tg is lower than in WT, increasing thereafter, to reach significantly higher levels than WT at 9 months. In the aging Tg hippocampus a progressive decrease in GPX1 protein, below WT levels, is observed (Figure 5c). Immunohistochemical data are consistent with WB data (Figure 5d).
The molecular and morphological expression patterns of the two enzymes responsible for the dismutation of the superoxide anion to hydrogen peroxide – SOD1 and SOD2 – are shown in Figures 6 and 7, respectively. As to SOD1 (Figure 6a, b), in 3-month-old Tg hippocampus its levels are significantly lower than in WT, but progressively increase thereafter, reaching at 9 months significantly higher levels than its WT counterpart. SOD1 expression remains stable in the Tg at 12 months of age, when WT levels are instead maximally increased, and sharply decreases at 18 months in both genotypes.
Figure 6.

SOD1 protein levels and distribution in WT and Tg hippocampus. (a) Densitometric values of SOD1 WB performed on hippocampal protein extracts of 3-, 6-, 9-, 12- and 18-month-old WT and Tg brains. Values are expressed as mean ± SD. *P <0.05; **P <0.01; ***P <0.001. (b) SOD1 immunolocalization in CA1 hippocampal field of 3-, 6-, 9-, 12- and 18-month-old WT and Tg brains. or, stratum oriens; pyr, stratum pyramidale; rad, stratum radiatum. Scale bars, 25 μm.
Figure 7.

SOD2 protein levels and distribution in WT and Tg hippocampus. (a) Densitometric values of SOD2 WB performed on hippocampal protein extracts of 3-, 6-, 9-, 12- and 18-month-old WT and Tg brains. Values are expressed as mean ± SD. **P <0.01; ***P <0.001. (b) SOD2 immunolocalization in CA1 hippocampal field of 3-, 6-, 9-, 12- and 18-month-old WT and Tg brains. or, stratum oriens; pyr, stratum pyramidale; rad, stratum radiatum. Scale bars, 25 μm. (c) Higher magnification showing SOD2 immunopositive glial cells around senile plaques in 18-month-old Tg hippocampus. Scale bar, 15 μm.
SOD2 expression pattern during normal aging (Figure 7a, b) closely resembles that of SOD1 in WT hippocampus, while in Tg mice no age-dependent variations are observed. Interestingly, SOD2 levels are significantly higher in 3- and in 18-month-old Tg hippocampus, compared to their WT counterparts. Remarkably, at 18 months, SOD2 mainly localizes to glial cells surrounding senile plaques in Tg hippocampus (Figure 7c).
Oxidative stress markers: acrolein and 8-OH(d)G
To evaluate possible oxidative stress occurring in the hippocampus during normal and pathological aging, markers of oxidative damage to biomolecules were investigated by immunohistochemistry. Antibodies to acrolein, marker of lipid peroxidation (Figure 8a), and to 8-OH(d)G, marker of DNA/RNA oxidative modifications (Figure 8b), were tested at 3 and 18 months, as representative of early and advanced AD stages.
Figure 8.

Immunolocalization of oxidative stress markers at early and advanced AD stages. (a, b) Acrolein and 8-OH(d)G in CA1 hippocampal field of 3- and 18-month-old WT and Tg mice. or, stratum oriens; pyr, stratum pyramidale; rad, stratum radiatum. Scale bars, 25 μm.
We found a similar immunoreactivity pattern for both markers, strongly related to the genotype, in that intense labeling in Tg CA1 pyramidal layer was observed, as early as 3 months, when WT neurons are faintly immunopositive in their cytoplasm. In 18-month-old hippocampus, oxidative damage markers are detected in both WT and Tg CA1 pyramidal cells, even though the former are consistently less immunoreactive than the latter. Intriguingly, the intensity of 8-OH(d)G immunoreaction at 18 months in Tg CA1 neurons appears lower than at 3 months, suggesting a lower degree of oxidative damage to nucleic acids in late AD.
PPARα and PGC-1α
Since the expression and activity of PPARα, major regulator of peroxisome biogenesis, is influenced by oxidative stress [28], we also addressed the putative involvement of this transcription factor at the onset and at late stages of AD. PPARα immunostaining of CA1 pyramidal cell layer shows significantly higher levels in 3-month-old Tg hippocampus, compared to its WT counterpart (Figure 9a). Triple immunofluorescence experiments of PPARα in combination with the neuronal marker NeuN and the astroglial marker GFAP, confirm high expression of the receptor in 3-month-old Tg neuronal cell nuclei and demonstrate its presence also in glial cell nuclei (Figure 9b). Pre-embedding immunoelectron microscopy further confirms the massive presence of PPARα within the nucleus of Tg hippocampal pyramidal neurons (Figure 9c).
Figure 9.

PPARα immunolocalization in 3-month-old WT and Tg mice. (a) PPARα immunohistochemistry in CA1 pyramidal cell layer shows a higher immunoreactivity in Tg hippocampus, than in its WT counterpart. or, stratum oriens; pyr, stratum pyramidale; rad, stratum radiatum. Scale bars, 25 μm. (b) Confocal image of triple immunofluorescence for PPARα (green) in combination with GFAP (red) and NeuN (blue) demonstrates the presence of PPARα-positive neurons and astrocytes. Note the brightness of green signal in the Tg section. Scale bars, 25 μm. (c) PPARα pre-embedding immunoelectron microscopy of CA1 pyramidal neurons shows the nuclear localization of the immunoreaction product, especially concentrated in the Tg nucleus. Arrows indicate the nuclear envelope. N, nucleus; n, nucleolus. Scale bars, 2 μm.
Given the growing evidence for a PGC-1α-mediated transcriptional regulation in response to oxidative stress and in view of its roles in peroxisome biogenesis [31] and in promoting the nonamyloidogenic pathway of APP [40], we analyzed the immunohistochemical localization of this coactivator in the hippocampal CA1 region (Figure 10). At 3 months of age, PGC-1α appears localized in both the nucleus and the cytoplasm, including dendrites extending in the stratum radiatum, of WT and Tg hippocampus. Interestingly, at 6 months, immunoreactivity is highly concentrated in Tg neuronal nuclei, while in WT hippocampus nuclear localization is observed at 9 months. At the latest age considered, a general decrease in immunoreactivity is detected.
Figure 10.
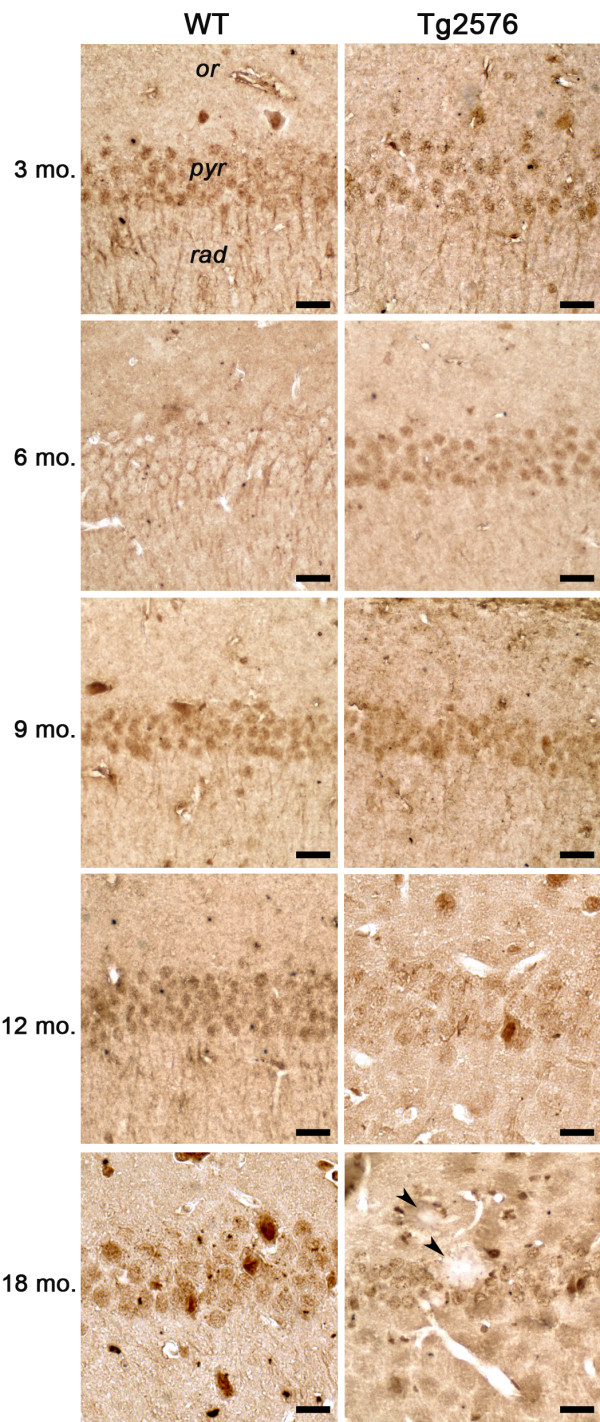
PGC-1α immunohistochemistry in CA1 hippocampal field of 3, 6-, 9-, 12- and 18-month-old WT and Tg mice. Arrowheads, Aβ plaques. or, stratum oriens; pyr, stratum pyramidale; rad, stratum radiatum. Scale bars, 25 μm.
Discussion
The aim of the present study was to investigate the role of peroxisomes in AD onset and progression. In a previous work [18], peroxisomal involvement in early AD was demonstrated in the Tg2576 mouse model, also used in this work. Differently from that study, in which male animals were analyzed, we here utilized females, since epidemiological and histopathological data indicate this as the most AD-affected gender in mice and humans [41,42]. We focused on the hippocampal formation, i.e., the primarily affected area of the diseased brain [43]. The expression of peroxisome-related proteins was studied by combined molecular and morphological approaches, at the onset (3 months) and during AD progression (6, 9, 12, 18 months) in Tg animals, compared to their WT counterparts.
Our results demonstrate that significant peroxisomal alterations occur in Tg mice, as early as 3 months. These early changes are consistent with behavioral, electrophysiological, ultrastructural and molecular modifications reported in this model at the same age [22]. Indeed, PMP70, long been considered marker of the numerical size of peroxisomal population [36] is upregulated, as assessed by WB. Consistently, PMP70 immunohistochemistry reveals especially intense immunostaining in the pyramidal layer of Tg hippocampal CA1 subdivision, and immunoelectron microscopy shows numerous positive peroxisomes in the somata of these neurons.
Differently from PMP70, the biogenesis marker Pex14p, also used to evaluate peroxisome number [35], is unchanged in 3-month-old Tg animals. The absence of correlation between PMP70 and Pex14p levels in early AD could either indicate specific concentration of PMP70 in a numerically unchanged peroxisomal population, or impoverishment of Pex14p in proliferated peroxisomes. In either case, the altered content in these proteins likely results in profound modifications of peroxisomal functions. Interestingly, Pex14p has recently been involved in microtubule-based peroxisome motility [44], suggesting that at this early AD stage, peroxisomes, endowed with a relatively low Pex14p/PMP70 ratio are altered in their transport along neurites. To this respect, it is worth mentioning that a recent work from Berger’s group demonstrates an increased peroxisomal volume density in neuronal cell bodies of human AD brain, associated with impaired peroxisome trafficking [16].
The specific increase in early AD of PMP70, presumably involved in fatty acyl-CoA import across peroxisomal membranes [37], could reflect the need for a more efficient acyl-CoA β-oxidation. In agreement with this hypothesis, AOX, rate-limiting enzyme of peroxisomal β-oxidation pathway, is upregulated in 3-month-old Tg hippocampus. This increase could thus represent a compensatory response to early Aβ-mediated mitochondrial insult, to cope with the impaired energy metabolism, occurring in AD [45,46]. Indeed, abnormalities in glucose metabolism/mitochondrial function are invariant features of early AD [46-49]. Consistently, positron emission tomography studies in AD patients report abnormal cerebral glucose utilization, particularly in the hippocampus, decades prior to the onset of histopathological and clinical features [50]. A consequent shift in brain metabolism, from a primarily aerobic glycolysis pathway to a ketogenic/fatty acid β-oxidation pathway is thought to occur [51]. In this context, we suggest that peroxisomal β-oxidation contributes to such metabolic change, providing mitochondria with acetyl-CoA and fatty acyl substrates. Induction of peroxisomal β-oxidation pathway may also be neuroprotective, by enhancing the clearance of potentially toxic VLCFAs and by promoting the biosynthesis of the neuroprotective docosahexaenoic acid (DHA) [15]. Relevantly, elevated levels of VLCFA have been found in human AD brain lesions [16] and the ability of these fatty acids to trigger oxidative stress and mitochondrial dysfunction on neural cells has recently been demonstrated [52,53].
A biogenetic/metabolic relationship between peroxisomes and mitochondria has long been established and recently emphasized [13,30,31]. In yeast, experimentally induced or age-associated mitochondrial dysfunction activates the so-called retrograde (RTG) signaling pathway, leading to the transcription of RTG-target genes, including those required for peroxisome biogenesis and function [13]. Our results showing increased PPARα nuclear immunoreactivity allow us to speculate that this gene be included among the above mentioned RTG-target genes. The increased PPARα expression could in turn be responsible for the induction of its target genes PMP70 and AOX [54,55]. Even though the molecular triggers of PPARα activation remain to be determined in our model, endogenous production of PPARα ligands, e.g., oxidized lipid molecules, is likely to occur due to Aβ-mediated ROS generation.
While, in principle, AOX induction in 3-month-old Tg hippocampus may represent a positive response to enhanced need for lipid substrates to be processed in mitochondria, this induction may also result in increased H2O2 production by the oxidase. Moreover, mitochondrial respiration supported by fatty acids generates substantial rates of ROS production [56], thus contributing to cellular redox status alteration. In this context, even the observed early increase of SOD2 expression, in agreement with reports demonstrating induction of mitochondrial genes in young Tg mice [57], may contribute itself to redox imbalance, through enhanced conversion of superoxide anion to H2O2.
Notably, GPX1, the major cytosolic H2O2–scavenging enzyme, is significantly down-regulated in our early AD samples, further exacerbating imbalance between generation and removal of hydrogen peroxide. GPX1 seems especially critical in AD, since its deletion increases susceptibility of neurons to Aβ-mediated damage, while its overexpression protects against neurodegeneration [58,59]. The postulated pro-oxidant environment likely results in post-translational modifications of GPX1 itself, leading to its irreversible inactivation [60]. Even the low levels of SOD1 detected in the young Tg hippocampus may be explained taking into account that this cytosolic protein is selectively damaged by H2O2[61]. Importantly, SOD1 deficiency in Tg2576 mice has been associated with oxidative damage, accelerated Aβ oligomerization and memory impairment [62].
Therefore, our data strongly support the idea that oxidative stress is the primary culprit in AD pathogenesis [63]. Consistently, 8-OH(d)G and acrolein, markers of oxidative modification to biomolecules [64,65], show DNA/RNA oxidation and lipoperoxidation in the hippocampus of young Tg mice. Interestingly, 8-OH(d)G immunostaining is predominantly localized in the cytoplasm, suggesting that mitochondrial nucleic acids are primarily affected. To our knowledge, this is the first report of such a modification occurring at 3 months in this mouse strain. In this context, it is worth mentioning that acrolein has been suggested not only as a marker of lipoperoxidation, but also as initiator of oxidative stress [65].
Between 3 and 6 months of age, most peroxisomal markers decrease in WT hippocampal formation, indicating a numerical reduction and/or a metabolic change of the organelles in the physiological course of maturity. By contrast, in Tg mice of the same age, when several hallmarks of AD pathology make their appearance [21], marker-specific patterns are observed. In particular, while high levels of CAT and Pex14p are maintained, PMP70 and AOX are significantly decreased, suggesting metabolic, rather than numerical, modifications of peroxisomes. These changes, indicating decreased efficiency of peroxisomal β-oxidation, presumably lead to VLCFA accumulation, consistent with what reported in human AD [16]. Interestingly, pharmacological inhibition of peroxisomal β-oxidation, resulting in VLCFA accumulation, is positively correlated with Aβ40 levels in rat cerebral cortex [66].
The relatively high CAT levels observed in 6-month-old Tg hippocampus are conceivably sustained by transcription factors activated in response to oxidative stress. To this respect, the cellular redox state sensor PGC-1α may play a role in regulating CAT expression [67]. Our immunohistochemical data support this relationship, in that at 6 months intense PGC-1α nuclear staining, suggestive of its induction and activity, is observed in CA1 pyramidal neurons, exclusively in Tg. The decrease in PGC-1α nuclear immunostaining seen at later AD stages, also parallels the behavior of CAT.
The same transcriptional co-activator, which has been involved in aging and age-associated diseases [68], may be responsible for the pattern of other antioxidant enzymes. Indeed, the increased PGC-1α nuclear immunoreactivity observed around 9–12 months in Wt neurons is paralleled by the raise in SOD1 and SOD2 levels, occurring in 12-month-old normal hippocampus. Conversely, the dramatically low expression of PGC-1α at 12 months in AD neurons, when we first observe hippocampal amyloid deposits (not shown), is concomitant with the lower levels of SOD1, SOD2 and, particularly, GPX1, compared to WT. Accordingly, altered glutathione redox status has been associated to the onset of amyloid plaques in a mouse model of AD [69] and decreased activities of SOD1 and GPX1 have been reported in symptomatic human AD [62,70]. By contrast, SOD2 overexpression in Tg mice results in diminished plaque formation [71].
In 18-month-old mouse hippocampus, genotype-dependent variations are observed for most peroxisomal and antioxidant proteins. At this stage, gliosis, known to occur during brain aging and exacerbated in AD [72-74], should be taken into account, when interpreting WB quantitative data. In fact, astrogliosis may be at least in part responsible for the peaks of PMP70 and Pex14p observed in old Tg mice. An important contribution to the overall brain peroxisomal population by astrocytes, highlighted by our GFAP/PMP70 double immunofluorescence images, is consistent with the notion that peroxisomes are abundant in this cell type [75-77]. Nevertheless, our immunohistochemical data on 18-month-old hippocampus clearly show enhanced positivity to PMP70 and Pex14p also in CA1 pyramidal neurons, markedly in the Tg, suggesting a global increase of peroxisome number in the hippocampal tissue, contributed by both neuronal and glial cells. Differently from the above membrane proteins, peroxisomal matrix enzymes, namely CAT, AOX and THL are not increased or even decreased, in 18-month-old hippocampus, possibly owing to age-related low efficiency of peroxisomal protein import [13] and in keeping with the impairment of brain peroxisomal acyl-CoA β-oxidation in physiological and pathological human aging [15,16].
Oxidative stress is known to occur in age-related neurodegeneration [78,79]. Our results on 18-month-old WT hippocampus show significantly lower levels of SOD1 and SOD2 proteins, while GPX1 is not decreased, with respect to previous ages. Thus, it seems that oxidative damage in the aging hippocampus may result from defective scavenging of superoxide anion, rather than hydrogen peroxide. In 18-month-old Tg hippocampus, GPX1 levels are significantly lower than in the corresponding WT, indicating impaired removal of H2O2, thus exacerbating the physiological age-related alteration of the cellular redox status. Concerning SODs, while SOD1 follows the decreasing trend of the age-matched WT hippocampus, SOD2 is not decreased in 18-month-old Tg, being significantly higher than in its WT counterpart. This finding can be explained by immunohistochemical data, showing particularly high concentration of SOD2 in glial cells surrounding amyloid plaques. Consistently, SOD2 - but not SOD1, CAT, or GPX1 - is induced in astroglia by activated microglia in an in vitro model of neuroinflammation [80].
A pro-oxidant cellular environment in the aged hippocampus is further demonstrated by the presence of oxidative damage markers (acrolein and 8-OH(d)G), particularly concentrated in Tg neurons. However, while in WT hippocampus both markers clearly show age-dependent increase, the same is not true for the Tg, where 8-OH(d)G immunoreactivity appears reduced in 18-month-old hippocampus, compared to the young. This observation is in agreement with data from other Authors indicating a negative correlation between 8-OH(d)G levels and histological amyloid burden [81,82].
Conclusions
Overall, our results confirm and extend our previous observations on the early involvement of peroxisomes in AD pathogenesis [18]. In particular, we strongly suggest that these organelles constitute a first line of defense in support of mitochondria, primary targets of intracellular Aβ/ROS-mediated damage. This protective role specifically involves increased peroxisomal fatty acyl β-oxidation, likely providing mitochondria with acetyl-CoA and shortened acyl-CoAs. This conclusion may shed new light into the physiological significance of peroxisome metabolism in mature neurons, so far mainly related to the synthesis of plasma membrane components and to neurotransmission [7-10,83,84].
In brain aging, when oxidative stress sensu strictu begins, peroxisomes become target of ROS, their functions irreversibly decline, thus contributing themselves to the “pro-oxidant loop” [13]. Deepening knowledge of these processes, which are associated to aging and accelerated/exacerbated in AD, will add significantly to the dissection of pathogenic mechanisms of the disease. At the same time, the early response shown by peroxisomes supports the potential use of some of their proteins as biomarkers, to recognize the first signs of presymptomatic AD and to follow the effectiveness of novel therapeutic approaches, possibly involving peroxisomal induction. Indeed, preventing or treating the disease earlier than in its mild to moderate stages presently appears a major task, as stressed by the latest commentaries on the topic [85,86].
Methods
Animals
Heterozygous female Tg mice [20] and WT littermates were used for all experiments. Male mice (C57B6 × SJL), hemizygous for human AβPP695 carrying the double mutation K670N and M671L (FADSwedish mutation), were purchased from Taconic Farms, Inc. (Germantown, NY, USA). The Tg colony is maintained and the genotyping is performed as previously described [18].
For immunoblotting analyses, Tg and WT female mice aging 3, 6, 9, 12, and 18 months were used. Six animals from each group were killed by cervical dislocation, brains were rapidly excised on an ice-cold plate, and the hippocampus was dissected out before pooling samples (three pools of two hippocampi each). For morphological studies, three Tg mice and three WT littermates for each age considered were deeply anesthetized with urethane (1 g/kg body weight, injected i.p.), before rapid killing by transcardial perfusion with the fixative solution.
Experiments were performed in accordance with the European Community’s Council Directive 86/609/EEC. Formal approval of these experiments was obtained from the Italian Ministry of Health (D.L.vo 116/92; Prot. No. 155-VI-1.1.). All efforts were made to minimize the number of animals used and their suffering.
Molecular analyses
Total homogenate preparation from hippocampal tissue
Protein extraction was performed by homogenizing tissue in lysis buffer (320 mM sucrose, 50 mM NaCl, 50 mM Tris–HCl, pH 7.5, 1% Triton X-100, 0.5 mM sodium orthovanadate, 5 mM β-glycerophosphate, 1% protease inhibitor), incubating on ice for 30 min. Homogenates were then centrifuged at 13,000 g for 10 min. The total protein content of the resulting supernatant was determined using a spectrophotometric assay, according to Bradford [87]. Samples were then diluted 3:4 in 200 mM Tris–HCl, pH 6.8, containing 40% glycerol, 20% β-mercaptoethanol, 4% sodium dodecyl phosphate (SDS), bromophenol blue.
Immunoblotting analyses
Western blotting (WB) experiments were performed as previously described [18]. Membranes were probed at 4°C overnight with the following primary antibodies: 1:10000 rabbit polyclonal anti-Pex14p (generous gift of Prof. R. Erdmann, Ruhr-Universität Bochum, Bochum, Germany); 1:1000 rabbit polyclonal anti-PMP70 (Sigma-Aldrich, St. Louis, MO, USA); 1:200 rabbit polyclonal anti-glial fibrillary acidic protein (anti-GFAP, Sigma-Aldrich); 1:1000 rabbit polyclonal anti-AOX (generous gift of Prof. A. Völkl, University of Heidelberg, Heidelberg, Germany); 1:1000 rabbit polyclonal anti-THL (generous gift of Prof. P. Van Veldhoven, Katholieke Universiteit Leuven, Leuven, Belgium); 1:2000 rabbit polyclonal anti-CAT (Rockland, Gilbertsville, PA, USA); 1:3000 rabbit polyclonal anti-GPX1 (Abnova, Taipei City, Taiwan); 1:1000 rabbit polyclonal anti-SOD1 (Abcam, Cambridge Science Park, Cambridge, UK); 1:10000 anti-SOD2 (Abcam); 1:3000 mouse monoclonal anti-β-actin (Sigma-Aldrich). This was followed by incubation with 1:2000 HRP-conjugated goat anti-rabbit or anti-mouse IgG secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) in blocking solution, for 1 h at 4°C. Immunoreactive bands were visualized by a chemiluminescence detection kit (ECL Plus™ Western Blotting Detection Reagents, Amersham GE Healthcare, Little Chalfont, UK). The relative densities of immunoreactivity were determined by densitometry using the software ImageJ (NIH, Bethesda, MD, USA) and normalized with respect to β-actin. Data are mean of five different experiments.
Morphological analyses
Brain tissue preparation
Mice were perfusion-fixed and brains were paraffin-embedded, as previously described [18]. The left halves from 3-month-old Tg and WT brains were used for immunoelectron microscopy.
Immunohistochemistry
Serial, 5-μm-thick sagittal brain sections from Tg and WT animals were deparaffinized and processed for immunohistochemistry, as previously described [18]. The following primary antibodies were used: 1:500 rabbit polyclonal anti-Pex14p (generous gift of Prof. R. Erdmann); 1:200 rabbit polyclonal anti-PMP70 (Sigma-Aldrich); 1:300 rabbit polyclonal anti-GFAP (Dako, Glostrup, Denmark); 1:100 rabbit polyclonal anti-AOX (generous gift of Prof. A. Volkl, Heidelberg); 1:200 rabbit polyclonal anti-CAT (Rockland); 1:100 rabbit polyclonal anti-SOD1 and 1:1000 anti-SOD2 (Abcam); 1:500 rabbit polyclonal anti-acrolein (generous gift of Prof. K. Uchida, Nagoya University, Nagoya, Japan); 1:200 rabbit polyclonal anti-PPARα (ABR Affinity BioReagents, Golden, CO, USA); 1:50 mouse monoclonal anti-PGC-1α (Calbiochem), and 1:500 mouse monoclonal anti-8OH(d)G (QED Bioscience Inc., San Diego, CA, USA),. In control sections, the primary antibody was omitted. Biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA, USA), or biotinylated goat anti-mouse IgG (Vector) were used as secondary antibodies. Immuno-complexes were visualized, and slides were dehydrated and mounted, as previously reported [18]. Sections were observed under an Olympus BX 51 microscope, equipped with a Leica DFC 420 camera; electronic images were captured by a Leica Application Suite system, and composed in an Adobe Photoshop CS5 format.
Immunofluorescence
For double immunofluorescence experiments, sagittal paraffin sections from 18-month-old Tg and WT brains were deparaffinized and rehydrated, as above. Sections were incubated overnight at 4°C with a mixture of rabbit polyclonal anti-PMP70 (1:100, Sigma-Aldrich) and either mouse monoclonal anti-GFAP (1:200, Chemicon, Temecula, CA, USA), or mouse monoclonal anti-Neuronal Nuclei (anti-NeuN 1:100, Chemicon). Sections were thoroughly rinsed with PBS, then incubated for 1 h at RT with a mixture of 1:100 Alexa488 conjugated goat anti-rabbit IgG (Invitrogen) and 1:500 Alexa555 conjugated goat anti-mouse IgG (Invitrogen). All antibodies were diluted with PBS containing 4% BSA. Triple immunofluorescence experiments were performed on 3-month-old Tg and WT brains, using a mixture of primary antibodies (1:100 rabbit polyclonal anti-PPARα, 1:200 sheep polyclonal anti-GFAP, 1:100 mouse monoclonal anti-NeuN) and of secondary antibodies (1:100 Alexa633 conjugated goat anti-rabbit IgG, 1:500 Alexa488 conjugated goat anti-mouse IgG, and 1:100 Alexa546 conjugated goat anti-sheep IgG). Controls were performed by omitting the primary antibody. Slides were observed in a Leica TCS SP5 confocal microscope and electronic images, captured by a Leica Application Suite system, were composed in an Adobe Photoshop CS5 format.
Immunoelectron microscopy
Samples from 3-month-old Tg and WT brains were sagittally cut by a vibratome, and 100-μm-thick sections, were processed for pre-embedding immunolocalization, according to the previously described procedure [18]. Sections were incubated with either 1:500 anti-PMP70 or with 1:150 anti-PPARα, as primary antibodies, diluted in PBS. After immunocomplexes visualization, slices were osmium-postfixed, dehydrated, and flat-embedded in Epon as previously described [18]. CA1 hippocampal fields of the resin-embedded sections, identified in a light microscope, were remounted on Epon blanks, and further sectioned on a Reichert Ultracut S ultramicrotome. Ultrathin sections were briefly contrasted with uranyl acetate and lead citrate, and observed in a Philips CM120 electron microscope equipped with a Philips Megaview III camera. Electronic images were captured by AnalySys 2.0 software and composed in Adobe Photoshop CS5 format.
Statistical analysis
Statistical evaluation for WB experiments was performed using GraphPad Prism 4 software. For comparison between genotypes and among different ages, statistical analysis of WB densitometric values was accomplished using two-way analysis of variance (ANOVA) followed by the Bonferroni test to detect significant differences between groups. Means from independent experiments were then expressed as means ± SD. For all statistical analyses, P <0.05 was considered as statistically significant.
Abbreviations
8-OH(d)G: 8-hydroxy(deoxy)guanosine;Aβ: Amyloid β;AD: Alzheimer’s disease;AOX: Acyl-CoA oxidase 1;CAT: Catalase;GFAP: Glial fibrillary acidic protein;GPX1: Selenium-dependent glutathione peroxidase;NeuN: Neuronal nuclei;Pex14p: Peroxin 14p;PGC-1α: Peroxisome proliferator-activated receptor γ coativator 1α;PMP70: Peroxisomal membrane protein of 70 kDa;PPAR: Peroxisome proliferator-activated receptor;PVDF: Polyvinylidene fluoride;RT: Room temperature;ROS: Reactive oxygen species;SDS: Sodium dodecyl phosphate;SOD1: CU,Zn-superoxide dismutase;SOD2: Mn-superoxide dismutase;Tg: Tg2576 transgenic mouse model of Alzheimer’s disease;THL: 3-ketoacyl-CoA thiolase;VLCFA: Very long chain fatty acid;WB: Western blotting;WT: Wild-type
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FF made substantial contribution to conception and design of the study, carried out the morphological and molecular experiments and has been involved in drafting the manuscript. SS participated in performing immunoblotting experiments and statistical analyses. MD participated in interpretation of data and revised the manuscript critically. CB has been involved in morphological analysis and interpretation of data. LC participated in performing and interpreting immunofluorescence experiments. AC participated in the design of the study. FC revised the manuscript critically. MPC participated in design and coordination of the study and has been involved in revising the manuscript critically. SM conceived and designed the study, helped to draft the manuscript and gave final approval of the version to be published. All authors read and approved the final manuscript.
Contributor Information
Francesca Fanelli, Email: f.fanelli@unicampus.it.
Sara Sepe, Email: sara.sepe84@gmail.com.
Marcello D’Amelio, Email: m.damelio@hsantalucia.it.
Cinzia Bernardi, Email: cinziabernardi@libero.it.
Loredana Cristiano, Email: loredana.cristiano@univaq.it.
AnnaMaria Cimini, Email: cimini@univaq.it.
Francesco Cecconi, Email: francesco.cecconi@uniroma2.it.
Maria Paola Ceru', Email: mariapaola.ceru@univaq.it.
Sandra Moreno, Email: sandra.moreno@uniroma3.it.
Acknowledgements
We wish to thank Prof. Ralph Erdmann (Ruhr-Universität Bochum, Bochum, Germany), for generously providing us with the anti-Pex14p, Prof. Alfred Volkl (University of Heidelberg, Germany) for anti-acyl CoA oxidase, Prof. Paul Van Veldhoven (Catholic University of Leuven, Belgium) for anti-thiolase, and Prof. Koji Uchida (Nagoya University, Aichi, Japan), for anti-acrolein.
We are indebted to Sara Porcellotti, Pamela Rosso, Federica Giardi and Anna Fracassi for technical and imaging assistance. The technical staff of UOC Pathological Anatomy of San Filippo Neri Hospital is also gratefully acknowledged for skilful assistance.
This work was partially supported by grants to SM from the University “Roma Tre” (“Internationalization and Research Training Project ” and “CAL”). MPC and AC are financially supported by RIA from University of L’Aquila and by FP-6 Integrated European Project LSHG-CT 768-512018. FC is financially supported by the Telethon Foundation (GGP10225), the Italian Ministry of University and Research (FIRB Accordi di Programma 2011) and the Italian Ministry of Health (“Ricerca Finalizzata” and “Ricerca Corrente”). MDA is financially supported by a grant from Alzheimer's Association (NIRG-11-204588).
References
- Bonda DJ, Wang X, Perry G, Nunomura A, Tabaton M, Zhu X, Smith MA. Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology. 2010;59:290–294. doi: 10.1016/j.neuropharm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Teplow DB. Alzheimer’s disease and the amyloid β-protein. Prog Mol Biol Transl Sci. 2012;107:101–124. doi: 10.1016/B978-0-12-385883-2.00012-6. [DOI] [PubMed] [Google Scholar]
- De Duve C, Baudhuin P. Peroxisomes (microbodies and related particles) Physiol Rev. 1966;46:323–357. doi: 10.1152/physrev.1966.46.2.323. [DOI] [PubMed] [Google Scholar]
- Poirier Y, Antonenkov VD, Glumoff T, Hiltunen JK. Peroxisomal beta-oxidation, a metabolic pathway with multiple functions. Biochim Biophys Acta. 2006;1763:1413–1426. doi: 10.1016/j.bbamcr.2006.08.034. [DOI] [PubMed] [Google Scholar]
- Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329. [DOI] [PubMed] [Google Scholar]
- Sprecher H, Luthria DL, Mohammed BS, Baykousheva SP. Reevaluation of the pathways for the biosynthesis of polyunsaturated fatty acids. J Lipid Res. 1995;36:2471–2477. [PubMed] [Google Scholar]
- Wanders RJ, Ferdinandusse S, Brites P, Kemp S. Peroxisomes, lipid metabolism and lipotoxicity. Biochim Biophys Acta. 2010;1801:272–280. doi: 10.1016/j.bbalip.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Drago I, Giacomello M, Pizzo P, Pozzan T. Calcium dynamics in the peroxisomal lumen of living cells. J Biol Chem. 2008;283:14384–14390. doi: 10.1074/jbc.M800600200. [DOI] [PubMed] [Google Scholar]
- Lasorsa FM, Pinton P, Palmieri L, Scarcia P, Rottensteiner H, Rizzuto R, Palmieri F. Peroxisomes as novel players in cell calcium homeostasis. J Biol Chem. 2008;283:15300–15308. doi: 10.1074/jbc.M800648200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko VI, Terlecky SR. Peroxisome metabolism and cellular aging. Traffic. 2011;12:252–259. doi: 10.1111/j.1600-0854.2010.01144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro D, Castro I, Fahimi HD, Schrader M. Peroxisome morphology in pathology. Histol Histopathol. 2012;27:661–676. doi: 10.14670/HH-27.661. [DOI] [PubMed] [Google Scholar]
- Lizard G, Rouaud O, Demarquoy J, Cherkaoui-Malki M, Iuliano L. Potential roles of peroxisomes in Alzheimer’s disease and in dementia of the Alzheimer’s type. J Alzheimers Dis. 2012;29:241–254. doi: 10.3233/JAD-2011-111163. [DOI] [PubMed] [Google Scholar]
- Kou J, Kovacs GG, Höftberger R, Kulik W, Brodde A, Forss-Petter S, Hönigschnabl S, Gleiss A, Brugger B, Wanders R, Just W, Budka H, Jungwirth S, Fischer P, Berger J. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 2011;122:271–283. doi: 10.1007/s00401-011-0836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MJ, Quintanilla RA, Toro A, Grandy R, Dinamarca MC, Godoy JA, Inestrosa NC. Peroxisomal proliferation protects from beta-amyloid neurodegeneration. J Biol Chem. 2005;280:41057–41068. doi: 10.1074/jbc.M505160200. [DOI] [PubMed] [Google Scholar]
- Cimini A, Moreno S, D’Amelio M, Cristiano L, D’Angelo B, Falone S, Benedetti E, Carrara P, Fanelli F, Cecconi F, Amicarelli F, Cerù MP. Early biochemical and morphological modifications in the brain of a transgenic mouse model of Alzheimer’s disease: a role for peroxisomes. J Alzheimer Dis. 2009;18:935–952. doi: 10.3233/JAD-2009-1199. [DOI] [PubMed] [Google Scholar]
- Cimini A, Benedetti E, D’Angelo B, Cristiano L, Falone S, Di Loreto S, Amicarelli F, Cerù MP. Neuronal response of peroxisomal and peroxisome-related proteins to chronic and acute Abeta injury. Curr Alzheimer Res. 2009;6:238–251. doi: 10.2174/156720509788486518. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, Moreno S, Bacci A, Ammassari-Teule M, Marie H, Cecconi F. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat Neurosci. 2011;14:69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- Lee SH, Kim KR, Ryu SY, Son S, Hong HS, Mook-Jung I, Lee SH, Ho WK. Impaired short-term plasticity in mossy fiber synapses caused by mitochondrial dysfunction of dentate granule cells is the earliest synaptic deficit in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32:5953–5963. doi: 10.1523/JNEUROSCI.0465-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochim Biophys Acta. 2006;1763:1755–1766. doi: 10.1016/j.bbamcr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Montagner A, Rando G, Degueurce G, Leuenberger N, Michalik L, Wahli W. New insights into the role of PPARs. Prostaglandins Leukot Essent Fatty Acids. 2011;85:235–243. doi: 10.1016/j.plefa.2011.04.016. [DOI] [PubMed] [Google Scholar]
- Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- Chung JH, Seo AY, Chung SW, Kim MK, Leeuwenburgh C, Yu BP, Chung HY. Molecular mechanism of PPAR in the regulation of age-related inflammation. Ageing Res Rev. 2008;7:126–136. doi: 10.1016/j.arr.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Campolongo P, Roozendaal B, Trezza V, Cuomo V, Astarita G, Fu J, McGaugh JL, Piomelli D. Fat-induced satiety factor oleoylethanolamide enhances memory consolidation. PNAS. 2009;106:8027–8031. doi: 10.1073/pnas.0903038106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delille HK, Alves R, Schrader M. Biogenesis of peroxisomes and mitochondria: linked by division. Histochem Cell Biol. 2009;131:441–446. doi: 10.1007/s00418-009-0561-9. [DOI] [PubMed] [Google Scholar]
- Bagattin A, Hugendubler L, Mueller E. Transcriptional coactivator PGC-1alpha promotes peroxisomal remodeling and biogenesis. Proc Natl Acad Sci USA. 2010;107:20376–20381. doi: 10.1073/pnas.1009176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh R, Fujiki Y. Functional domains and dynamic assembly of the Peroxin Pex14p, the entry site of matrix proteins. J Biol Chem. 2006;281:10196–10205. doi: 10.1074/jbc.M600158200. [DOI] [PubMed] [Google Scholar]
- Meinecke M, Cizmowski C, Schliebs W, Kruger V, Beck S, Wagner R, Erdmann R. The peroxisomal importomer constitutes a large and highly dynamic pore. Nat Cell Biol. 2010;12:273–277. doi: 10.1038/ncb2027. [DOI] [PubMed] [Google Scholar]
- van der Zand A, Gent J, Braakman I, Tabak HF. Biochemically distinct vesicles from the endoplasmic reticulum fuse to form peroxisomes. Cell. 2012;149:397–409. doi: 10.1016/j.cell.2012.01.054. [DOI] [PubMed] [Google Scholar]
- Ahlemeyer B, Neubert I, Kovacs WJ, Baumgart-Vogt E. Differential expression of peroxisomal matrix and membrane proteins during postnatal development of mouse brain. J Comp Neurol. 2007;505:1–17. doi: 10.1002/cne.21448. [DOI] [PubMed] [Google Scholar]
- Santos MJ, Kawada ME, Espeel M, Figueroa C, Alvarez A, Hidalgo U, Metz C. Characterization of human peroxisomal membrane proteins. J Biol Chem. 1994;269:24890–24896. [PubMed] [Google Scholar]
- Morita M, Imanaka T. Peroxisomal ABC transporters: Structure, function and role in disease. Biochim Biophys Acta. 2012;1822:1387–1396. doi: 10.1016/j.bbadis.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Moreno S, Mugnaini E, Cerù MP. Immunocytochemical localization of catalase in the central nervous system of the rat. J Histochem Cytochem. 1995;43:1253–1267. doi: 10.1177/43.12.8537642. [DOI] [PubMed] [Google Scholar]
- Moreno S, Nardacci R, Cerù MP. Regional and ultrastructural immunolocalization of copper-zinc superoxide dismutase in rat central nervous system. J Histochem Cytochem. 1997;45:1611–1622. doi: 10.1177/002215549704501204. [DOI] [PubMed] [Google Scholar]
- Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, Pasinetti GM. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009;66:352–361. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata-Fukae C, Li HF, Hoe HS, Gray AJ, Minami SS, Hamada K, Niikura T, Hua F, Tsukagoshi-Nagai H, Horikoshi-Sakuraba Y, Mughal M, Rebeck GW, LaFerla FM, Mattson MP, Iwata N, Saido TC, Klein WL, Duff KE, Aisen PS, Matsuoka Y. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 2008;1216:92–103. doi: 10.1016/j.brainres.2008.03.079. [DOI] [PubMed] [Google Scholar]
- Craig MC, Murphy DG. Estrogen therapy and Alzheimer’s dementia. Ann N Y Acad Sci. 2010;1205:245–253. doi: 10.1111/j.1749-6632.2010.05673.x. [DOI] [PubMed] [Google Scholar]
- Small SA, Schobel SA, Buxton RB, Witter MP, Barnes CA. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat Rev Neurosci. 2011;12:585–601. doi: 10.1038/nrn3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti P, Schliebs W, Schievelbusch T, Neuhaus A, David C, Kock K, Herrmann C, Meyer HE, Wiese S, Warscheid B, Theiss C, Erdmann R. PEX14 is required for microtubule-based peroxisome motility in human cells. J Cell Sci. 2011;124:1759–1768. doi: 10.1242/jcs.079368. [DOI] [PubMed] [Google Scholar]
- Pagani L, Eckert A. Amyloid-Beta interaction with mitochondria. Int J Alzheimers Dis. 2011;2011:1–12. doi: 10.4061/2011/925050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I. Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease. J Bioenerg Biomembr. 2009;41:425–431. doi: 10.1007/s10863-009-9243-5. [DOI] [PubMed] [Google Scholar]
- Gillardon F, Rist W, Kussmaul L, Vogel J, Berg M, Danzer K, Kraut N, Hengerer B. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics. 2007;7:605–616. doi: 10.1002/pmic.200600728. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Shi Q. A mitocentric view of Alzheimer’s disease suggests multi-faceted treatments. J Alzheimers Dis. 2010;2:S591–S607. doi: 10.3233/JAD-2010-100336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson RM, Kusne Y, Nowak LA, LaFerla FM, Reiman EM, Valla J. Regional cerebral glucose uptake in the 3xTG model of Alzheimer’s disease highlights common regional vulnerability across AD mouse models. Brain Res. 2010;1347:179–185. doi: 10.1016/j.brainres.2010.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:180–195. doi: 10.1196/annals.1427.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Rettberg JR, Klosinski LP, Cadenas E, Brinton RD. Shift in brain metabolism in late onset Alzheimer’s disease: implications for biomarkers and therapeutic interventions. Mol Aspects Med. 2011;32:247–257. doi: 10.1016/j.mam.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baarine M, Andréoletti P, Athias A, Nury T, Zarrouk A, Ragot K, Vejux A, Riedinger JM, Kattan Z, Bessede G, Trompier D, Savary S, Cherkaoui-Malki M, Lizard G. Evidence of oxidative stress in very long chain fatty acid - treated oligodendrocytes and potentialization of ROS production using RNA interference-directed knockdown of ABCD1 and ACOX1 peroxisomal proteins. Neuroscience. 2012;213:1–18. doi: 10.1016/j.neuroscience.2012.03.058. [DOI] [PubMed] [Google Scholar]
- Zarrouk A, Vejux A, Nury T, El Hajj HI, Haddad M, Cherkaoui-Malki M, Riedinger JM, Hammami M, Lizard G. Induction of mitochondrial changes associated with oxidative stress on very long chain fatty acids (C22:0, C24:0, or C26:0)-treated human neuronal cells (SK-NB-E) Oxid Med Cell Longev. 2012. [DOI] [PMC free article] [PubMed]
- Kane CD, Francone OL, Stevens KA. Differential regulation of the cynomolgus, human, and rat acyl-CoA oxidase promoters by PPARalpha. Gene. 2006;380:84–94. doi: 10.1016/j.gene.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Gray E, Ginty M, Kemp K, Scolding N, Wilkins A. Peroxisome proliferator-activated receptor-α agonists protect cortical neurons from inflammatory mediators and improve peroxisomal function. Eur J Neurosci. 2011;33:1421–1432. doi: 10.1111/j.1460-9568.2011.07637.x. [DOI] [PubMed] [Google Scholar]
- Anderson EJ, Yamazaki H, Neufer PD. Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J Biol Chem. 2007;282:31257–31266. doi: 10.1074/jbc.M706129200. [DOI] [PubMed] [Google Scholar]
- Reddy PH, McWeeney S, Park BS, Manczak M, Gutala RV, Partovi D, Jung Y, Yau V, Searles R, Mori M, Quinn J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet. 2004;13:1225–1240. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- Crack PJ, Cimdins K, Ali U, Hertzog PJ, Iannello RC. Lack of glutathione peroxidase-1 exacerbates αβ-mediated neurotoxicity in cortical neurons. J Neural Transm. 2006;113:645–657. doi: 10.1007/s00702-005-0352-y. [DOI] [PubMed] [Google Scholar]
- Barkats M, Millecamps S, Abrioux P, Geoffroy MC, Mallet J. Overexpression of glutathione peroxidase increases the resistance of neuronal cells to Abeta-mediated neurotoxicity. J Neurochem. 2000;75:1438–1446. doi: 10.1046/j.1471-4159.2000.0751438.x. [DOI] [PubMed] [Google Scholar]
- Lubos E, Loscalzo J, Handy DE. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2011;15:1957–1997. doi: 10.1089/ars.2010.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara N, Nakano M, Kato S, Yoshihara D, Ookawara T, Eguchi H, Taniguchi N, Suzuki K. Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J Biol Chem. 2007;282:35933–35944. doi: 10.1074/jbc.M702941200. [DOI] [PubMed] [Google Scholar]
- Murakami K, Murata N, Noda Y, Tahara S, Kaneko T, Kinoshita N, Hatsuta H, Murayama S, Barnham KJ, Irie K, Shirasawa T, Shimizu T. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease. J Biol Chem. 2011;286:44557–44568. doi: 10.1074/jbc.M111.279208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Tamaoki T, Motohashi N, Nakamura M, McKeel DW Jr, Tabaton M, Lee HG, Smith MA, Perry G, Zhu X. The earliest stage of cognitive impairment in transition from normal aging to Alzheimer disease is marked by prominent RNA oxidation in vulnerable neurons. J Neuropathol Exp Neurol. 2012;71:233–241. doi: 10.1097/NEN.0b013e318248e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Nam DT, Arseneault M, Ramassamy C. Role of by-products of lipid oxidation in Alzheimer’s disease brain: a focus on acrolein. J Alzheimers Dis. 2010;21:741–756. doi: 10.3233/JAD-2010-100405. [DOI] [PubMed] [Google Scholar]
- Dang TN, Arseneault M, Murthy V, Ramassamy C. Potential role of acrolein in neurodegeneration and in Alzheimer’s disease. Curr Mol Pharmacol. 2010;3:66–78. [PubMed] [Google Scholar]
- Shi R, Zhang Y, Shi Y, Shi S, Jiang L. Inhibition of peroxisomal β-oxidation by thioridazine increases the amount of VLCFAs and Aβ generation in the rat brain. Neurosci Lett. 2012;528:6–10. doi: 10.1016/j.neulet.2012.08.086. [DOI] [PubMed] [Google Scholar]
- Clark J, Simon DK. Transcribe to survive: transcriptional control of antioxidant defense programs for neuroprotection in Parkinson’s disease. Antioxid Redox Signal. 2009;11:509–528. doi: 10.1089/ars.2008.2241. [DOI] [PubMed] [Google Scholar]
- Wenz T. Mitochondria and PGC-1α in aging and Age-associated diseases. J Aging Res. 2011;2011:1–12. doi: 10.4061/2011/810619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Rodriguez C, Spaulding J, Aw TY, Feng J. Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer’s disease. J Alzheimers Dis. 2012;28:655–666. doi: 10.3233/JAD-2011-111244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado A, Encarnación López-Fernández M, Concepción Casado M, de La Torre R. Lipid peroxidation and antioxidant enzyme activities in vascular and Alzheimer dementias. Neurochem Res. 2008;33:450–458. doi: 10.1007/s11064-007-9453-3. [DOI] [PubMed] [Google Scholar]
- Massaad CA, Washington TM, Pautler RG, Klann E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2009;106:13576–13581. doi: 10.1073/pnas.0902714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amenta F, Bronzetti E, Sabbatini M, Vega JA. Astrocyte changes in aging cerebral cortex and hippocampus: a quantitative immunohistochemical study. Microsc Res Tech. 1998;43:29–33. doi: 10.1002/(SICI)1097-0029(19981001)43:1<29::AID-JEMT5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9:156–167. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Zhao R, Gao K, Wei Z, Yin MY, Lau LT, Chui D, Hoi Yu AC. Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2011;8:67–80. doi: 10.2174/156720511794604543. [DOI] [PubMed] [Google Scholar]
- Arnold G, Holtzman E. Microperoxisomes in the central nervous system of the postnatal rat. Brain Res. 1978;155:1–17. doi: 10.1016/0006-8993(78)90300-1. [DOI] [PubMed] [Google Scholar]
- Cimini A, Moreno S, Giorgi M, Serafini B, Cerú MP. Purification of peroxisomal fraction from rat brain. Neurochem Int. 1993;23:249–260. doi: 10.1016/0197-0186(93)90116-M. [DOI] [PubMed] [Google Scholar]
- Cimini A, Singh I, Farioli-Vecchioli S, Cristiano L, Cerú MP. Presence of heterogeneous peroxisomal populations in the rat nervous tissue. Biochim Biophys Acta. 1998;1425:13–26. doi: 10.1016/S0304-4165(98)00049-X. [DOI] [PubMed] [Google Scholar]
- Gemma C, Vila J, Bachstetter A, Bickford PC. In: Brain aging: models, methods, and mechanisms. Riddle DR, editor. Boca Raton (FL): CRC Press; 2007. Oxidative stress and the aging brain: from theory to prevention; p. Chapter 15. Frontiers in Neuroscience. [PubMed] [Google Scholar]
- Filomeni G, Bolaños JP, Mastroberardino PG. Redox status and bioenergetics liaison in cancer and neurodegeneration. Int J Cell Bio. 2012;2012:659645. doi: 10.1155/2012/659645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röhl C, Armbrust E, Kolbe K, Lucius R, Maser E, Venz S, Gülden M. Activated microglia modulate astroglial enzymes involved in oxidative and inflammatory stress and increase the resistance of astrocytes to oxidative stress in vitro. Glia. 2008;56:1114–1126. doi: 10.1002/glia.20683. [DOI] [PubMed] [Google Scholar]
- Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, Huang X, Farrag YW, Perry G, Bush AI. Evidence that the beta-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of abeta by zinc. J Biol Chem. 2000;275:19439–19442. doi: 10.1074/jbc.C000165200. [DOI] [PubMed] [Google Scholar]
- Weidner AM, Bradley MA, Beckett TL, Niedowicz DM, Dowling AL, Matveev SV, LeVine H 3rd, Lovell MA, Murphy MP. RNA oxidation adducts 8-OHG and 8-OHA change with Aβ42 levels in late-stage Alzheimer’s disease. PLoS One. 2011;6:1–6. doi: 10.1371/journal.pone.0024930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S, Nardacci R, Cimini A, Cerù MP. Immunocytochemical localization of D-amino acid oxidase in rat brain. J Neurocytol. 1999;28:169–185. doi: 10.1023/A:1007064504007. [DOI] [PubMed] [Google Scholar]
- Farioli-Vecchioli S, Moreno S, Cerù MP. Immunocytochemical localization of acyl-CoA oxidase in the rat central nervous system. J Neurocytol. 2001;30:21–33. doi: 10.1023/A:1011913223541. [DOI] [PubMed] [Google Scholar]
- Callaway E. Alzheimer’s drugs take a new tack. Nature. 2012;489:13–14. doi: 10.1038/489013a. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Preventing Alzheimer’s disease. Science. 2012;337:1488–1492. doi: 10.1126/science.1228541. [DOI] [PubMed] [Google Scholar]
- Bradford MM. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]