

1. Introduction
In 1996, Leesong et al. published the structure of FabA (PDB ID:1MKA), the dehydratase–isomerase enzyme involved in the bacterial type II fatty acid synthase (FAS) system from E. coli.1 The authors described a central, long α-helix “hot dog” surrounded by a bent β-sheet “bun” and so dubbed this the “hot dog fold” (Figure 1). Since that time, roughly 60 proteins have been crystallized and found to have the hot dog fold.2 The core fold topology can be described as an antiparallel β-sheet ordered 1-3-4-5-2. The long hot dog helix is located between strands β1 and β2 (Figure 2D). Some enzymes contain additional β-strands in their “bun”.
Figure 1. The Hot Dog Fold.
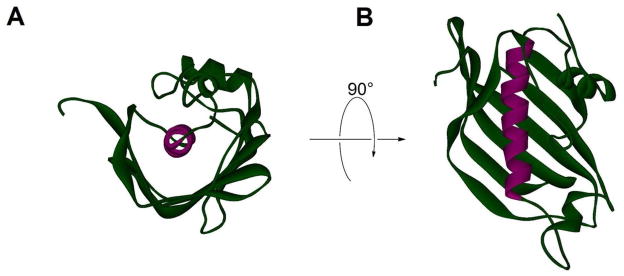
(A) E. coli dehydratase–isomerase (Fab A, PDB#1MKA) in green with the hot dog helix (αHD) viewed head-on and colored purple. A “bun” of β-sheet wraps around αHD. (B) FabA rotated around the x axis 90° to look down on the length of the “hot dog”.
Figure 2. Three Types of Hot Dog Folds.

(A) The FabA dimer as an example of a single hot dog (SHD) enzyme. FabA contains two active sites that reside at the dimer interface (red circles). (B) C. tropicalis MFE-2 hydratase 2 domain (PDB#1PN4), an example of a double hot dog (DHD) fold, at the same scale and orientation as in A. One of the hot dog helices (shown in blue) is distorted. DHD enzymes and domains only have a single active site, and it resides at the pseudodimer interface (red circle). (C) T. lanuginosus DH domain (PDB#2UVA), a fungal type I domain, as an example of a triple hot dog (THD) fold. The left image is at the same scale and orientation as A and B; the right image has been rotated 90° to show how the middle hot dog motif is “behind” the other two. The hot dog helices are colored in orange, and the active site is indicated with a red circle. (D), (E), and (F) Topology diagrams for the core SHD, DHD, and THD folds, respectively, utilizing the naming convention introduced in the text. (Topology diagrams generated with TopDraw: Bond, C.S. Bioinformatics 2003, 19, 311.)
This enzyme fold serves as a scaffold to execute a variety of reactions involving fatty acid or polyketide thioesters and is found in enzymes of both fatty acid catabolism and anabolism as well as in enzymes of natural product biosynthetic pathways. Every enzyme in this family containing a single hot dog (SHD) fold dimerizes (Figure 2A and D), and two identical active sites are found at the dimeric interface, with key residues in each active site residing on both monomers. Depending on the enzyme and species, these homodimers (protomers) themselves often associate into dimers of dimers or trimers of dimers. (For an excellent review of the various oligomeric states of hot dog enzymes, see Pidugu et al.2) Some hot dog enzymes have two hot dog motifs — thought to arise from gene duplication — and these double hot dog (DHD) enzymes (or domains) have two SHD subdomains that are fused together. In DHD structures, one of the active sites, which both reside at the pseudodimeric interface, has become inactive (Figure 2B and E). The hot dog helix of the C-terminal domain is often kinked; the one in the N-terminal domain remains linear. The oligomeric states of these pseudodimeric enzymes also vary. There are even instances of triple hot dog (THD) enzymes; fungal FAS dehydratase domains, for example, contain such a fold.3,4 In a THD enzyme, the third hot dog region is a domain insertion between the first and second hot dog motifs, which associate with each other in similar manner to the two motifs of a DHD enzyme, including the bent, C-terminal helix. The added domain is highly distorted, such that the hot dog helix is much shorter. It no longer locates within the β-sheet bun and is instead found alongside the β-sheet (Figure 2C and F). THD enzymes also only have a single active site at the pseudodimeric interface of the “normal” hot dog regions. The significance of only one hot dog helix remaining linear in both DHD and THD folds will be discussed later in this review.
The secondary structural arrangements of idealized SHD, DHD, and THD folds are compared in the topology diagrams depicted in Figure 2D, E, and F. Because different enzymes have extra helices and strands in addition to the core hot dog and bun shown in the topology diagrams, for the purposes of comparing hot dog folds, we have formulated the following system: Core β-strands are labeled with an Arabic numeral in the same order as they occur in the sequence. Strands found in the second subdomain of DHD folds are treated as if they reside on an SHD with the further designation of a prime symbol. The strands of the inserted third hot dog motif of the THDs are designated with a double prime. Any extra strands in addition to these may be designated with an “x” followed by an Arabic numeral. The hot dog helices of the first, second, or third motifs are labeled αHD, αHD′, αHD″, respectively; additional helices are named by “x” followed by a capital letter. This review will use this notation throughout.
The hot dog superfamily includes over a thousand enzymes or domains and has been divided into at least 17 subfamilies based on primary sequence analysis — although this analysis did not capture the majority of DHD and THD enzymes, including animal and fungal FAS domains.5 Hot dog folds are found or predicted in archaea, bacteria, and eukaryotes. Functionally, however, the majority of the superfamily can be primarily divided into two groups: those enzymes performing either dehydration or hydration of thioester substrates and those performing thioester hydrolysis.
2. Dehydratase/Hydratase Group
2.1. Dehydratases: Reaction Overview and History
Dehydratase (DH) enzymes or enzyme domains (occasionally termed “dehydrases”) are crucial to the biosynthesis of fatty acids and polyketide-derived natural products. In fatty acid biosynthesis, a β-ketothioester intermediate is first reduced to an alcohol by a β-ketothioester reductase (KR) to provide a β-hydroxythioester with a common absolute (R) stereochemistry. The DH then formally removes water from the extended chain to give an α, β-unsaturated thioester (Scheme 1), which is further reduced by an enoyl reductase (ER) to a saturated fatty acyl thioester.6
Scheme 1.
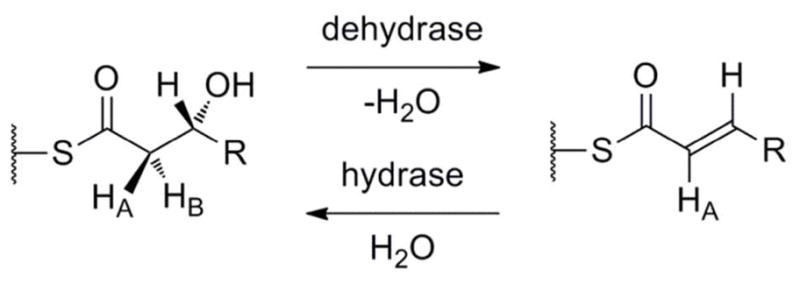
The Reactions Catalyzed by Dehydratases and Hydratases.
It has been known since the late 1960s from isotopic labeling studies that removal of the α-hydrogen in FAS DH reactions is the rate-limiting step for this enzyme. This α-hydrogen is also lost by proton exchange with the solvent.7 Further labeling studies showed that the pro-2S hydrogen (HB in Scheme 1) is the one removed, making the overall reaction a syn elimination of the (3R) hydroxyl group,8,9 which implies — but does not prove — a one-base mechanism.10
The situation with polyketide synthase (PKS) DHs is more complicated. PKSs are related to FASs and share many of the same catalytic steps; however, variations in oxidation states and alkylations lead to a wide range of natural products.11 For example, many PKS systems bear DHs that dehydrate methylated carbon chains, derived from propionyl-CoA starter units, methylmalonyl-CoA extender units, or the action of methyl transferases (MeTs), which can methylate even-numbered carbons.12 Furthermore, the end products of some PKSs contain unsaturated main carbon chains in the cis configuration, instead of trans.13 It is known that, unlike in the case of FAS systems where KRs produce 3R intermediates, not all PKS KRs give hydroxy intermediates of the same relative stereochemistry,14 and it has been hypothesized that DHs acting on “alternative” 3S intermediates give eliminations in which the main carbon chain becomes cis instead of trans.15 (In other cases, exogenous enzymes are indicted.16)
In regards to alkylated DH products, bioinformatic analyses alongside experimental studies performed on the modular PKS system picromycin synthase (PICS) had suggested that, while these modular PKS KRs can give either L- or D-β-hydroxythioacyl branched methyl intermediates — corresponding to non-methylated (S)- and (R)-β-hydroxy fatty acyl intermediates, respectively (Figure 3) — PICS DHs seem to only work with DD-β-hydroxy-α-methylacyl substrates, leading to trans double bonds.17 (Note that for methylated PKS intermediates, it is generally more useful to use the relative stereochemistry indicators L and D than absolute indicators (S) and (R), because the presence or absence of methyl groups on intermediates can change the Cahn–Ingold–Prelog priority designation of absolute stereochemistry between stereochemically related compounds.) It may be that, when such substrates are L-α-methylated, the DHs cannot catalyze elimination, presumably because these substrates have no “pro-L” hydrogen to remove (Figure 3), as others have speculated.18
Figure 3. Fischer Projections of Potential DH Substrates.
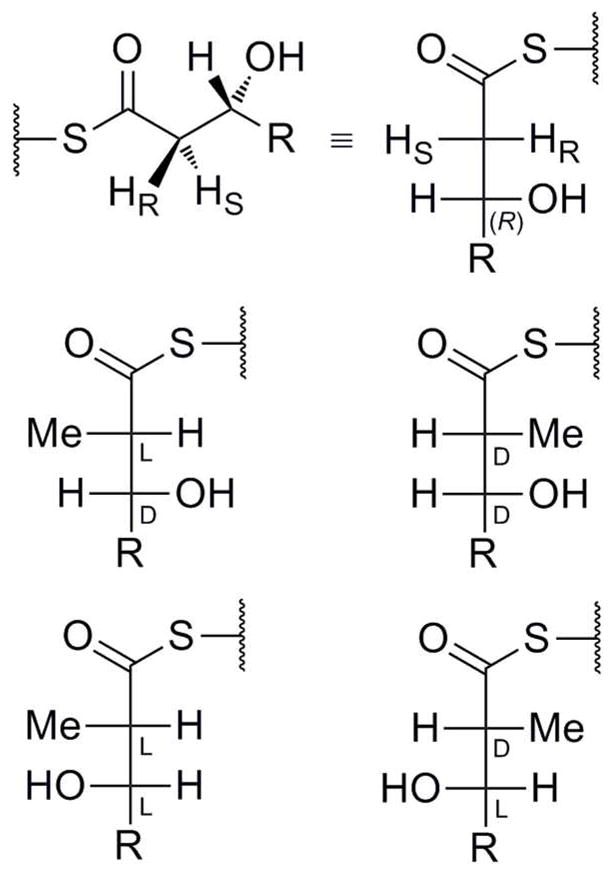
Only D- -METHYLATED compounds (right column) are thought to be substrates for hot dog fold DHs. Of these, L- -HYDROXYLATED compounds (bottom right) are postulated to lead to cis products.
Others have continued to argue that cis double bonds are, in fact, the result of some PKS DHs. These cis bonds, however, likely derive from L-β-hydroxy substrates.19
2.2. Hydratases: Reaction Overview and History
The (R)-specific hydratases (or, “hydrases”) perform the opposite reaction from DHs — β-addition, attack by hydroxide ion at the β position of a trans-α,β-unsaturated thioester (Scheme 1). Different biological systems make use of different stereochemistries of hydration. Ordinary length fatty acids are catabolized with (S)-specific hydratases (crotonases), whereas polyhydroxyalkanoate (PHA) biosynthesis and peroxisomal β-oxidation pathways of very long chain fatty acids (VLCFAs) utilize (R)-specific hydratases.20
Both (S) and (R) addition systems react on trans double bonds to give β-hydroxythioesters by syn addition. (S)-Hydratases are better studied than (R)-hydratases; in the former case, the substrate π-electrons are known to be highly polarized towards the carbonyl oxygen and away from the electrophilic β-carbon. This polarization results from electronic fields within the protein and does not result from substrate distortion upon binding in the active site.21 A similar polarization could also play a role in (R)-hydratases. As with dehydratases, hydratases are thought to require only one catalytic residue and are argued to use concerted mechanisms.22
2.3. DH/H Active Sites
In prokaryotic organisms, DHs and hydratases tend to be free enzymes (Type II) or components of giant modular synthases; in eukaryotic systems, they are domains within large iterative synthases.
In Table 1 are compiled, to the best of our knowledge, all non-hypothetical enzymes in the dehydratase/hydratase (DH/H) group for which there is structural information currently available. Structures were superimposed by particular active site residues (not by sequence alignment) using the Discovery Studio Suite. Because of the low sequence similarity among the enzymes, this method results in a more meaningful comparison, as severe structural differences far from the active site no longer play an affect in the alignment. These superimpositions were used to generate the figures in this section and Table 2. The residues chosen to superimpose were the critical histidine (described below) of each protein (column 3 of Table 2) and four or five residues beginning with the residue backbone hydrogen-bonded to that histidine (column 4 of Table 2). Table 2 compares the key active site residues for those enzymes.
Table 1.
Dehydratases and Hydratases of the Hot Dog Superfamily with Published Structural Data.a
| Enzyme | Species | Pathway | Oligomeric Stateb | Substrate(s) | Pub. Year(s) | PDB #sc | Active Site Cocrystalsd |
|---|---|---|---|---|---|---|---|
| Arrangement A DHs | |||||||
| FabA | E. coli | FAS | D | (R)-3-HODec-ACP | 1996 | 1MKA | none |
| 1MKB | 3-decynoyl-SNAC | ||||||
| PaFabZ | P. aeruginosa | FAS | H1 | (R)-3-HO-acyl-ACPs | 2004 | 1U1Z | none |
| PfFabZ | P. falciparum | FAS | H1 | (R)-3-HO-acyl-ACPs | 2005 | 1Z6B | none |
| 2006 | 1ZHG | none | |||||
| 2007 | 2OKH | none | |||||
| 2OKI | none | ||||||
| 2011 | 3AZ8 | NAS21 | |||||
| 3AZ9 | NAS91 | ||||||
| 3AZA | NAS91-10 | ||||||
| 3AZB | NAS91-11 | ||||||
| HpFabZ | H. pylori | FAS | H1 | (R)-3-HO-acyl-ACPs | 2008 | 2GLL | chloride |
| 2GLM | none | ||||||
| 2GLP | none | ||||||
| 2GLV* | none | ||||||
| 2008 | 3B7J | none | |||||
| 2008 | 3CF8 | none | |||||
| 3CF9 | none | ||||||
| 3D04 | none | ||||||
| 2009 | 3ED0 | none | |||||
| 2009 | 3DOY | none | |||||
| 3DOZ | none | ||||||
| 3DP0 | none | ||||||
| 3DP1 | none | ||||||
| 3DP2 | none | ||||||
| 3DP3 | none | ||||||
| CjFabZ | C. jejunti | FAS | H1 | (R)-3-HO-acyl-ACPs | 2009 | 3D6X | none |
| DEBS DH | S. erythraea | Modular PKS | DdhC | 2008 | 3EL6 | none | |
| CurF DH | L. majuscula | Modular PKS | DdhC | 2010 | 3KG6 | none | |
| CurH DH | L. majuscula | Modular PKS | DdhC | 2010 | 3KG7 | none | |
| CurJ DH | L. majuscula | Modular PKS | DdhC | 2010 | 3KG8 | none | |
| CurK DH | L. majuscula | Modular PKS | DdhC | 2010 | 3KG9 | none | |
| FAS DH | S. scrofa | Animal FAS | DdhD | (R)-3-HO-acyl-ACPs | 2008 | 2VZ8 | none |
| 2VZ9 | none | ||||||
|
| |||||||
| Arrangement J DHs & Hydratases | |||||||
| FAS DH | T. lanuginosus | Fungal FAS | Mth | (R)-3-HO-acyl-ACPs | 2007 | 2UVA | none |
| FAS DH | S. cerevisiae | Fungal FAS | Mth | (R)-3-HO-acyl-ACPs | 2007 | 2UV8 | none |
| 2007 | 2PFF | none | |||||
| PhaJ | A. caviae | PHA Biosynthesis | D | 2003 | 1IQ6 | none | |
| MFE-2 Hydratase 2 | C. tropicalis | β-Oxidation | DdhA | 2004 | 1PN2 | none | |
| 1PN4* | (R)-3-HO-Dec-CoA | ||||||
| MFE-2 Hydratase 2 | H. sapiens | β-Oxidation | DdhA | 2005 | 1S9C | none | |
| MFE-2 Hydratase 2 | D. melanogaster | β-Oxidation | DdhA | 2011 | 3OML | none | |
Abbreviations used: FAS, fatty acid synthesis; PKS, polyketide synthesis; PHA, polyhydroxyalkanoate; Dec, decanoyl; CoA, coenzyme A; ACP, acyl carrier protein.
Terms defined as in Pidugu, et al.: D, dimer; H1, trimer of dimers; DdhA, DHD dimer corresponding to a face-to-face dimer of dimers (TA). Terms introduced in this review: DdhC & DdhD, DHD side-by-side dimers; Mth, THD monomer.
An asterisk indicates a mutant.
Buffers and/or salt ions are not necessarily noted nor are inhibitors not bound within the active site. The NAS compounds are a series of competitive inhibitors designed for PfFabZ.
Table 2.
Structural Alignment and Proposed Function of the Active Site Residues of the Dehydratases/Hydratases in Table 1.a
| Enzyme | H-Bond to W3 | H-Bond to W3 | Catalytic Residue | BB H-Bond to Catalytic Residue | Oxyanion Holeb | H-Bond to W2 | H-Bond to W3 | H-Bond to W2c | H-Bond to Asp/Glu n-4 |
|---|---|---|---|---|---|---|---|---|---|
| Arrangement A DHs | |||||||||
| FabA | — | — | His70A (cis) | Val76A | Gly79A | Cys80A | — | Asp84B | Gln88B |
| PaFabZ | — | — | His49A (cis) | Ile55A | Gly58A | Val59A | His13B | Glu63B | Gln67B |
| PfFabZ | — | — | His133A (cis) | Ile139A | Gly142A | Val143A | His98B | Glu147B | Gln151B |
| HpFabZ | — | — | His58A (cis) | Ile64A | Gly67A | Val68A | His23B | Glu72B | Gln76B |
| CjFabZ | — | — | His48A (cis) | Ile54A | Gly57A | Val58A | His13B | Glu62B | Gln66B |
| DEBS DH | — | — | His2409 (trans) | Leu2416 | Gly2419 | Ser2420 | Tyr2533 | Asp2571 | Gln2575 |
| CurF DH | — | — | His1720 (trans) | Leu1727 | Ser1730 | Thr1731 | Tyr1851 | Asp1893 | Gln1897 |
| CurH DH | — | — | His971 (trans) | Val978 | Val982 | Ser983 | Tyr1094 | Asp1136 | Arg1140 |
| CurJ DH | — | — | His978 (trans) | Val985 | Gly988 | Ala989 | (Leu1115) | Asp1156 | Gln1160 |
| CurK DH | — | — | His996 (trans) | Leu1003 | Ala1006 | Thr1007 | Tyr1127 | Asp1169 | Gln1173 |
| FAS DH | — | — | His878 (trans) | Leu885 | Gly888 | Thr889 | Tyr1003 | Asp1033 | His1037 |
|
| |||||||||
| Arrangement J DHs & Hydratases | |||||||||
| TlFAS DH | Asp1586 | Asn1588 | His1591 (trans) | Ile1606 | Gly1609 | Met1610 | — | H2O* | — |
| ScFAS DH | Asp1559 | Asn1561 | His1564 (trans) | Ile1579 | Gly1582 | Met1583 | — | H2O* | — |
| PhaJ | Asp31A | Asn33A | His36A (trans) | Ile51A | Gly54A | Met55A | — | Ser62B | — |
| CtMFE-2 Hydratase 2 | Asp808 | Asn810 | His813 (trans) | Ile828 | Gly831 | Met832 | — | H2O | — |
| HsMFE-2 Hydratase 2 | Asp510 | Asn512 | His515 (trans) | Ile530 | Gly533 | Leu534 | — | H2O* | — |
| DmMFE-2 Hydratase 2 | Asp496 | Asn498 | His501 (trans) | Ile516 | Gly519 | Leu520 | — | H2O | — |
A or B after residue numbers indicates subunit for SHD enzymes/domains. Bold font indicates fully conserved residues; italic font indicates similar residues. W2 and W3 are water molecules referred to in the text and shown in Figures 3, 4, and 6. Parentheses indicate that a residue is located at the same location as other residues in the same column but not sharing the same function. BB, backbone.
Positive end of helix αHD.
An asterisk indicates that a water was not present in the crystal structures but is assumed to be involved based on structural superimposition with PhaJ and the other MFE-2 hydratase 2s.
2.3.1. Conserved Residues and Crystallographic Water Molecules
All known enzymes in this group contain a fully conserved catalytic histidine residue (Table 2) located before the start of the central “hot dog” helix (αHD). In the bacterial DH enzymes,1,23–33 which are all SHD enzymes, this residue has a rare cis peptide bond (Figure 4A and B); whereas, both the eukaryotic DHs3,4,34–36 and the hydratases20,37–39 contain the common trans bond (Figure 4C and Figure 5). This histidine is polarized by hydrogen bonding to the backbone carbonyl oxygen of a residue located downstream (taken in this review to mean in the C-terminal direction; e.g., FabA Val76A, Figure 4A). Owing to this polarization by a hydrogen-bond acceptor, the histidine is deprotonated at Nε, and in fact, it has been shown to remain deprotonated even at pH 5.40 For DH enzymes and domains, this histidine is theorized to deprotonate the substrate α-carbon, initiating the net elimination of water to give a double bond.1 π-Stacking with nearby aromatic rings is almost always observed for this histidine, but the residues involved vary (e.g., Phe71A in FabA vs. His53A in PhAJ).
Figure 4. Dehydratase Active Site Arrangement A.
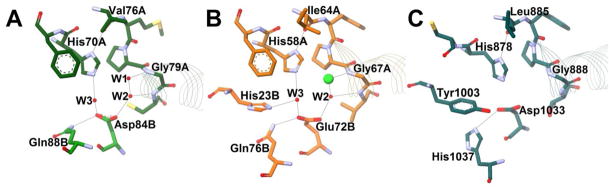
Potential hydrogen bonds are shown with grey lines, red spheres represent resolved water molecules, and residues are shown as sticks and colored by element. The beginnings of the hot dog helices are shown with faint line ribbons. The images are drawn to scale and superimposable. (A) The active site of E. coli FabA (PDB#1MKB), an example of an SHD DH, in green. (B) The active site of H. pylori FabZ (PDB#2GLL), another SHD DH, in orange. The green sphere represents a chloride ion. (C) The active site of S. scrofa (porcine) FAS DH domain (PDB#2VZ8), a DHD DH, in dark cyan. The crystal structure for this enzyme did not include resolved water molecules.
Figure 5. Dehydratase/Hydratase Active Site Arrangement J.
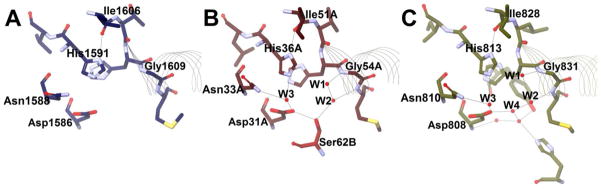
Potential hydrogen bonds are shown with grey lines, red spheres represent water molecules, and residues are shown as sticks and colored by element. The beginnings of the hot dog helices are shown with faint line ribbons. The images are drawn to scale and superimposable. (A) The active site of T. lanuginosus FAS DH domain (PDB#2UVA), an example of a THD DH, in blue. The crystal structure for this enzyme did not include waters. (B) The active site of A. caviae PhaJ (PDB#1IQ6), an SHD hydratase, in maroon. (C) The active site of C. tropicalis Mfe2p hydratase domain (PDB#1PN2), a DHD DH, in olive.
A highly conserved glycine residue (e.g., FabA Gly79A, Figure 4A; Table 2) is found three (or, in one known case, four) residues downstream of the above-mentioned histidine-polarizing residue, at the N-terminal end of αHD. The placement of this residue at the tip of the main helix results in extra polarity from the helical dipole moment, and its amide hydrogen has been implicated in the mechanisms as contributing to the polarization of the thioester carbonyl. Multiple crystal structures show a captured chloride ion bound to this amide hydrogen (Figure 4B), confirming the electropositive nature of this region. In other structures, a water molecule occupies the space of the chloride ion; in such structures, this water will be referenced as W1.
The residue immediately downstream of this conserved glycine or its substitute (and still located at the electropositive end of αHD) is involved in hydrogen bonding to a water molecule (W2). W2 always hydrogen bonds with the chloride or water (W1) bound to the conserved glycine or its substitute but also has other hydrogen bonding partners, depending on the enzyme. W2 is present in every dehydratase/hydratase structure of high enough resolution to model water molecules (Figure 4A and B and Figure 5B, and C), except in cases where a competitive inhibitor is bound.
A third water molecule (W3) is often observed hydrogen-bonded to the catalytic histidine and/or one or two other residues in the active site. In one of the FabZ structures (PDB#1Z6B), an oxygen atom of the buffer cacodylic acid (dimethylarsenic acid) occupies this space instead.24
The remainder of the active site residues in the DH/H group fall into two different arrangements, as revealed by superimposition of the active sites. “Arrangement A” (named for FabA, the first enzyme found with this arrangement) is shared by most of the DH structures and is shown in Figure 4. “Arrangement J” (for PhaJ, the first with this arrangement) is utilized by both hydratases and DHs found in the fungi and mycobacteria and is shown in Figure 5.
2.3.2. Active Site Arrangement A
In Arrangement A, there is a conserved aspartate or glutamate residue in the active site (FabA Asp84B; Table 2), located on the opposite hot dog helix αHD′. Mutation of this residue abolishes activity. It was first implicated as a catalytic acid by Leesong et al., when they published their FabA structure with a bound inhibitor.1 They proposed that Asp84 of FabA protonates the leaving hydroxide to water, a mechanistic proposal that will be discussed further below. One of the oxygen atoms of this carboxylate residue is within hydrogen bonding distance of W2. The other oxygen is bound to yet another water molecule, W3, the one within hydrogen bonding distance to the catalytic histidine.
Arrangement A contains another conserved residue (also on αHD′) — a glutamine, histidine, or arginine residue (FabA Gln88B; Table 2), which is thought to anchor the aforementioned carboxylate residue in the proper orientation for catalysis via a hydrogen bond (Figure 4). Finally, a less conserved residue is often found in Arrangement A that can hydrogen bond to W3, along with the catalytic histidine and the aspartate/glutamate. Although this residue is absent in FabA, in FabZ, the standard bacterial FAS DH, this residue is another histidine (His23B, Figure 4B; Table 2) located on loop αxA′–βx1′; in animal FAS and modular PKS DHs, this role is fulfilled by a tyrosine (Tyr1003, Figure 4C).
2.3.3. Active Site Arrangement J
The first hydratase structures, which exemplify active site Arrangement J, were published in 2003,20,41,42 and it was observed that the active sites differed from those of the DH structures known at that time. Like Arrangement A, Arrangement J also contains a key aspartate residue (e.g. PhaJ Asp31A, Figure 5B; Table 2), yet it falls in a different location (five residues upstream of the catalytic histidine instead of on αHD′) and does not superimpose with the aspartate/glutamate found in Arrangement A. This aspartate still hydrogen bonds to W3, but from a different oxygen atom than seen in Arrangement A. Depending on the enzyme, the aspartate also bonds to a seryl hydroxyl group (from Ser62B in PhaJ) or W4, a part of a complex water network, as observed in the Mfe2p-H2 domain (Figure 5C).
The replacement of serine by a water in a larger network, such as in the Mfe2p-H2 domain, may occur to accommodate longer chain-length substrates. PhaJ is involved in polyhydroxyalkanoate (PHA) biosynthesis and accepts short chain thioesters, such as butenoyl- and hexenoyl-CoAs. The peroxisomal β-oxidation hydratases like Mfe2p-H2, on the other hand, require room for chains longer than 20 carbons in length. The serine residue or W4 hydrogen bonds to W2.
Two residues downstream from the aspartate in Arrangement J lies a conserved asparagine residue (Asn33A in PhaJ, Figure 5B; Table 2). While falling in a different location in the primary sequence, spatially this residue is positioned near His23B in HpFabZ and Tyr1003 in animal FAS DH. Likewise, it hydrogen bonds to W3.
Interestingly, although sequentially in entirely different regions of their respective enzymes, several specific atoms of the key residues of the two active site arrangements do nearly superimpose. A) One of the side chain oxygens of the key aspartate/glutamate residue of Arrangement A superimposes with an aspartate Oδ of Arrangement J. B) The second side chain oxygen of the aspartate/glutamate residue in Arrangement A superimposes with Oγ of Ser62 in PhaJ and W4 in the other enzymes with Arrangement J. C) When present, the third residues bound to W3 (His23B in HpFabZ vs. Asn33A in PhaJ) superimpose (Figure 6).
Figure 6. Active Site Comparison.

(A) A stereo view of the superimposition of DH/H active site Arrangement A (orange, HpFabZ) with active site Arrangement J (purple, PhaJ). Structurally conserved waters W1, W2, and W3 are labeled. (B) A two-dimensional representation of active site Arrangements A and J. As described in the text, key heteroatoms (shown in bold) in each of the active site arrangements superimpose, as can be observed in (A). In FabA, the leftmost imidazole ring is absent; in animal FAS DH, it is replaced by a phenol. In fungal FAS DHs and MFE2 hydratases, the seryl hydroxyl is substituted with a water molecule.
2.4. Mechanism of Hydration and Dehydration
Thus, although the active sites appear different at first, they present the same spatial arrangement of heteroatoms to perform the same chemistry. The discovery that fungal3,4 and mycobacterial43 FAS DHs have Arrangement J confirms that active site rearrangement alone cannot predict whether an enzyme will function as a DH or hydratase. Mechanistically, this should come as no surprise, since hydration is the microscopic reverse of dehydration. In fact, at least in some cases, the driving force for dehydration reactions is the presence of an ER downstream in the biosynthetic pathway to saturate the double bond and draw this equilibrium reaction in the synthetic direction.44,45
3-Decynoyl-N-acetylcysteamine (1, Figure 7C) is the prototypical mechanism-based (“suicide”) inhibitor, and for a long time it has been utilized in the inactivation of enzymes containing catalytic histidine residues.46 Scheme 2 outlines the mechanism of inactivation, where for the classical inhibitor 1, R is a hexyl group, Z represents a heteroatom capable of hydrogen bonding, and NAC is N-acetylcysteamine. The catalytic histidine deprotonates at the α-carbon to give an enolate stabilized by the protein oxyanion hole, which then collapses to an allene with the proton returned from the histidinium. This reactive allene thioester is now attacked nucleophilically by the same catalytic histidine, which leads initially to the β,γ-alkenoyl covalent adduct with the (E) configuration as shown. Tautomerization to one of the isomers of the more thermodynamically stable α,β-alkenoyl adducts slowly occurs, most likely catalyzed by solvent.47
Figure 7. Cocrystal Comparison of FabA and Mfe2p hydratase domain.

Potential hydrogen bonds are shown with grey lines, red spheres represent water molecules, and residues are shown as sticks and colored by element. The beginnings of the hot dog helices are shown with faint line ribbons. The images are drawn to scale and superimposable. (A) The active site of E. coli FabA (PDB#1MKA) in green, including the covalent adduct resulting from addition of mechanism-based inhibitor 3-decynoyl-N-acetylcysteamine (1) shown in black. Note that the adduct has distorted the orientation of catalytic His70A relative to the orientation seen in Figure 4A. (B) The active site of a H813Q mutant of C. tropicalis Mfe2p hydratase domain (PDB#1PN4) in olive, including the product (R)-3-hydroxydecanoyl-CoA (2) shown in black. Some of the CoA portion of the product has not been drawn for the purpose of figure clarity. (C) The structures of compounds 1 and 2.
Scheme 2.
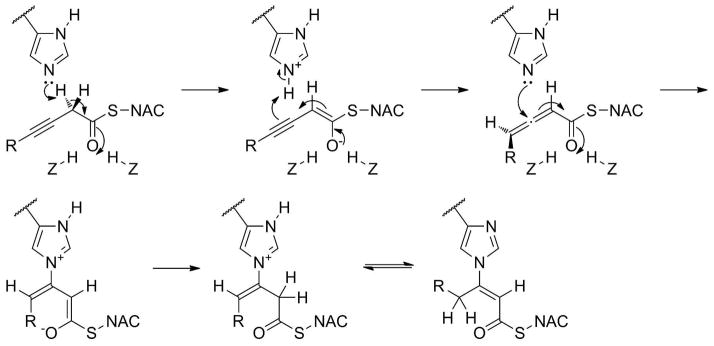
Mechanism-Based Inhibition by 3-Decynoyl-N-Acetylcysteamine.
Leesong et al., in the original hot dog structure paper, also obtained a structure of the adduct of 1 bound in the active site of FabA. Based on the orientation of the plane of the double bond and carbonyl (Figure 7A), they proposed that W2 represented the leaving water from the dehydration. Furthermore, they proposed that Asp84 protonates the leaving hydroxide as water and that His70, acting as base, abstracts the α-proton. There are a number of problems with this suggestion, however. First, upon reacting covalently with the inhibitor, His70 has rotated 180° and broken its hydrogen bond with the backbone carbonyl of Val76 (Figure 7A). Thus, the active site of FabA is severely distorted from its natural conformation, making an argument for catalysis based on structure difficult. Perhaps most importantly, this acid–base mechanism also conflicts with the earlier works referenced above that strongly suggest a one-base mechanism.8–10 Despite these factors, most DH structural papers published to date continue to assume that the aspartate/glutamate residue is acting as a catalytic acid and not some other role and that W2 is the “stored” water of dehydration.
In 2004, Koski et al. co-crystallized the product (R)-3-hydroxydecanoyl-CoA (2, Figure 7C) within the active site of a mutated eukaryotic hydratase 2 domain (Mfe2p-H2) and found an entirely different arrangement of the cocrystal compared to that found in FabA.37 The carbonyl of the product was rotated about 90° relative to its location in the FabA inhibitor structure and was pointing between W2 and Gly831 at the end of αHD — a typical oxyanion hole arrangement. This orientation also placed the hydroxyl group of the product within H-bonding distance to the key histidine and superimposable with W3 in the other apo structures (Figure 7B). While FabA and Mfe2p-H2 have different active site arrangements, as described above, W2 and W3 are in the same locations in both arrangement types (Figure 6). This would seem to indicate that W3, rather than W2, is the “stored” water molecule. It also indicates that an acid/base mechanism is not in fact in play but that histidine acts as both base and acid (as histidinium) or that a concerted syn addition occurs, both of which agree with previous biochemical and stereochemical experiments on this class of enzymes.8,9,48
Thus, Koski et al. proposed the mechanism outlined in Scheme 3.37 The substrate is polarized by the effect of the N-terminal end of αHD, particularly through hydrogen bonding to a glycine backbone amide and a water molecule (W2). This polarization activates C-3 for nucleophilic attack. The histidine deprotonates the attacking water, which is further positioned by two other residues. This conjugate attack leads to an enolate, which collapses back and is reprotonated at C-2 by the histidinium from the same side, resulting in syn addition. Alternatively, the entire sequence may be concerted, involving a five-membered transition state. The same mechanism in reverse would occur in the DH enzymes and domains. The differences between the two active sites have little effect on the mechanistic analysis here, as only the histidine, αHD glycine, and W2 are directly involved; the other residues seem only necessary for positioning of the substrate and W3.
Scheme 3.
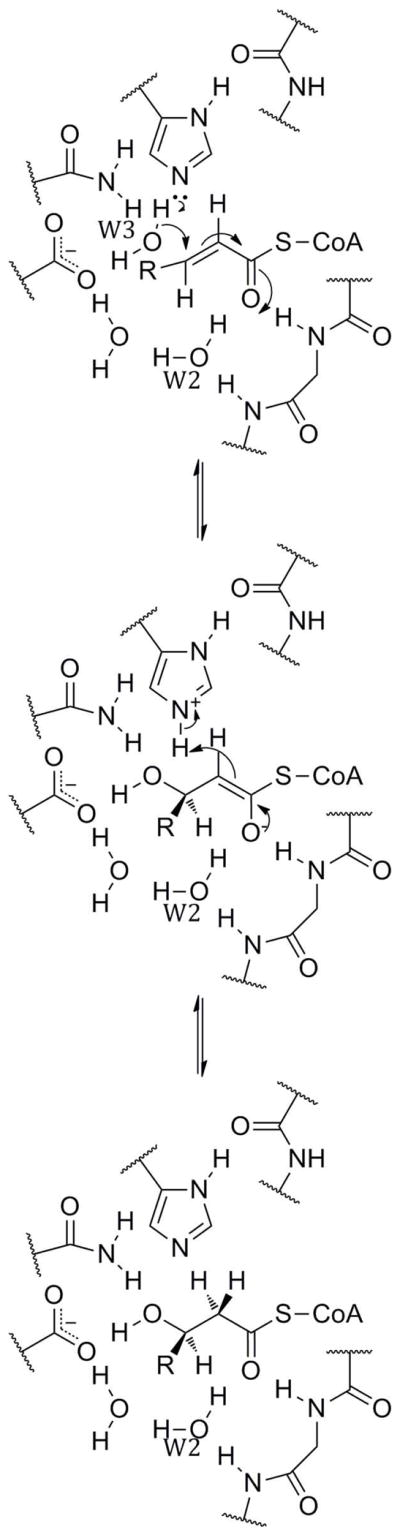
Proposed Mechanism of (R)-Specific Hydratases.
As was mentioned above, some modular PKS DHs result in cis double bonds at the C2 position. There have been five crystal structures published for modular PKS DHs to date, one of the erythromycin biosynthetic pathway PKS 6-deoxyerythronolide synthase (DEBS)18 and the four DHs from curacin A biosynthesis (CurF, CurH, CurJ, and CurK).34 Curacin A contains a cis double bond, but it is still unknown whether it is the CurF or CurH DH that installs this cis bond. While some of the residues differ slightly (Table 2), there seem to be no significant active site changes among the four Cur DH structures. Likewise, among the modular PKS DH sequences known, no differences have been found to suggest active site residues have moved for cis DHs in other systems. As has already been discussed, it has been suggested that the stereochemistry of the β-hydroxyl group determines the stereochemistry of the double bond formed within the DH — that is, that the double bond stereochemistry is ultimately determined by the KRs — and this seems a fair hypothesis (Figure 8).15,18,49 In contrast, Akey et al. attempted to model an alternative mechanism in their Cur crystal structures; however, their models were made with the constraint that the substrate hydroxyl form a hydrogen bond to the key aspartate, i.e. on the assumption that the earlier Leesong et al. mechanism is correct. Akey et al. explain that their models predict removal of different α-hydrogens for trans or cis products. If so, the pro-2R hydrogen would lead to the cis product and require a net anti elimination. Labeling studies with (S)-3-hydroxythioesters, therefore, may be able to distinguish between the Leesong mechanism and the Koski mechanism, which predicts pro-2S hydrogen removal and syn elimination in both cases.
Figure 8. Proposed Binding Modes Leading to Either Trans or Cis Double Bonds.
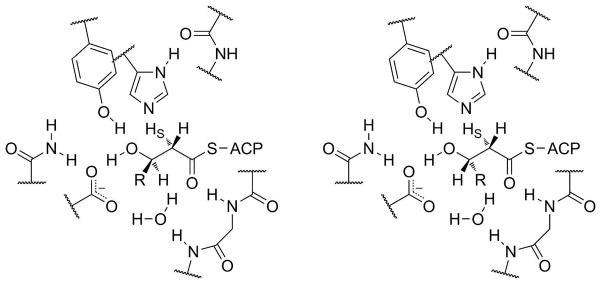
Syn elimination of an (R)-3-hydroxyacyl-ACP (left) occurs by removal of the pro-2S hydrogen and results in a trans double bond. Syn elimination of an (S)-3-hydroxyacyl-S-ACP (right) by removal of the pro-2S hydrogen is expected to result in a cis double bond. In both cases, the hydroxyl group is held in place by interactions with tyrosine and aspartate residues. (The side chains shown are those found in DEBS DH.)
However, not all PKS products with cis double bonds derive from KR domains predicted to give (S)-3-hydroxythioesters. In at least one such case, the DHs from modules 2 and 3 in the borrelidin PKS system have been shown to produce trans double bonds from (R)-3-hydroxythioesters, even though the final product is cis at the double bond formed by BorDH3.13 This report provides further evidence that hot dog DHs only perform syn eliminations and that some other mechanism for isomerization (such as a downstream enzyme, as proposed for hypothemycin biosynthesis50) to the cis double bond must occur if the KR provides the usual D-β-hydroxylated starting material.
2.5. Dual-Function Dehydratases
The first hot dog enzyme crystallized, FabA, also functions as an isomerase, catalyzing the allylic rearrangement of the dehydrated product trans-2-decenoyl-ACP (3) to cis-3-decenoyl-ACP (4) as part of the anaerobic pathway to install unsaturation into fatty acids (Scheme 4). Schwab and Klassen showed that the pro-4R hydrogen is removed in the reaction to form the resonance-stabilized anion. Reprotonation at the si face of C-2 affords 4. Once again, this is indicative of a single-base mechanism and not an acid–base mechanism.51 Notably, it can be observed from the crystal structures (Figure 7) that the catalytic histidine should be able to reach the pro-4R proton but not the pro-4S proton. This mechanism is outlined in Scheme 4. An acid–base mechanism would require Asp84 to deprotonate the pro-4R hydrogen, yet the structures show Asp84 closer to the pro-4S proton.
Scheme 4.
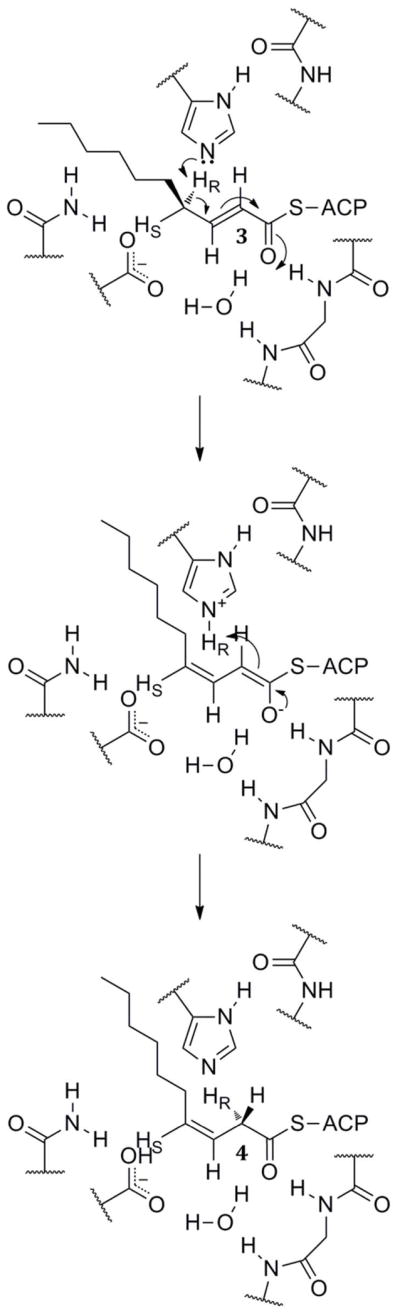
The Allylic Isomerization Reaction of FabA.
It is an obvious conundrum that FabA performs isomerization while FabZ does not. Kimber et al. investigated this issue by a series of “residue swapping” experiments involving D84E FabA and E68D FabZ. The variant of FabA still retained the ability to perform both reactions; the FabZ mutant retained its ability to perform dehydration but did not gain the ability to perform the isomerization. They achieved similar results with P29H FabA/H13P FabZ and C80V FabA/V59C FabZ residue swaps. Therefore, they argued that the shape of the alkyl-binding region in FabZ disallows the cis conformation allowed in FabA (Figure 9).23 It is of note that FabA shows optimal activity for 10-carbon acyl substrates, whereas FabZ functions efficiently on a wide range of substrate lengths up to sixteen carbons.
Figure 9. Comparison of Active Site Tunnels Between FabA and FabZ.
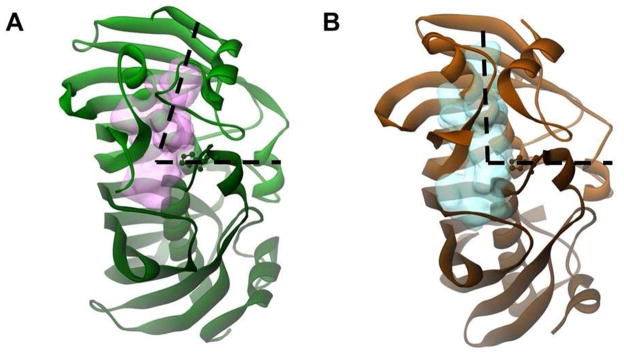
The enzyme backbones are drawn with solid ribbons, the catalytic histidines are shown with scaled balls and sticks. (A) Fab A (PDB#1MKA), the dehydratase–isomerase, in green with the active site volume indicated by a faint pink cloud. (B) P. aeruginosa Fab Z (PDB#1U1Z), the standard dehydratase, in orange with the active site volume indicated by a faint blue cloud. Note the shorter binding tunnel in FabA and the slight difference in the angle of the two tunnels (indicated with dashed lines), hypothesized to allow for C-3 cis double bond formation in FabA.
It is possible that some PKS DHs are actually α-epimerases for α-substituted β-keto substrates;18 however, it is probably too early to propose a mechanism for such a reaction, lacking further experimental data. Thus far, enzymes in the DH/H group seem capable of performing exclusively suprafacial reactions, yet epimerization is an antarafacial event.
3. Thioesterase Group
3.1. Reaction Overview and Comparisons
Two years after the first hot dog structure was published, Benning et al. solved the structure of the second hot dog fold enzyme and the first member of the hot dog thioesterase (TE) group.52 Thioesterases, in general, catalyze the hydrolysis of thioesters to carboxylic acids (Scheme 5) and are widespread in biosynthetic pathways. They function on fatty acid or polyketide acyl thioesters, as well as on benzyl and phenylacetyl thioesters and the peptidyl thioesters of NRPSs.
Scheme 5.
The Reaction Catalyzed by Thioesterases.
The majority of thioesterases are members of the α/β hydrolase family and are typically found at the C-terminus as domains of a much larger NRPS or type I FAS or PKS enzyme. α/β Hydrolase family thioesterases contain the classic catalytic triad of nucleophile–histidine–aspartate and often perform macrocyclizations instead of or in addition to hydrolysis.53 In contrast, the vast majority of hot dog fold thioesterases differ in that they are found as free (Type II) thioesterases. Also, hot dog thioesterases neither utilize the classic catalytic triad nor (with one known exception) have the catalytic histidine of the hot dog DH/H group. In fact, the polarization effect of the hot dog helix may be the only active site feature of the DH/H group that carries over to the hot dog thioesterases (Figure 10). Many of the hot dog TEs discovered to date contain the conserved glycine residue at the tip of αHD, and the backbone amide of this residue, glycine or not, is similarly implicated in most of the proposed mechanisms as functioning as one part of the oxyanion hole.
Figure 10. Comparison Between Hot Dog Dehydratase/Hydratase and Thioesterase Active Sites.

Potential hydrogen bonds are shown with grey lines, red spheres represent water molecules, and residues are shown as sticks and colored by element. The beginnings of the hot dog helices are shown with faint line ribbons. The images are drawn to scale and superimposable. Below each structural image is a two-dimensional representation of each active site with conserved features in bold. (A) The active site of E. coli FabA (PDB#1MKB), an example of the dehydratase/hydratase group. The side chains of residues 71 through 74 and 76 are not shown for the purpose of figure clarity. (B) The active site of T. thermophilus HB8 PaaI (PDB#1WLU), an example of the thioesterase group. The side chains of residues 34 and 35 are not shown for the purpose of figure clarity. Note the major difference in the β1–αHD loop between the two enzymes. Note also the conservation of W1 and the conserved glycine residue to which it binds.
3.2. TE Active Sites
Like in the case of the DH/H group, multiple active site arrangements occur, and in fact, bioinformatic studies indicate more subfamilies of hot dog thioesterases than of dehydratases/hydratases.2,5 Listed in Table 3 are, to the best of our knowledge, all non-hypothetical enzymes in the thioesterase group for which there is currently structural information available. Laid out in Table 4 is a comparison of key active site residues for those enzymes. As previously performed for the DH/H group, structures were superimposed by residue (not sequence alignment) using the Discovery Studio Suite, and these superimpositions were used to generate the figures in this section and Table 4. The residues chosen to superimpose varied depending on the enzymes being compared, but this selection of residues always included the residues found at the positive end of αHD.
Table 3.
Thioesterases of the Hot Dog Superfamily with Published Structural Data.a
| Enzyme | Species | Pathway | Oligomeric Stateb | Substrate(s) | Pub. Year(s) | PDB #sc | Active Site Cocrystalsd |
|---|---|---|---|---|---|---|---|
| Arrangement Ψ | |||||||
| 4HBT-I | Pseudomonas sp. strain CBS-3 | 4-HOBz Degradation | TA | 4-HOBz-CoA | 1998 | 1BVQ | none |
| 2002 | 1LO7* | 4-HOBz-CoA | |||||
| 1LO8 | 4-HOBn-CoA | ||||||
| 1LO9 | 4-HOPa-CoA | ||||||
| YbgC | H. pylori | Tol-Pal | TA | LC acyl-CoAs | 2008 | 2PZH | none |
|
| |||||||
| Arrangement R | |||||||
| 4HBT-II | Arthrobacter sp. strain SU | 4-HOBz Degradation | TB | 4-HOBz-CoA | 2003 | 1Q4S | 4-HOBz & CoA |
| 1Q4T | 4-HOBn-CoA | ||||||
| 1Q4U | 4-HOPa-CoA | ||||||
| EntH | E. coli | NRPS | TB | HOBz-PCPs | 2009 | 1VH9 | none |
| TtPaaI | T. thermophilus HB8 | PhAcOH Degradation | TB | PhAc-CoAs | 2005 | 1J1Y | none |
| 2005 | 1WLU | glycerol | |||||
| 1WLV | CoA | ||||||
| 1WM6 | none | ||||||
| 1WN3 | Hex-CoA | ||||||
| EcPaaI | E. coli | PhAcOH Degradation | TB | PhAc-CoAs | 2006 | 2FS2 | none |
| THEM2 | H. sapiens | unknown | TB | M/LC acyl-CoAs | 2006 | 2F0X | sulfate |
| 2009 | 3F5O | 2-oxoundecyl-CoA | |||||
| YciA | H. influenza | unknown | H2 | acyl-CoAs | 2008 | 1YLI | none |
| 3BJK | CoA | ||||||
| Cj0915 | C. jejuni | unknown | [H2] | acyl-CoAs | 2009 | 3D6L | none |
| Acot7 | M. musculus | unknown | Trdh | arachidonoyl-CoA | 2007 | 2V1Oe | CoA |
| 2Q2Bf | none | ||||||
| TE-II | E. coli | FAS | DdhB | MC acyl-CoA | 2000 | 1C8U | none |
|
| |||||||
| FlK | S. cattleya | FAc Detoxification | D | FAc-CoA | 2010 | 3KX7 | none |
| 3KX8 | none | ||||||
| 3KV7 | AcOH | ||||||
| 3KV8 | FAcOH | ||||||
| 3KVZ | FAc-CNAC | ||||||
| 3KW1 | FAc-ONAC | ||||||
| 3KUV* | AcOH | ||||||
| 3KUW* | FAcOH | ||||||
| 3KVU* | Ac-CoA | ||||||
| 3KVI* | FAcOH | ||||||
| 2010 | 3P2Q | none | |||||
| 3P2R | FAcOH | ||||||
| 3P2S | none | ||||||
| 3P3F* | none | ||||||
| 3P3I* | FAcOH & CoA | ||||||
| CalE7g | M. echinospora | IPKS | TA | unconfirmed | 2009 | 2W3X | none |
| DynE7g | M. chernisa | IPKS | TA | unconfirmed | 2010 | 2XFL | none |
| 2XEM | |||||||
| FcotT | M. tuberculosis | unknown | H3 | LC acyl-CoA | 2007 | 2PFC | dodecenoate |
| 2012 | 3B18 | dodecanoate | |||||
Abbreviations used: Bz, benzoyl; Bn, benzyl; Pa, phenacyl; Ac, acetyl; Hex, hexanoyl; CNAC, N-acetylaminopropyl; ONAC, N-acetylaminoethoxy; CoA, coenzyme A; PCP, peptidyl carrier protein; ACP, acyl carrier protein; LC, long-chain; MC, medium chain.
Terms defined as in Pidugu, et al.: D, dimer; TA, face-to-face dimer of dimers; TB, back-to-back dimer of dimers; H2 & H3, trimers of dimers; DdhB, DHD dimer corresponding to TB; Trdh, DHD trimer corresponding to H2.
An asterisk indicates a mutant.
Buffers and/or salt ions are not necessarily noted.
N-terminal domain only.
C-terminal domain only.
These enzymes may additionally perform decarboxylations.
Table 4.
Structural Alignment and Proposed Function of the Active Site Residues of the Thioesterases in Table 3.a
| Enzyme | *Catalytic Residue, †LG Assistance, or ‡Oxyanion Hole | BB H-Acceptor from W2 | Oxyanion Holeb | H-Bonds to W2 | BB H-Donor to W2 | BB H-Bonds to W3 | Catalytic Residue | Positioning of Hydrolytic Water | BB H-Bonds to Pant Amide N-H |
|---|---|---|---|---|---|---|---|---|---|
| Arrangement Ψ | |||||||||
| 4HBT-I | Asp17A* | Val22A | Tyr24A | Tyr27A | Ala72A | Val110A | (Asp32Bc) | (Arg36B) | Ile61B |
| YbgC | Asp11A* | Val16A | His18Ad | Tyr21A | Ala60A | Val102A | (Glu26B) | (Ser30B) | Ile49B |
|
| |||||||||
| Arrangement R | |||||||||
| 4HBT-II | Gln58A† | Val63A | Gly65A | Tyr68A | Val104A | Cys137A | Glu73B | Thr77B | Gly93Be |
| EntH | Gln48A | Leu53A | Gly55A | H2Of | Val93A | Cys126A | Glu63B | Ser67B | Gly82Be |
| TtPaaI | Asn33A‡ | Ala38A | Gly40A | (Leu43A) | Leu74A | Val107A | Asp48B | (Ala52B) | Ala63B |
| EcPaaI | Asn46A‡ | Cys51A | Gly53A | (Leu56A) | Gly88A | Val122A | Asp61B | (Ala65B) | Ala77B |
| THEM2 | Asn50A‡ | Leu55A | Gly57A | H2O | Ala94A | Ile129A | Asp65B | (Thr69B) | Ser83Bg |
| YciA | Asn29A | Ile34A | Gly36A | (Ile39A) | Ile73A | (Val115A) | Asp44B | (Ala48B) | Thr61B |
| Cj0915 | Asn19A | Ile24A | Gly26A | (Ile29A) | Val63A | Val105A | Asp34B | (Ala38B) | Thr51B[g] |
| Acot7 | Asn24 | Val29 | Gly31 | (Ile34) | Met74 | Thr113 | Asp213 | (Gly217) | Thr230[g] |
| TE-II | — | Val34 | Gly36 | (Val39) | Gly68 | Ile101 | Asp204 | — | Thr228g |
|
| |||||||||
| FlK | (Val23A) | Phe40A | Thr42Ah | (Met45A) | Thr80A | (Ile113A) | (Glu50B) | (Val54B) | (Gly69B)i |
|
| |||||||||
| CalE7 | Asn19A | Val24A | Tyr26A | Tyr29A | Leu71A | Val107A | (Gly34B) | (Glu38B) | Thr60B |
| DynE7 | Asn17A | Val22A | Phe24A | Tyr27A | Gly69A | Leu106A | (Gly32B)j | (Glu36B) | Thr58B |
|
| |||||||||
| FcotT | — | (Phe73A) | Ala75A | Leu78A | (Leu131A) | — | (Asn83B) | Tyr87B | (Ile120B) |
A or B after residue numbers indicates subunit for SHD enzymes/domains. Bold font indicates fully conserved residues; italic font indicates similar residues. W2 and W3 are water molecules thought to serve as a binding site for one of the pantetheinyl amide carbonyls and shown in Figures 10, 11, and 12. Parentheses indicate that a residue is located at the same location as other residues in the same column but not sharing the same function. LG, leaving group; BB, backbone; Pant, pantetheinyl.
Positive end of helix αHD.
This residue was found crucial for substrate binding.
The backbone amide and side chain of this residue are both thought to stabilize the oxyanion.
The amide N-H of this residue may also assist in positioning of the hydrolytic water molecule.
This water was not present in the crystal structure but is assumed to be involved based on structural superimposition with THEM2. This water is bound to Thr60A in THEM2, corresponding to Ser58A in EntH.
The side-chain hydroxyl of this residue may also assist in positioning of the hydrolytic water molecule.
The side chain hydroxyl of this residue is thought to act as the catalytic base for this enzyme.
The amide N–H of this residue, along with Arg120B is thought to interact with the polar substrate C–F bond.
The side chain of this residue binds to the hypothetical product ketone carbonyl.
3.2.1. 4HBT-I and -II: The Prototypical Hot Dog TE Active Site Arrangements
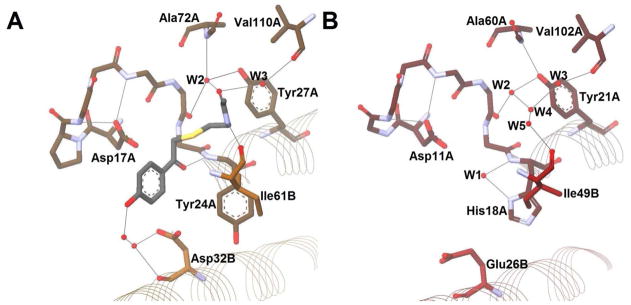
As stated above, the first hot dog thioesterase crystal structure was published in 1998 by the Dunaway-Mariano and Holden groups.52 The enzyme was Pseudomonas sp. strain CBS-3 4-hydroxybenzoyl-CoA thioesterase (4HBT-I), involved in the unusual degradation pathway of 4-chlorobenzoate. Mutation experiments have revealed Asp17A to be the catalytic residue (Figure 11A).54,55 This aspartate is located near the same relative location in space as the catalytic histidine in the DHs/hydratases, on loop β1–αHD. This loop is shorter in the majority of hot dog thioesterases than in the DH/H group (Figure 10). On the opposite side of the active site, in the middle of hot dog helix αHD′ from the other monomer, Asp32B was found to play a role in substrate binding, through the intermediacy of two bridging water molecules (Figure 11A).56 This group has also obtained structures of 4HBT-I with two inhibitors bound and a D17N mutant with substrate. From these structures and thorough kinetic and labeling studies, they have made a strong case for an acyl enzyme anhydride intermediate (Scheme 6A), which, while not common, is not unprecedented.54,57 In these structures, the substrate/inhibitor carbonyl is hydrogen bonded to the backbone amide N–H bond of Tyr24A, located at the positive end of αHD and corresponding to the conserved glycine of the DH/H group. Raman spectroscopic studies, however, reveal only moderate changes in substrate C=O polarization in the enzyme relative to spectra in buffer, implying that carbonyl polarization alone does not drive catalytic efficiency for this particular enzyme.55 In related structures, W1 often substitutes for the substrate thioester carbonyl, as in the DH/H group (Figure 10).
Figure 11. The Active Sites of the Two Hydroxybenzoyl-CoA Thioesterases.

(A) 4HBT-I (PDB#1LO9) shown in orange. The structure includes inhibitor 4-hydroxyphenacyl-CoA shown in black. (B) 4HBT-II (PDB#1Q4S) shown in yellow. The structure includes the products 4-hydroxybenzoate and coenzyme A shown in black. For both (A) and (B) only the N-acetylcysteamine portion of CoA has been drawn and side chains of some residues, particularly of residues involved in loop β1–αHD, are not shown for the purpose of figure clarity. Below each structural image is a two-dimensional representation of selected residues of each active site.
Scheme 6.
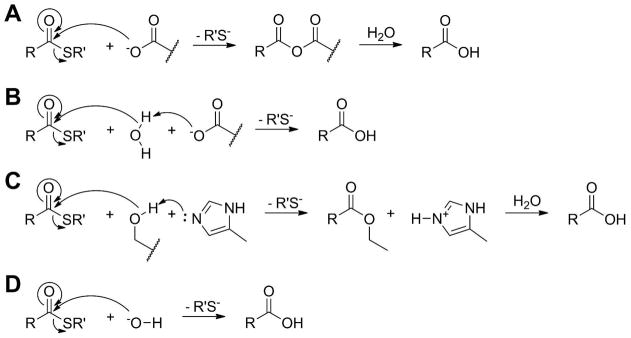
Some Proposed Mechanisms of Thioesterases.
The same research group published a set of structures for another 4-hydroxybenzoyl-CoA TE, from Arthrobacter sp. strain SU,58 termed 4HBT-II. While both 4HBT enzymes are involved in the same degradation pathway and act on exactly the same substrate, surprisingly, the catalytic residue (Glu73B) was found on the opposite side of the active site. This glutamate residue occupies the corresponding position in the primary sequence where Asp32B resides in 4HBT-I; however, the orientations of helix αHD′ relative to helix αHD differ between the two enzymes, being more parallel in 4HBT-II. This global structure change pulls Glu73B in 4HBT-II far closer to the thioester carbonyl site than Asp32B is in 4HBT-I (Figure 11).
Furthermore, the residue corresponding to Asp17A in 4HTB-I has been replaced by a glutamine (Gln58A) in 4HBT-II (Figure 11, Table 4). In addition to the apo enzyme, structures with two inhibitors and the products were also published for 4HBT-II. As with 4HBT-I, the electrophilic carbonyls of the bound structures were hydrogen bonded to the N-terminal end of αHD, in this case to a glycine (Gly65A) as with the DH/H group. In one of the 4HBT-II structures, Gln58A is also in close proximity to the sulfur atom of the leaving thiol.
In both enzymes, the structure of loop β1–αHD is identical, and residues Asp17A (4HBT-I) and Gln58A (4HBT-II) are held in place by backbone interactions with the amides two and four residues upstream (Figure 11).
The oligomeric states of the two enzymes differ (Table 3), as do the adenine-binding sites; however, the pantetheinyl-binding sites of the two enzymes are very similar. The carbonyl oxygen of the amide connecting the pantothenic acid and cysteamine portions of the cofactor is positioned roughly between two crystallographic waters, W2 and W3, which sit in a relatively hydrophobic environment. W2 is held in place by three hydrogen bonds (Figure 11, Table 4). The first is to the backbone carbonyl two residues upstream from the polarized helix residue — that is, Val22A and Val63A for 4HBT-I and II, respectively. The second bonding partner for W2 is located on loop β2–β3 from the backbone amide N–H bond of Ala72A and Val104A, respectively. The third is to the tyrosine hydroxyl located three residues downstream from the polarized helix residue — that is, Tyr27A and Tyr68A for 4HBT-I and II, respectively. W3 hydrogen bonds to the carbonyl oxygen of Val110A and Cys137A, respectively, on strand β5 (Figure 11, Table 4). In related structures without substrates, inhibitors, or products cocrystalized, W4 often substitutes for the pantetheinyl carbonyl, (e.g., as in Figure 12B and Figure 13C). The amide N–H of the cysteamine moiety of pantetheine hydrogen bonds to the carbonyl of Ile61B and Gly93B in 4HBT-I and II, respectively (Figure 11, Table 4). Similarly, W5 often substitutes for this cysteaminyl amide nitrogen, (as in Figure 12B and Figure 13C). All these residues/waters structurally superimpose among the related enzymes. (Figure 11, Figure 12, and Figure 13).
Figure 12. Hot Dog Thioesterase Active Site Arrangement Ψ.
Potential hydrogen bonds are shown with grey lines, red spheres represent water molecules, and residues are shown as sticks and colored by element. Hot dog helices are shown with faint line ribbons. The images are drawn to scale and superimposable. The side chains of some residues, particularly of residues involved in loop β1–αHD, are not shown for the purpose of figure clarity. (A) The active site of Pseudomonas sp. strain CBS-3 4-HBT-I (PDB#1LO9), an example of an SHD TE, in orange, including inhibitor 4-hydroxyphenacyl-CoA shown in black. Only the N-acetylcysteamine portion of CoA has been drawn for the purpose of figure clarity. (B) The active site of H. pylroi YbgCF (PDB#2PZH), another SHD TE, in red.
Figure 13. Hot Dog Thioesterase Active Site Arrangement R.

Potential hydrogen bonds are shown with grey lines, red spheres represent water molecules, and residues are shown as sticks and colored by element. Hot dog helices are shown with faint line ribbons. The images are drawn to scale and superimposable. The side chains of some residues, particularly of residues involved in loop β1–αHD, are not shown for the purpose of figure clarity. (A) The active site of Arthrobacter sp. strain SU 4-HBT (PDB#1Q4S), an example of an SHD TE, in yellow, including the products 4-hydroxybenzoate and coenzyme A shown in black. Only the N-acetylcysteamine portion of CoA has been drawn for the purpose of figure clarity. (B) The active site of E. coli EntH (PDB#1VH9), another SHD TE, in light green. (C) The active site of T. thermophilus HB8 (PDB#1J1Y), another SHD TE, in blue-green, including a glycerol molecule in black. (D) The active site of H. sapiens THEM2 (PDB#2F0X), another SHD TE, in green, including a sulfate ion. (E) The active site of H. influenzae YciA (PDB#3BJK), another SHD TE, in cyan, including the product coenzyme A shown in black. Only the N-acetylcysteamine portion of CoA has been drawn for the purpose of figure clarity. (F) The active site of E. coli TE-II, also known as TesB (PDB#1C8U), an example of a DHD TE, in blue. Note the bent second helix typical of DHD enzymes.
While, as just described, the pantetheine arm and the reactive carbonyl are positioned similarly by both 4HBT enzymes, the other residues in the active site direct the rest of the substrate to orient differently. This, together with the alternate positions of Asp32B (4HBT-I) and Glu73B (4HBT-II), implies that the hydrolysis reaction occurs on a different face of the substrate in each enzyme — the si face for 4HBT-I and the re face for 4HBT-II (Scheme 7).
Scheme 7.

Si vs. Re Attack in 4-Hydroxybenzoyl-CoA Thioesterases.
Since the discovery of the two 4HBTs, the majority of hot dog fold TEs have been found similar to one or the other of these “prototypical” enzymes. Those TEs with an active site comparable to 4HBT-I can be termed “Arrangement Ψ”, after Pseudomonas 4HBT. The other arrangement can be called “Arrangement R”, after Arthrobacter 4HBT (4HBT-II).
3.2.2. Active Site Arrangement Ψ
The structure of YbgC from H. pylori59 shows that it has an active site extremely similar to 4HBT-I (Figure 12). HpYbgC acts on long-chain acyl-CoAs, whereas H. influenza YbgC prefers short-chain fatty acyl groups,60 but neither hydrolyzes a benzoyl substrate as does 4HBT-I. Even so, the catalytic machinery among the enzymes is shared, with the only notable change being a conservative replacement with Glu26B in YbgC for Asp32B in 4HBT-I (Table 4). Another difference between the two enzymes is that there appear to be two members for the oxyanion hole in YbgC — the backbone amide of His18A and the imidazole side chain of the same residue — whereas 4HBT-I seems only to rely on the backbone amide of Tyr24A (Figure 12). Within this proposed oxyanion hole sits W1, the water molecule thought to mimic the carbonyl oxygen of the substrate, as described previously.
In addition to YbgC, several other enzymes — such as the gentisyl-CoA TE from B. halodurans C-12561 and the benzoyl-CoA TE from A. evansii62 — show high sequence similarity to 4HBT-I and likely contain an Arrangement Ψ active site. While bioinformatic studies have placed the enzyme HP0496 from the Tol-Pal system into a different subfamily from 4HBT-I,2,5 it is expected to share at Arrangement Ψ as well, being a YbgC homolog.
3.2.3. Active Site Arrangement R
Arrangement R (Figure 13, Table 3) currently contains a larger number of structures for which papers have been published. With only one exception, all enzymes with this arrangement have a motif (Q/N)XXXXXΦGGXXXXXX(D/E), where Φ is an aromatic amino acid and the underlined glycine is the one acting in the oxyanion hole.
In the case of prototypical member 4HBT-II, Asn96B, located on the “bun” (on β2′), appears to act with Glu73B as part of a dyad (Figure 13A). In the product-containing crystal structure of this enzyme, the carboxylate oxygen is hydrogen bonded to catalytic Glu73B, Thr77B, and Gly93B, and this could be the location for the attacking water if 4HBT-II utilizes a general base mechanism (Scheme 6B, Scheme 7). Mutation of Thr77 to alanine resulted in a three-fold reduction in kcat with no reduction in KM.63 Finally, recent Raman spectroscopy studies suggest that 4HBT-II holds the substrate thioester group in a syn gauche orientation, which weakens the thioester bond.64
EntH (also called YbdB) is a type II thioesterase involved in a “proofreading” capacity for the NRPS-utilizing biosynthesis of enterobactin.65,66 EntH reacts with hydroxybenzoyl groups bound to peptidyl-carrier proteins, as opposed to coenzyme A, but apart from that, it appears structurally very similar to 4HBT-II, except for the following notable changes: the tyrosine bound to W2 has been replaced by a serine (Ser58A, Figure 13, Table 4) and Asn96B, (which hydrogen bonds to the catalytic glutamate in 4HBT-II,) by Leu85B.67 Interestingly, without a corresponding asparagine, the catalytic glutamate in this case (Glu63B) now receives assistance from His89A on β2 (Figure 13B). It turns out that the catalytic residue in Arrangement R usually acts in concert with at least a second residue but that that residue is not structurally conserved. Unfortunately, the resolution of the EntH structure limits determination to a relatively small number of water molecules; W2 is present, but W1, W3, W4, and W5 are not. In the EntH structure, Gln48A is also in a different conformation, being displaced by a water (Figure 13B), but one can assume that this is an artifact from crystallization. EntH also includes Ser67B, corresponding to Thr77B in 4HBT-II (Figure 13, Table 4). An S67A mutation reduced activity by 140-fold.67 4HBT-II Thr77 and EntH Ser67 may assist in positioning a nucleophilic water molecule.
An example of an enzyme with this arrangement where the catalytic residue may act without a polarizing interaction is PaaI, which is involved in the phenylacetate degradation pathway. Structures for PaaI have been solved for the enzymes from both T. thermophilus68 and E. coli.69 In the former case, structures with either CoA or hexanoyl-CoA were also solved. Modest changes from Gln58A and Glu73B (4HBT-II) to Asn33A and Asp48B (TtPaaI), respectively, are present (Figure 13C, Table 4), but the catalytic mechanism is proposed to be the same,69 except that no other residue is within 3.5 Å of the side chain of the catalytic aspartate (Figure 13C). In the PaaI structures, W2 has also lost one of its three binding partners with the replacement of tyrosine by a nonpolar residue (Table 4), yet W2 is still present in the structures, as are W1, W3, W4, and W5, implying the same binding site for the pantetheine moiety as well as the same oxyanion hole. W1 also hydrogen bonds with Asn33A (Figure 13C), which suggests that the asparagine/glutamine at this position in Arrangement R may function as the other half of the oxyanion hole for these enzymes. In one of the structures, a glycerol molecule sits within the active site (Figure 13C). One of its terminal hydroxyls is bound between W1 and catalytic Asp48B, in a similar position as the carboxylate oxygen of the product of 4HBT-II (Figure 12A), lending support to a general base mechanism (Scheme 6B).
The other enzymes found with this arrangement act on aliphatic acyl-CoAs. Human thioesterase superfamily member 2 (THEM2) has been shown to hydrolyze medium-to-long–chain fatty acids, although its role is not entirely known. The Dunaway-Mariano group has published two structures of this enzyme.63,70 Like PaaI, THEM2 makes use of an asparagine and an aspartate (Figure 13D, Table 4). In place of tyrosine, THEM2 uses Thr60A to hydrogen bond to W2 indirectly through another water molecule (Figure 13D) — which might also occur in the case of EntH, which has a serine at this position (Figure 13B). Hydrogen-bonded to catalytic Asp65B is His134B, which is in turn hydrogen bonded to the backbone carbonyl of Asp85B, both of which are located on the “bun” (Figure 13D). The Cheng et al. structure70 fortuitously has a captured sulfate ion, which may mimic the tetrahedral intermediate. One of the oxygens of the sulfate points into the oxyanion hole to interact with Asn50A and Gly57A. A second sulfate oxygen hydrogen bonds with both catalytic Asp65B and the side chain of Ser83A (Figure 13D), which also is partly responsible for pantetheine binding through its carbonyl. The S83A mutation lowers kcat by ten-fold. In the Cao et al. structure63, there is a water molecule found here. Again, this is where the nucleophilic water is thought to be positioned for re face attack. The third oxygen (pointing vertically in Figure 13D) substitutes for where the sulfur atom would reside in the intermediate of the reaction, leaving the fourth to point in the direction of the phenylacetyl group of the natural substrate. This structure, then, further adds support to a general base mechanism for this active site arrangement (Scheme 6B).
H. influenza YciA,71,72 C. jejuni Cj0915,73 and mouse acyl-CoA thioesterase 7 (Acot7)74,75 are related enzymes acting on acyl-CoAs and containing Arrangement R. YciA is shown in Figure 13E along with the byproduct coenzyme A. YciA is unique in binding to CoA more tightly than to its substrates, and the crystal structure shows the sulfur atom of CoA interacting with the carbonyl of Lys71A, a feature not present in other Arrangement R enzymes. The cysteamine moiety amide, however, still binds in the same way as in the other enzymes. The catalytic residue is Asp44B. Interestingly, it appears involved in two hydrogen bonds, one to each oxygen of the side chain, from Thr61B and Gly37A — neither of which is a basic residue.
Acot7 is interesting in that it has a double hot dog fold. For Acot7, it is a tyrosine (Tyr290) that hydrogen bonds with catalytic Asp213.
Another DHD acyl-CoA thioesterase, E. coli thioesterase II (TE-II), or TesB, was the first double hot dog enzyme of any kind discovered.76 It has a distinct β1–αHD loop from other members with Arrangement Ψ or R, such that the conserved asparagine/glutamine residue has no structural equivalent in TE-II (Figure 13F, Table 4). The N-terminal helix region, pantetheine-binding region, and catalytic aspartate (Asp204), however, are structurally conserved. The partner for Asp204 is Gln278, found on the “bun”. Gln278 is also hydrogen bonded to Thr228. Thr228 corresponds to Ser83A of THEM2 (Table 4), and, as in THEM2, it is found in the structure to bind to the supposed nucleophilic water along with Asp204. Site-directed mutagenesis has confirmed the importance of residues Asp204, Gln278, and Thr228 to TE-II catalysis.76
3.2.4. Other Active Site Arrangements
Until recently, it was thought that all hot dog fold thioesterases fell into the two arrangements described above; however, more active site arrangements have been discovered — and with them further mechanistic proposals.
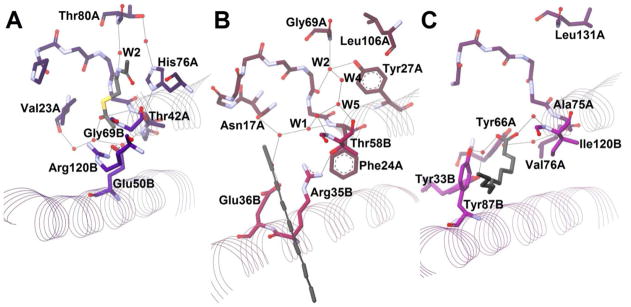
In 2010, two separate groups released a large collection of structures of S. cattleya FlK, a TE involved in the detoxification pathway of fluoroacetyl-CoA.77,78 While the residues of subunit B superimpose well with Arrangement R, subunit A is very dissimilar (Figure 14A). Loop β1–αHD has a modified structure, and it contains neither the aspartate of Arrangement Ψ nor the glutamine/asparagine of Arrangement R, instead having a valine (Val23A) at that site. The oxyanion still appears to be at the tip of the hot dog, but it is neither an aromatic residue (as in Arrangement Ψ) nor a glycine (as in Arrangement R), being instead a threonine (Thr42A). W2 is still present, but W3 is not, and indeed, the pantetheine portion of the substrate seems to bind somewhat differently in structures where it is present. In this arrangement, Glu50B (corresponding to Glu73B of 4HBT-II) does not appear to act as catalytic base and instead seems involved in some other, as of yet undetermined, function. Instead a new triad is present of water–His76A–Thr42A. Thus, Thr42A has a dual function; its side chain hydroxyl group acts as catalytic nucleophile (Scheme 6C), while its backbone amide helps stabilize the oxyanion. Great effort has gone into the attempt to determine how FlK can have such a strong preference for fluoroacetyl-CoA over acetyl-CoA. Gly69B, (which in the other enzymes would have served as the binding site for the pantetheinyl amide nitrogen through its carbonyl,) and Arg120B are thought to interact with the polar C–F bond.
Figure 14. Further Hot Dog Thioesterase Active Site Arrangements.
(A) The active site of S. cattleya FlK (PDB#3KVU), an example of an SHD TE, in plum, including acetyl-CoA shown in black. Only the N-acetylcysteamine portion of CoA has been drawn for the purpose of figure clarity. (B) The active site of M. chernisa DynE7 (PDB#2XEM), another SHD TE, in magenta, including the proposed product trans-3,5,7,9,11,13-pentadecahexaen-2-one (5) shown in black. (C) The active site of M. tuberculosis FcoT (PDB#2PFC), another SHD TE, in purple, including the product trans-dodecenoate shown in black.
Two closely related enzymes involved in enediyne biosynthesis were crystallized in 200979 and 2010.80 CalE7 and DynE7 are thioesterases thought to act in a proofreading fashion. Both enzymes act on acyl-carrier-protein–bound substrates instead of CoA-bound ones. Subunit A of these enzymes superimposes well with Arrangement Ψ. The oxyanion residue is also aromatic, as in Arrangement Ψ. The aspartate in loop β1–αHD, however, is replaced by an asparagine (Asn17A in DynE7, Figure 14B), as in Arrangement R (Table 4). W2 is bound to a tyrosine (Tyr27A in DynE7, Figure 14B) as in both 4HBTs. However, the conserved aspartate/glutamate in subunit B of the other thioesterases is replaced by a glycine (Table 4). Instead, three residues downstream, an arginine (Arg35B in DynE7) is present (Figure 14B). This arginine has been shown to be crucial for catalytic activity in each of these enzymes. The authors of the papers argue for its function as an oxyanion stabilizer based on computer modeling studies and the rationale that arginine does not have the proper predicted pH in its environment to act as catalytic base. However, they are the only authors not to invoke the polarized hot dog helix in their mechanism. Furthermore, if the pantetheine-binding site is the same as is usual, as appears to be the case, this implies that the thioester carbonyl would be delivered to the same locale. In the apo structure of DynE7, W1 is present and in the usual location of the oxyanion hole between Phe24A and Asn17A; in the product structure, on the other hand, while W1 is indeed still present, Asn17A has twisted out of its usual hydrogen bonds with loop β1–αHD (Figure 14B). Without an obvious base or nucleophile, they suggest hydroxide from the solvent may perform hydrolysis (Scheme 6D), since higher pH favors product formation,79 but they concede that more research is needed to find the answer.
Interpretation of the crystal data is further complicated by the fact that the cocrystal, trans-3,5,7,9,11,13-pentadecahexaen-2-one (5), arises not as the result of a thioester hydrolysis alone but of hydrolysis followed by decarboxylation. CalE7 and DynE7 may each perform a dual function of thioesterase/decarboxylase (Scheme 8). Whether the second reaction is catalyzed or spontaneous is not known.
Scheme 8.

Hydrolysis and Decarboxylation Leading to Compound 5.
A fifth active site arrangement is found in the enzyme fatty acyl-CoA thioesterase (FcoT, Rv0098) from M. tuberculosis.81,82 Like TE-II, FcoT, a long-chain fatty acid TE, has a β1–αHD region that does not superimpose with the common loop found in the TE group. On this loop region is Tyr66A, which was found necessary for catalysis. Two other tyrosines thought to be involved in catalysis are found on subunit B, Tyr33B and Tyr87B (Figure 14C). These residues may be involved in the activation of water as a nucleophile (akin to Scheme 6B but with a phenol or phenolate acting as base in place of the carboxylate). The N-terminal helix residue for this enzyme is uniquely an alanine (Ala75A, Table 4), and W1 is not present. The initial crystal structure included dodecenoate, but the carboxylate is not near the site of the usual oxyanion hole. If this is an accurate picture of substrate binding, it would appear that the usual oxyanion hole has shifted, yet one might still argue that polarization of αHD still plays a key role via the water network from Val76A. W2 and W3 are absent, but this may simply be an issue of poor resolution (2.3 Å). On the other hand, the backbone carbonyl that usually binds W3 is also absent, so perhaps even pantetheine-binding differs in this enzyme. As in the enediyne enzymes, the conserved glutamate/aspartate of subunit B is absent, replaced by Asn83B (Table 4), which does not seem to play an important role.
A homology model of DHD plant acyl-ACP thioesterase FatB from Arabidopsis thaliana suggests a sixth active site arrangement,83 but further evidence is needed to confirm this.
Finally, in 2010, Moriguchi et al. showed that what was thought by homology to be a DH domain within an iterative PKS 6-methylsalicylic acid synthase (MSAS) in actuality functions as the first example of an internal TE domain.84 No structure has yet been solved of this domain, but sequence analysis may suggest an active site arrangement similar to DH/H Arrangement A but with slight differences. For one, the hot dog glycine has been replaced by alanine (Ala982), as in the curacin PKS DHs (Table 2). There is also no glutamine or histidine four residues downstream from the key aspartate/glutamate; instead, a tryptophan is found at that position (Trp1133). One can easily imagine a mechanism in which histidine could act as general base (akin to Scheme 6B but with an imidazole acting as base in place of the carboxylate) with the conserved oxyanion hole, but more work remains to confirm this hypothesis.
Thus, unlike the case of the hot dog fold dehydratases/hydratases, where the two differing active site arrangements seem to share the same mechanism, all five observed arrangements for the hot dog thioesterases — and two postulated ones — seem to have distinct mechanisms from each other, yet they all benefit from the polarization of αHD to universally stabilize the transition state and reaction intermediates.
4. Hot Dog Enzymes with Other Functions
There have been numerous other enzymes within the hot dog family for which function has not yet been assigned, only one example being the enzyme Rv0216 from M. tuberculosis. Similar to how the internal TE within MSAS mentioned above was at first thought to be a DH, Rv0216 appeared from sequence comparisons to be a DHD hydratase; however, it was found to be inactive with crotonyl-CoA. The enzyme has been crystallized and its structure determined, yet its function still remains unknown.85 Residues in Rv0216 corresponding to catalytic Asp808 and Asn810 in C. tropicalis MFE-2 hydratase 2 domain (Figure 5C) are Asn232 and Ala234. However, the histidine of the hydratases remains conserved, as does the orientation of αHD.
FapR is a regulatory protein involved in lipid biosynthesis by repressing DNA transcription of several genes involved in fatty acid production. Excellent work by Schujman, et al. has shown that binding of malonyl-CoA by B. subtilis FapR triggers a conformational change in the protein that causes it to no longer bind to DNA and inhibit transcription. FapR was found to consist of an N-terminal DNA-binding region, an α-helical linker region, and a C-terminal hot dog domain. The dimerization of the SHD domain forms the core of the functional protein. In the absence of malonyl-CoA, the α-helical linkers from each monomer also dimerize, and this brings the N-terminal domains close together for DNA binding. The two malonyl-CoA–binding sites are at the SHD dimer interface, and the sites share many similarities with Arrangement R of hot dog thioesterases and also make use of a similar binding mode for the pantetheinyl portion of CoA. However, the conserved asparagine/glutamine of loop β1–αHD is replaced by Phe99A and the catalytic aspartate/glutamate by Asn115B. Crystal structures (Figure 15) show that Asn115B binds to the thioester carbonyl, and a salt bridge between Arg106 and the malonyl carboxylate may trigger loop conformational changes that drive the α-helical linkers away from each other, preventing DNA binding and leading ultimately to fatty acid anabolism in the presence of excess malonyl-CoA within the cell.86 In FapR, αHD does not polarize the thioester carbonyl; instead, its positive character serves as a binding region for the malonyl carboxylate.
Figure 15. FapR.
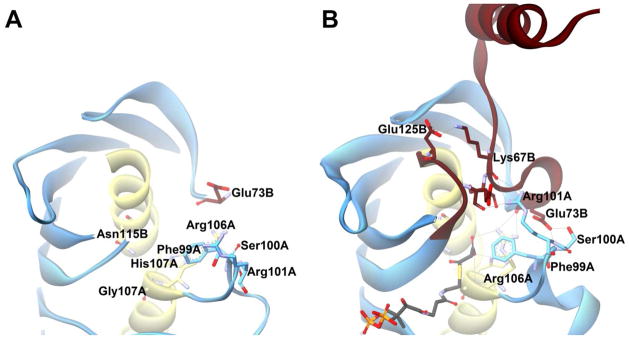
(A) The “thioesterase-like” domain of B. subtilis FabR in the apo form (PDB#2F41) in pale blue with the αHDs colored in pale yellow. Key active site residues are shown as sticks and colored by element. (B) The α-helical linker and “thioesterase-like” domains of FapR with malonyl-CoA (shown in black) bound. Regions colored in red are those that were unordered in the apo form. Potential hydrogen bonds are shown with grey lines.
Not all hot dog proteins were predicted to have this fold before their structures were determined. Cj0977 from food pathogen Campylobacter jejuni was known to be a virulence protein, but its function was unknown. Upon structural determination, it was discovered to be a homodimeric SHD protein with some similarities to FapR, including sharing an asparagine (Asn79) with that protein, but lacking the typical thioesterase residues. The binding site for the pantetheinyl group, however, does appear to be the same as in the thioesterases. Some mutations within the predicted binding pocket (Q83A and F70A) have eliminated virulence. The current hypothesis is that the C-terminal region of Cj0977 undergoes a structural change upon binding of some unknown acyl-CoA substrate, which allows the protein to bind to some other unknown protein in the virulence pathway, perhaps one involved in regulation.87
Another surprising addition to the hot dog family is the “product template” (PT) domain88 from the nonreducing iterative PKS (NR-PKS) of the aflatoxin biosynthetic pathway, which was found to have a DHD fold. The PT domain is responsible for two consecutive carbon–carbon cyclizations by intramolecular aldol condensations between C4 and C9 and between C2 and C11 of a 20-carbon linear polyketide substrate (Scheme 9). At first, the active site appeared similar to DH Arrangement A, having the conserved histidine, aspartate, and glutamine. W2 is also still present. On closer examination, however, significant differences were observed. As mentioned above, in both dehydratases and hydratases, the histidine is polarized by hydrogen bonding to the backbone carbonyl oxygen of a residue located downstream on the same loop. In PksA PT, the histidine (His1345) instead has rotated, and its polarization derives from a different source, a hydrogen bond to the conserved aspartate (Asp1543), meaning it functions as part of a true dyad. Perhaps more surprising is that the conserved glycine of the entire DH/H group is a proline (Pro1355), which eliminates the possibility of PksA PT sharing the same oxyanion hole with the DHs and hydratases. A complicated water network, still polarized by αHD, is proposed to serve as the new oxyanion hole for the reaction (Figure 16). The altered orientation of His1345 initially positions the basic nitrogen toward C4 for the first reaction, instead of C2, as in the case of the DHs and hydratases, while carbonyl 9 occupies the oxyanion hole. After first ring cyclization, the substrate must reorient so that the same catalytic machinery can operate on C2 and carbonyl 11 for the second cyclization.89
Scheme 9.
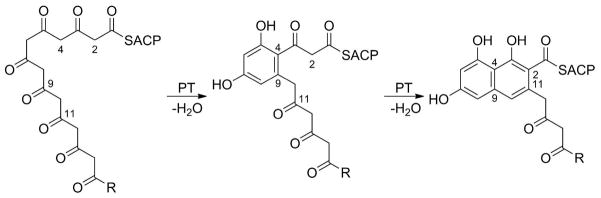
Reaction Catalyzed by the PksA “Product Template” Domain (R = C5H11).
Figure 16. Comparison Between Hot Dog Dehydratase/Hydratase and PT Domain Active Sites.

Potential hydrogen bonds are shown with grey lines, red spheres represent water molecules, and residues are shown as sticks and colored by element. The beginnings of the hot dog helices are shown with faint line ribbons. The images are drawn to scale and superimposable. (A) The active site of E. coli FabA (PDB#1MKB), an example of the dehydratase/hydratase group. The side chain of residues 76 is not shown for the purpose of figure clarity. (B) The active site of A. parasiticus PksA PT domain (PDB#3HRR). The side chain of residue 1351 is not shown for the purpose of figure clarity. Note the different polarization of histidine and the replacement of glycine with proline at the N-terminal end of the hot dog helix.
Not all PT domains from NR-PKSs perform the same cyclizations. Homologous PT domains from other systems are known to initially close rings between C2 and C7 or between C6 and C11.90 How different PTs direct different cyclization patterns is currently not known.
Also of interest is the unique dimerization of the PT domain. In mammalian type I FAS systems, the dimerization of the DH domains makes up the central core of the “modifying portion” of the megasynthase.35 In similar manner, the DH domains of modular PKSs and the PT domains of NR-PKSs likewise are thought to act as part of the core for these systems. However, the dimerization interfaces of these domains differ substantially; mammalian FAS and modular PKS DHs can be considered variations of side-by-side dimers, whereas the PTs are head-to-head dimers. More importantly, the attachment points for the linkers do not match. In the FAS and modular PKS DHs, the N-terminal linkers meet together near the dimer interface and lead to the “condensing portion” of the enzyme. The C-terminal linkers extend from the other side of the DH in opposite directions. In PksA PT, it is the C-terminal ends that are together at the dimer interface, whereas, the N-termini extend in opposite directions from the sides of the domain (Figure 17). Thus, it seems that, despite obvious structural similarity among their domains, the core architecture of the various FAS and PKS megasynthases may well differ.
Figure 17. Comparison of Dimerization Modes Among Megasynthase Core Domains.
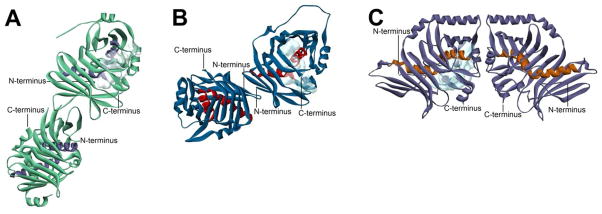
Solid ribbon representations of the core domains from three distinct classes of megasynthase with termini indicated and active site volumes from one monomer shown with a light blue cloud. The images are drawn to scale and superimposable at the monomers with the active site volume shown. (A) L. majuscule CurK DH domain (PDB#3KG9), an example of a modular PKS core, showing a side-by-side dimeric arrangement. (This domain is given the oligomeric state designation of DdhC in Table 1.) (B) S. scrofa FAS DH domain (PDB#2VZ8), an example of a mammalian type I FAS core, showing a tilted side-by-side dimeric arrangement. (This domain is given the oligomeric state designation of DdhD in Table 1.) (C) A. parasiticus PksA PT domain (PDB#3HRQ), an example of a nonreducing IPKS core, showing a unique head-to-head dimeric arrangement. (This domain is given the oligomeric state designation of DdhE in Table 1.)
5. Summary and Outlook
The hot dog fold has proven a versatile scaffold for reactions involving thioester substrates. In nearly every case, part of the enzyme secondary structure — a linear hot dog helix — is responsible for polarizing a carbonyl and/or stabilizing an oxyanion formed in the course of the reaction. Since the 1970s, it has been known that α-helixes bear a considerable dipole moment.91 Because the dipoles of each amino acid residue in the helix are nearly parallel to the helix axis, the dipoles are almost additive; a 10-residue helix may thus bear a dipole moment of greater than 30 Debye (D). Moreover, this calculation ignores the effect of further polarization from hydrogen bonding within the helix.92,93 It is estimated that this dipole confers the effect of between 0.5 and 0.75 of a isolated charge to each end of the helix. Helices of seven residues (two turns) generate an electric field with a potential of 0.5 V at a distance of 5 Å from the end of the helix, which would supply a binding energy contribution of ~12 kcal/mol to a bound ion of opposite charge at that distance.93 It is no surprise then, to find that helices have evolved to function in a catalytic manner as in the hot dog fold. It may also be noted that in the DHD enzymes, the helix no longer being used for catalysis is “bent” or shortened, which would disrupt or weaken its strong dipole moment, as it is no longer needed to generate an electric potential in a vestigial active site.
Within the hot dog superfamily are found excellent examples of both convergent and divergent evolution. In the case of the dehydratase/hydratase group, two distinct active site arrangements of amino acids have convergently evolved to perform a reaction in the forward or reverse direction from a spatially identical arrangement of heteroatoms in space. In the case of the thioesterase group, at least five varying active site arrangements have diverged to accommodate thioester hydrolysis of a wide range of substrate types. Not being limited to these reaction types, the list of hot dog proteins with novel functions — be they isomerizations, regulatory functions, or ring closings — continues to grow.
Acknowledgments
We thank A. G. Newman, Katherine Belecki, and A. L. Vagstad for critical reading of the manuscript and helpful suggestions, Prof. K. Weissman for exquisitely informed comments for preparation of the final version, and the NIH for financial support (ES001670).
Biographies
 Jason W. Labonte was born in Maine and earned his B.S. degree in Biochemistry from Grove City College, PA. He received his Ph.D. under Professor Craig A. Townsend at Johns Hopkins University. Beginning his graduate career as a synthetic chemist studying the biosynthesis of clavulanic acid, he progressively grew more interested in computational modeling of active sites, focusing particularly on the active sites of the product template and thioesterase domains of polyketide synthases. He is currently a postdoctoral fellow with Professor Jeffrey J. Gray, writing computational algorithms for the determination of fusion protein structures with the Rosetta modeling suite. Labonte is an avid backpacker, having summited several volcanoes in Ecuador and spending multiple weekends on the nearby Appalachian Trail. When not coding or hiking, he also enjoys learning dead languages or reading philosophy. Labonte lives in Baltimore City with his wife, an artist.
Jason W. Labonte was born in Maine and earned his B.S. degree in Biochemistry from Grove City College, PA. He received his Ph.D. under Professor Craig A. Townsend at Johns Hopkins University. Beginning his graduate career as a synthetic chemist studying the biosynthesis of clavulanic acid, he progressively grew more interested in computational modeling of active sites, focusing particularly on the active sites of the product template and thioesterase domains of polyketide synthases. He is currently a postdoctoral fellow with Professor Jeffrey J. Gray, writing computational algorithms for the determination of fusion protein structures with the Rosetta modeling suite. Labonte is an avid backpacker, having summited several volcanoes in Ecuador and spending multiple weekends on the nearby Appalachian Trail. When not coding or hiking, he also enjoys learning dead languages or reading philosophy. Labonte lives in Baltimore City with his wife, an artist.
 Craig A. Townsend is the Alsoph H. Corwin Professor of Chemistry and holds joint appointments in the Departments of Biology and Biophysics. He was born in Chicago and received a B.A. with Honors from Williams College and his Ph.D. in organic chemistry from Yale University where he worked with A. Ian Scott on the biosynthesis of vitamin B12. He was then an International Exchange Postdoctoral Fellow of the Swiss National Science Foundation in the laboratory of Duilio Arigoni at the ETH in Zurich and joined the faculty at Johns Hopkins in 1976. His research interests are broadly in the chemistry of natural products and the interface of organic chemistry, biology and medicine: synthetic, stereochemical and mechanistic studies of principal biosynthetic enzymes and their engineering for synthetic ends; chemoenzymatic synthesis; enzymology and molecular biology of polyketides and beta-lactam antibiotics; drug design and the clinical applications of fatty acid synthesis inhibition in the treatment of cancer and infectious diseases, and targets of lipid metabolism for metabolic diseases and obesity. When not working with his diversely interested and interesting research group, he enjoys travel, running, swimming and the visual arts.
Craig A. Townsend is the Alsoph H. Corwin Professor of Chemistry and holds joint appointments in the Departments of Biology and Biophysics. He was born in Chicago and received a B.A. with Honors from Williams College and his Ph.D. in organic chemistry from Yale University where he worked with A. Ian Scott on the biosynthesis of vitamin B12. He was then an International Exchange Postdoctoral Fellow of the Swiss National Science Foundation in the laboratory of Duilio Arigoni at the ETH in Zurich and joined the faculty at Johns Hopkins in 1976. His research interests are broadly in the chemistry of natural products and the interface of organic chemistry, biology and medicine: synthetic, stereochemical and mechanistic studies of principal biosynthetic enzymes and their engineering for synthetic ends; chemoenzymatic synthesis; enzymology and molecular biology of polyketides and beta-lactam antibiotics; drug design and the clinical applications of fatty acid synthesis inhibition in the treatment of cancer and infectious diseases, and targets of lipid metabolism for metabolic diseases and obesity. When not working with his diversely interested and interesting research group, he enjoys travel, running, swimming and the visual arts.
References
- 1.Leesong M, Henderson BS, Gillig JR, Schwab JM, Smith JL. Structure. 1996;4:253. doi: 10.1016/s0969-2126(96)00030-5. [DOI] [PubMed] [Google Scholar]
- 2.Pidugu LS, Maity K, Ramaswamy K, Surolia N, Suguna K. BMC Struct Biol. 2009;9:37. doi: 10.1186/1472-6807-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenni S, Leibundgut M, Boehringer D, Frick C, Mikolasek B, Ban N. Science. 2007;316:254. doi: 10.1126/science.1138248. [DOI] [PubMed] [Google Scholar]
- 4.Leibundgut M, Jenni S, Frick C, Ban N. Science. 2007;316:288. doi: 10.1126/science.1138249. [DOI] [PubMed] [Google Scholar]
- 5.Dillon SC, Bateman A. BMC Bioinf. 2004;5:109. doi: 10.1186/1471-2105-5-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wakil SJ. Biochemistry. 1989;28:4523. doi: 10.1021/bi00437a001. [DOI] [PubMed] [Google Scholar]
- 7.Rando RR, Bloch K. J Biol Chem. 1968;243:5627. [PubMed] [Google Scholar]
- 8.Sedgwick B, Morris C, French SJ. J Chem Soc Chem Commun. 1978;5:193. [Google Scholar]
- 9.Schwab JM, Habib A, Klassen JB. J Am Chem Soc. 1986;108:5304. [Google Scholar]
- 10.Hanson KR, Rose IA. Acc Chem Res. 1975;8:1. [Google Scholar]
- 11.Staunton J, Weissman KJ. Nat Prod Rep. 2001;18:380. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 12.Cox RJ. Org Biomol Chem. 2007;5:2010. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]
- 13.Vergnolle O, Hahn F, Baerga-Ortiz A, Leadlay PF, Andexer JN. Chem Bio Chem. 2011;12:1011. doi: 10.1002/cbic.201100011. [DOI] [PubMed] [Google Scholar]
- 14.Kwan DH, Schulz F. Molecules. 2011;16:6092. doi: 10.3390/molecules16076092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. Biochemistry. 2003;42:72. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]
- 16.Palaniappan N, Alhamadsheh MM, Reynolds KA. J Am Chem Soc. 2008;130:12236. doi: 10.1021/ja8044162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Zaleski TJ, Valenzano C, Khosla C, Cane DE. J Am Chem Soc. 2005;127:17393. doi: 10.1021/ja055672+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keatinge-Clay A. J Mol Biol. 2008;384:941. doi: 10.1016/j.jmb.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alhamadsheh MM, Palaniappan N, Daschouduri S, Reynolds KA. J Am Chem Soc. 2007;129:1910. doi: 10.1021/ja068818t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hisano T, Tsuge T, Fukui T, Iwata T, Miki K, Doi Y. J Biol Chem. 2003;278:617. doi: 10.1074/jbc.M205484200. [DOI] [PubMed] [Google Scholar]
- 21.Wu WJ, Anderson VE, Raleigh DP, Tonge PJ. Biochemistry. 1997;36:2211. doi: 10.1021/bi962549+. [DOI] [PubMed] [Google Scholar]
- 22.Bahnson BJ, Anderson VE. Biochemistry. 1991;30:5894. doi: 10.1021/bi00238a013. [DOI] [PubMed] [Google Scholar]
- 23.Kimber MS, Martin F, Lu Y, Houston S, Vedadi M, Dharamsi A, Fiebig KM, Schmid M, Rock CO. J Biol Chem. 2004;279:52593. doi: 10.1074/jbc.M408105200. [DOI] [PubMed] [Google Scholar]
- 24.Kostrewa D, Winkler FK, Folkers G, Scapozza L, Perozzo R. Protein Sci. 2005;14:1570. doi: 10.1110/ps.051373005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swarnamukhi PL, Sharma SK, Bajaj P, Surolia N, Surolia A, Suguna K. FEBS Lett. 2006;580:2653. doi: 10.1016/j.febslet.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 26.Swarnamukhi PL, Sharma SK, Padala P, Surolia N, Surolia A, Suguna K. Acta Crystallogr D Biol Crystallogr. 2007;63:458. doi: 10.1107/S0907444907003228. [DOI] [PubMed] [Google Scholar]
- 27.Maity K, Venkata BS, Kapoor N, Surolia N, Surolia A, Suguna K. J Struct Biol. 2011;176:238. doi: 10.1016/j.jsb.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Liu W, Hu T, Du L, Luo C, Chen K, Shen X, Jiang H. J Biol Chem. 2008;283:5370. doi: 10.1074/jbc.M705566200. [DOI] [PubMed] [Google Scholar]
- 29.Kong YH, Zhang L, Yang ZY, Han C, Hu LH, Jiang HL, Shen X. Acta Pharmacol Sin. 2008;29:870. doi: 10.1111/j.1745-7254.2008.00808.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Kong Y, Wu D, Zhang H, Wu J, Chen J, Ding J, Hu L, Jiang H, Shen X. Protein Sci. 2008;17:1971. doi: 10.1110/ps.036186.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, Zhang L, Zhang Y, Zhang H, Du J, Ding J, Guo Y, Jiang H, Shen X. BMC Microbiol. 2009;9:91. doi: 10.1186/1471-2180-9-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He L, Zhang L, Liu X, Li X, Zheng M, Li H, Yu K, Chen K, Shen X, Jiang H, Liu H. J Med Chem. 2009;52:2465. doi: 10.1021/jm8015602. [DOI] [PubMed] [Google Scholar]
- 33.Kirkpatrick AS, Yokoyama T, Choi KJ, Yeo H. Biochem Biophys Res Commun. 2009;380:407. doi: 10.1016/j.bbrc.2009.01.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akey DL, Razelun JR, Tehranisa J, Sherman DH, Gerwick WH, Smith JL. Structure. 2010;18:94. doi: 10.1016/j.str.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maier T, Leibundgut M, Ban N. Science. 2008;321:1315. doi: 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- 36.Lomakin IB, Xiong Y, Steitz TA. Cell. 2007;129:319. doi: 10.1016/j.cell.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 37.Koski MK, Haapalainen AM, Hiltunen JK, Glumoff T. J Biol Chem. 2004;279:24666. doi: 10.1074/jbc.M400293200. [DOI] [PubMed] [Google Scholar]
- 38.Koski MK, Haapalainen AM, Hiltunen JK, Glumoff T. J Mol Biol. 2005;345:1157. doi: 10.1016/j.jmb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 39.Haataja TJ, Koski MK, Hiltunen JK, Glumoff T. Biochem J. 2011;435:771. doi: 10.1042/BJ20101661. [DOI] [PubMed] [Google Scholar]
- 40.Annand RR, Kozlowski JF, Davisson VJ, Schwab JM. J Am Chem Soc. 1993;115:1088. [Google Scholar]
- 41.Haapalainen AM, Koski MK, Qin YM, Hiltunen JK, Glumoff T. Structure. 2003;11:87. doi: 10.1016/s0969-2126(02)00931-0. [DOI] [PubMed] [Google Scholar]
- 42.Koski MK, Haapalainen AM, Hiltunen JK, Glumoff T. Acta Crystallogr D Biol Crystallogr. 2003;59:1302. doi: 10.1107/s090744490300982x. [DOI] [PubMed] [Google Scholar]
- 43.Sacco E, Covarrubias AS, O’Hare HM, Carroll P, Eynard N, Jones TA, Parish T, Daffe M, Backbro K, Quemard A. Proc Natl Acad Sci USA. 2007;104:14628. doi: 10.1073/pnas.0704132104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sacco E, Legendre V, Laval F, Zerbib D, Montrozier H, Eynard N, Guilhot C, Daffe M, Quemard A. Biochim Biophys Acta. 2007;1774:303. doi: 10.1016/j.bbapap.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 45.Rock CO, Cronan JE. Biochim Biophys Acta. 1996;1302:1. doi: 10.1016/0005-2760(96)00056-2. [DOI] [PubMed] [Google Scholar]
- 46.Schwab JM, Henderson BS. Chem Rev. 1990;90:1203. [Google Scholar]
- 47.Schwab JM, Ho CK, Li WB, Townsend CA, Salituro GM. J Am Chem Soc. 1986;108:5309. [Google Scholar]
- 48.Schwab JM, Klassen JB, Habib A. J Chem Soc Chem Commun. 1986;4:357. [Google Scholar]
- 49.Caffrey P. Chem Bio Chem. 2003;4:654. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 50.Reeves CD, Hu Z, Reid R, Kealey JT. Appl Environ Microbiol. 2008;74:5121. doi: 10.1128/AEM.00478-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwab JM, Klassen JB. J Am Chem Soc. 1984;106:7217. [Google Scholar]
- 52.Benning MM, Wesenberg G, Liu R, Taylor KL, Dunaway-Mariano D, Holden HM. J Biol Chem. 1998;273:33572. doi: 10.1074/jbc.273.50.33572. [DOI] [PubMed] [Google Scholar]
- 53.Holmquist M. Curr Protein Pept Sci. 2000;1:209. doi: 10.2174/1389203003381405. [DOI] [PubMed] [Google Scholar]
- 54.Thoden JB, Holden HM, Zhuang Z, Dunaway-Mariano D. J Biol Chem. 2002;277:27468. doi: 10.1074/jbc.M203904200. [DOI] [PubMed] [Google Scholar]
- 55.Zhuang Z, Song F, Zhang W, Taylor K, Archambault A, Dunaway-Mariano D, Dong J, Carey PR. Biochemistry. 2002;41:11152. doi: 10.1021/bi0262303. [DOI] [PubMed] [Google Scholar]
- 56.Song F, Zhuang Z, Dunaway-Mariano D. Bioorg Chem. 2007;35:1. doi: 10.1016/j.bioorg.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 57.Zhuang Z, Latham J, Song F, Zhang W, Trujillo M, Dunaway-Mariano D. Biochemistry. 2011;51:786. doi: 10.1021/bi2013917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thoden JB, Zhuang Z, Dunaway-Mariano D, Holden HM. J Biol Chem. 2003;278:43709. doi: 10.1074/jbc.M308198200. [DOI] [PubMed] [Google Scholar]
- 59.Angelini A, Cendron L, Goncalves S, Zanotti G, Terradot L. Proteins. 2008;72:1212. doi: 10.1002/prot.22014. [DOI] [PubMed] [Google Scholar]
- 60.Zhuang Z, Song F, Martin BM, Dunaway-Mariano D. FEBS Lett. 2002;516:161. doi: 10.1016/s0014-5793(02)02533-4. [DOI] [PubMed] [Google Scholar]
- 61.Zhuang Z, Song F, Takami H, Dunaway-Mariano D. J Bacteriol. 2004;186:393. doi: 10.1128/JB.186.2.393-399.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ismail W. Arch Microbiol. 2008;190:451. doi: 10.1007/s00203-008-0393-3. [DOI] [PubMed] [Google Scholar]
- 63.Cao J, Xu H, Zhao H, Gong W, Dunaway-Mariano D. Biochemistry. 2009;48:1293. doi: 10.1021/bi801879z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dong J, Zhuang ZH, Song F, Dunaway-Mariano D, Carey PR. J Raman Spectrosc. 2012;43:65. doi: 10.1002/jrs.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen D, Wu R, Bryan TL, Dunaway-Mariano D. Biochemistry. 2009;48:511. doi: 10.1021/bi802207t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Badger J, Sauder JM, Adams JM, Antonysamy S, Bain K, Bergseid MG, Buchanan SG, Buchanan MD, Batiyenko Y, Christopher JA, Emtage S, Eroshkina A, Feil I, Furlong EB, Gajiwala KS, Gao X, He D, Hendle J, Huber A, Hoda K, Kearins P, Kissinger C, Laubert B, Lewis HA, Lin J, Loomis K, Lorimer D, Louie G, Maletic M, Marsh CD, Miller I, Molinari J, Muller-Dieckmann HJ, Newman JM, Noland BW, Pagarigan B, Park F, Peat TS, Post KW, Radojicic S, Ramos A, Romero R, Rutter ME, Sanderson WE, Schwinn KD, Tresser J, Winhoven J, Wright TA, Wu L, Xu J, Harris TJ. Proteins. 2005;60:787. doi: 10.1002/prot.20541. [DOI] [PubMed] [Google Scholar]
- 67.Guo ZF, Sun Y, Zheng S, Guo Z. Biochemistry. 2009;48:1712. doi: 10.1021/bi802165x. [DOI] [PubMed] [Google Scholar]
- 68.Kunishima N, Asada Y, Sugahara M, Ishijima J, Nodake Y, Miyano M, Kuramitsu S, Yokoyama S. J Mol Biol. 2005;352:212. doi: 10.1016/j.jmb.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 69.Song F, Zhuang Z, Finci L, Dunaway-Mariano D, Kniewel R, Buglino JA, Solorzano V, Wu J, Lima CD. J Biol Chem. 2006;281:11028. doi: 10.1074/jbc.M513896200. [DOI] [PubMed] [Google Scholar]
- 70.Cheng Z, Song F, Shan X, Wei Z, Wang Y, Dunaway-Mariano D, Gong W. Biochem Biophys Res Commun. 2006;349:172. doi: 10.1016/j.bbrc.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 71.Zhuang Z, Song F, Zhao H, Li L, Cao J, Eisenstein E, Herzberg O, Dunaway-Mariano D. Biochemistry. 2008;47:2789. doi: 10.1021/bi702334h. [DOI] [PubMed] [Google Scholar]
- 72.Willis MA, Zhuang Z, Song F, Howard A, Dunaway-Mariano D, Herzberg O. Biochemistry. 2008;47:2797. doi: 10.1021/bi702336d. [DOI] [PubMed] [Google Scholar]
- 73.Yokoyama T, Choi KJ, Bosch AM, Yeo HJ. Biochim Biophys Acta. 2009;1794:1073. doi: 10.1016/j.bbapap.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Serek R, Forwood JK, Hume DA, Martin JL, Kobe B. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:133. doi: 10.1107/S1744309106000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Forwood JK, Thakur AS, Guncar G, Marfori M, Mouradov D, Meng W, Robinson J, Huber T, Kellie S, Martin JL, Hume DA, Kobe B. Proc Natl Acad Sci USA. 2007;104:10382. doi: 10.1073/pnas.0700974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li J, Derewenda U, Dauter Z, Smith S, Derewenda ZS. Nat Struct Biol. 2000;7:555. doi: 10.1038/76776. [DOI] [PubMed] [Google Scholar]
- 77.Dias MV, Huang F, Chirgadze DY, Tosin M, Spiteller D, Dry EF, Leadlay PF, Spencer JB, Blundell TL. J Biol Chem. 2010;285:22495. doi: 10.1074/jbc.M110.107177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Weeks AM, Coyle SM, Jinek M, Doudna JA, Chang MC. Biochemistry. 2010;49:9269. doi: 10.1021/bi101102u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kotaka M, Kong R, Qureshi I, Ho QS, Sun H, Liew CW, Goh LP, Cheung P, Mu Y, Lescar J, Liang ZX. J Biol Chem. 2009;284:15739. doi: 10.1074/jbc.M809669200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liew CW, Sharff A, Kotaka M, Kong R, Sun H, Qureshi I, Bricogne G, Liang ZX, Lescar J. J Mol Biol. 2010;404:291. doi: 10.1016/j.jmb.2010.09.041. [DOI] [PubMed] [Google Scholar]
- 81.Wang F, Langley R, Gulten G, Wang L, Sacchettini JC. Chem Biol. 2007;14:543. doi: 10.1016/j.chembiol.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 82.Maity K, Bajaj P, Surolia N, Surolia A, Suguna K. J Biomol Struct Dyn. 2012;29:973. doi: 10.1080/07391102.2012.10507417. [DOI] [PubMed] [Google Scholar]
- 83.Mayer KM, Shanklin J. J Biol Chem. 2005;280:3621. doi: 10.1074/jbc.M411351200. [DOI] [PubMed] [Google Scholar]
- 84.Moriguchi T, Kezuka Y, Nonaka T, Ebizuka Y, Fujii I. J Biol Chem. 2010;285:15637. doi: 10.1074/jbc.M110.107391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Castell A, Johansson P, Unge T, Jones TA, Bäckbro K. Protein Sci. 2005;14:1850. doi: 10.1110/ps.051442305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schujman GE, Guerin M, Buschiazzo A, Schaeffer F, Llarrull LI, Reh G, Vila AJ, Alzari PM, de Mendoza D. EMBO J. 2006;25:4074. doi: 10.1038/sj.emboj.7601284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yokoyama T, Paek S, Ewing CP, Guerry P, Yeo HJ. J Mol Biol. 2008;384:364. doi: 10.1016/j.jmb.2008.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Crawford JM, Thomas PM, Scheerer JR, Vagstad AL, Kelleher NL, Townsend CA. Science. 2008;320:243. doi: 10.1126/science.1154711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Crawford JM, Korman TP, Labonte JW, Vagstad AL, Hill EA, Kamari-Bidkorpeh O, Tsai SC, Townsend CA. Nature. 2009;461:1139. doi: 10.1038/nature08475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li Y, Xu W, Tang Y. J Biol Chem. 2010;285:22764. doi: 10.1074/jbc.M110.128504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wada A. Adv Biophys. 1976;9:1. [PubMed] [Google Scholar]
- 92.Hol WGJ. Adv Biophys. 1985;19:133. doi: 10.1016/0065-227x(85)90053-x. [DOI] [PubMed] [Google Scholar]
- 93.Hol WGJ, van Duijnen PT, Berendsen HJC. Nature. 1978;273:443. doi: 10.1038/273443a0. [DOI] [PubMed] [Google Scholar]