

Abstract
Fibrocytes are mesenchymal cells that arise from monocyte precursors. They are present in injured organs and have both the inflammatory features of macrophages and the tissue remodelling properties of fibroblasts. Chronic inflammatory stimuli mediate the differentiation, trafficking and accumulation of these cells in fibrosing conditions associated with autoimmunity, cardiovascular disease and asthma. This Opinion article discusses the immunological mediators controlling fibrocyte differentiation and recruitment, describes the association of fibrocytes with chronic inflammatory diseases and compares the potential roles of fibrocytes in these disorders with those of macrophages and fibroblasts. It is hoped that this information prompts new opportunities for the study of these unique cells.
Fibrocytes are monocyte-derived cells that have features of both macrophages and fibroblasts. Although their biology has come under study only recently, the existence of a fibrocyte-like cell population was first proposed more than 150 years ago1. However, it was not until 1994 that the term ‘fibrocyte’ was first used to describe a circulating monocyte-derived cell that is capable of expressing a fibroblastic phenotype2. Fibrocytes were described as being unique in their co-expression of haematopoietic and progenitor cell markers (CD45 and CD34, respectively), together with the production of extracellular matrix (ECM) proteins. These cells were observed to adopt a spindle shape when adherent and were identified in wound exudates. Subsequent studies have greatly expanded our knowledge of the markers and functions attributed to this cell population3–5. Although these cells comprise only a small fraction of circulating leukocytes in normal humans, increased numbers of fibrocytes are present in human pathologies that are characterized by both chronic macrophage-driven inflammation and persistent fibroblast activation. Such disorders include pulmonary parenchymal and airway disease6–8, nephrogenic systemic fibrosis9, cardiovascular disease10, pulmonary hypertension11, autoimmune disorders12,13 and even normal ageing12. Furthermore, animal modelling implicates fibrocytes in the development of tissue fibrosis — the formation of excessive fibrous connective tissue that disrupts normal tissue function, a process in which fibroblasts (and their activated counterparts, myofibroblasts) have historically been considered the central cell type involved — including fibrosis involving the kidney14, liver15, heart16 and lungs17.
The functional relationship between fibrocytes and the related effector cell populations of macrophages and fibroblasts has not yet been fully explored. This question is important because the potential for overlap in the identification and function of these different cells and a lack of understanding of the subtle differences between them might impede a full understanding of the role of fibrocytes in chronic inflammation. To explore this issue, a basic understanding of macrophages and fibroblasts in the context of chronic inflammation is required.
A paradigm has emerged suggesting that macrophage-driven inflammation contributes to both tissue injury and repair18,19. In this model, ‘classically activated’ macrophages promote tissue injury through their secretion of pro-inflammatory mediators and reactive oxygen species (ROS), whereas ‘alternatively activated’ macrophages dampen inflammation and promote wound healing, in part through the recruitment and activation of fibroblasts19. Fibroblasts have organ-specific functions in promoting tissue homeostasis, including ECM and cytokine production20. In addition, in some settings fibroblasts express α-smooth muscle actin (αSMA) and have wound contractile and repair properties; these cells are known as activated myofibroblasts and are traditionally considered to be the ultimate effector cells in diverse forms of tissue remodelling20 (FIG. 1).
Figure 1. Tissue injury, repair and remodelling.
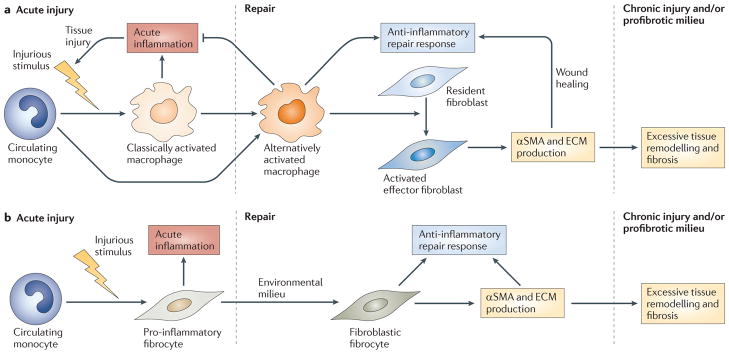
a | Current models suggest that in response to injurious stimuli, classically activated macrophages infiltrate diseased organs and mediate a programme of acute inflammation. As injury ceases and repair begins, the macrophage phenotype shifts towards that of alternative activation to dampen inflammation and promote repair. These macrophages stimulate resident fibroblasts to adopt an activated effector state characterized by the expression of α-smooth muscle actin (αSMA) and enhanced extracellular matrix (ECM) production. In the setting of severe or persistent injury, or a profibrotic milieu, this response shifts towards excessive remodelling and fibrosis. b | This model of many cells acting together is contradicted by the finding that fibrocytes have properties of both macrophages and fibroblasts. Thus, an alternative model of repair is proposed in which fibrocytes traffic to injured organs, where they participate in the inflammatory events that are also attributed to macrophages. As damage subsides, fibrocytes respond to local cues to downregulate their inflammatory responses and adopt a fibroblastic phenotype to promote repair and, in some pathological conditions, remodelling and fibrosis.
The discovery of fibrocytes adds to this paradigm by suggesting that, rather than resulting solely from two discrete populations (macrophages and fibroblasts) acting together, tissue repair and remodelling might also be influenced by a highly plastic cell population of fibrocytes (FIG. 1), with the ability to adopt the phenotype of macrophages or fibroblasts. To better understand the validity of this hypothesis, and to highlight the potential role of fibrocytes in human pathology, this Opinion article compares fibrocyte biology with that of macrophages and fibroblasts in chronic inflammatory settings. The utility of various approaches that are used to distinguish fibrocytes from macrophages and fibroblasts is presented, together with a discussion of the pathways that control fibrocyte accumulation and function in the normal host. The proposed role of fibrocytes is then compared with that of macrophages and/or fibroblasts in three common chronic inflammatory diseases: autoimmunity, cardiovascular disease and asthma. Other important areas of interest such as renal fibrosis and wound healing will not be covered in this brief Opinion article. We conclude with perspectives and suggestions of areas for future study in this new field.
Identification of fibrocytes
For several years, the minimum criteria for identifying fibrocytes in culture, tissue sections or the circulation were collagen production together with the expression of CD34 and/or CD45. However, this approach might be insufficient in some settings owing to the overlap in expression of these markers by fibrocytes, macrophages and fibroblasts5. Thus, additional assessments, including cell surface marker expression, morphology and ECM production might be required to accurately identify fibrocytes in their biological context. Specific factors that distinguish fibrocytes from macrophages and fibroblasts are detailed below and in TABLE 1.
Table 1.
Properties used to identify fibrocytes, macrophages and fibroblasts
| Property | Fibrocyte | Macrophage | Fibroblast | Refs |
|---|---|---|---|---|
| Morphology in sections or culture | Large, spindle-shaped when adherent | Large, rounded | Large, spindle-shaped | 2,5* |
| Functions | ||||
| Cytokine production | ++ | ++ | + | 4,21,27 |
| Immune cell trafficking | ++ | ++ | + | 4,32 |
| ECM production | ++ | ± | ++ | 4,5,24 |
| αSMA production | + | − | ++ | 2 |
| Lipid metabolism | + | + | − | 27 |
| Antigen presentation | ++ | ++ | − | 3,22,27 |
| Angiogenesis | + | + | + | 46 |
| MMP production | + | + | + | 45 |
| Chitinase production | + | + | − | 51 |
| Adhesion and motility markers | ||||
| CD9, CD11a, CD11b, CD11c, CD43, CD164, galectin 3, LSP1 | + or ++ | + or ++ | − | 2,5,87 |
| CD34 | + | − | − | 2,5 |
| CD29, CD44, CD81, ICAM1, CD49 complex, CD81 | + | + | + | 5,11,32 |
| Cell surface enzymes | ||||
| CD10, CD172a, CD45 | + | + | − | 2,5 |
| CD13, prolyl 4-hydroxylase | + | + | + | 2,5,88 |
| FAP | + | − | + | 5 |
| Scavenging receptors and molecules involved in host defence | ||||
| CD14, CD68, CD163, CD206, CD209, CD35, CD36 | ± | + | − | 5,11,12 |
| Fcγ receptors | ||||
| CD16, CD32a, CD32b, CD32c | + or ++ | + or ++ | − | 5,22 |
| Chemokine receptors | ||||
| CCR4, CCR5, CCR7, CXCR1, CXCR4, CX3CR1 | + or ++ | + | − | 5,25,89,90 |
| Cell-surface molecules involved in antigen presentation | ||||
| CD54, CD80, CD86, MHC class I and II | + | + | − | 3,22 |
| Extracellular matrix proteins | ||||
| Collagens I, III and IV, vimentin, tenascin | + | ± | ++ | 2,5,24 |
| Fibronectin, αSMA | ± | − | ++ | 2,5,15 |
| Collagen V | ++ | − | + | 24 |
| Glycosaminoglycans | ||||
| Perlecan, versican, hyaluronan | ++ or + | − | + | 5,24 |
| Decorin, biglycan | + | − | ++ | 24 |
| Miscellaneous | ||||
| Semaphorin 7A | + | + | − | 32 |
| CD115 | − | + | − | 5 |
| CD90 | ± | − | + | 5,63 |
| CD105 | + | + | + | 5 |
++ high level of expression; +, moderate level of expression; ±, conflicting reports or equivocal evidence of expression level; −, no expression; αSMA, α-smooth muscle actin; CCR, CC-chemokine receptor; CXCR, CXC-chemokine receptor; CX3CR1, CX3C-chemokine receptor 1; ECM, extracellular matrix; FAP, fibroblast activation protein; ICAM1, intercellular adhesion molecule 1; LSP1, lymphocyte-specific protein 1; MMP, matrix metalloproteinase.
The fibrocytes in REF. 5 were cultured in serum-free conditions; it is possible that fibrocytes arising in the presence of serum have other characteristics.
Fibrocyte markers
Consistent with their bone marrow origin, fibrocytes express several haematopoietic cell markers, including CD45 (REF. 2) and lymphocyte-specific protein 1 (LSP1)21. Like macrophages, fibrocytes express the adhesion proteins CD11b, CD11c and CD11d4; antigen-presenting factors such as MHC class I and II molecules, CD80 and CD86 (REF. 3); cell surface enzymes such as CD10 and CD13 (REF. 5); and in some circumstances scavenger receptors such as CD163 (REFS 5,22). Circulating and cultured fibrocytes also express CD34 (REF. 2), a protein that is often expressed by pluripotent cells. Expression of CD34 allows fibrocytes to be discriminated from macrophages and fibroblasts, which do not appreciably express CD34 (REFS 7,23). Like fibroblasts, fibrocytes express collagens4,23 and glycosaminoglycans24. However, compared with fibroblasts, fibrocytes have increased production of collagen V and lower levels of collagens I, III and IV. Similarly, the glycosaminoglycan profile of fibrocytes is distinct from that of fibroblasts, and is characterized by high-level expression of perlecan, versican and hyaluronan and low levels of biglycan and decorin24. This cell surface marker profile of fibrocytes would be expected to promote both basement membrane regrowth and inflammatory cell recruitment, rather than ongoing homeostatic effects, and is consistent with the proposed role of fibrocytes in repair.
Cell morphology
The co-expression of collagen and CD45 and/or CD34 accurately identifies fibrocytes in the peripheral blood (which contains neither mature macrophages nor fibroblasts)25. However, overlap in marker expression by these cell populations could result in the misclassification of fibrocytes in studies of cultured cells and tissue5. In this setting, morphological assessment becomes useful. Whereas macrophages and fibrocytes express similar inflammatory markers, both in vitro and in vivo, the spindle-shaped morphology adopted by adherent fibrocytes distinguishes them from macrophages, which are more spherical5. Similarly, whereas fibroblasts are also spindle shaped, they lack the haematopoietic cell and inflammatory markers that are expressed by fibrocytes. Thus, as shown in TABLE 1, the combination of a spindle-shaped adherent morphology, expression of CD45 (and/or CD34) and collagen production is sufficient for the identification of cultured or resident fibrocytes. FIGURE 2 shows representative electron micrograph and confocal microscopy images of tissue and circulating fibrocytes.
Figure 2. Characteristics of fibrocytes in tissues and in the circulation.
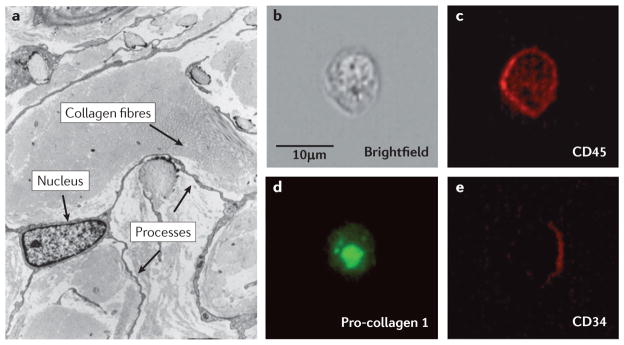
a | A representative electron micrograph of a CD34+ fibrocyte in the dermis of a patient with nephrogenic sclerosing fibrosis. The presence of a large nucleus, collagen fibres and extensive processes are indicated by the arrows. In three dimensions, this cell is spindle shaped. Image is reproduced, with permission, from REF. 91 © (2001) Lippincott Williams & Wilkins. b | A brightfield image of a fibrocyte obtained from the circulation of a normal human. c,d,e | Confocal images of this fibrocyte following staining for CD45 (bright red), intracellular pro-collagen I (green) and CD34 (dark red).
Fibrocyte origin and differentiation
Because fibrocyte outgrowth from peripheral blood mononuclear cells in vitro is enhanced by enriching for CD14+ cells, it is assumed that a subpopulation of monocytes gives rise to these cells. In this setting, the development of fibrocytes is regulated by signalling through the Fcγ receptor (FcγR)26 and is augmented by enriching monocytes for expression of pro-inflammatory markers such as CD11b, CD115 and GR1 (REF. 14). Fibrocytes can be isolated in vitro under serum-free or serum-containing conditions, although the presence of serum (such as might be encountered during in vivo injury) produces a more pro-inflammatory phenotype27. In vitro fibrocyte differentiation is augmented by fibrogenic cytokines such as interleukin-4 (IL-4) and IL-13, and is inhibited by inflammatory cytokines, such as interferon-γ (IFNγ), tumour necrosis factor (TNF) and IL-12 (REF. 28), and by innate immune stimulation with Toll-like receptor 2 (TLR2) agonists29 and serum amyloid P (SAP)30. The appearance of fibrocytes both in vivo and in vitro is markedly induced by exposure to transforming growth factor-β1 (TGFβ1) and endothelin 1 (ET1)23,31. In this setting, the profibrotic protein semaphorin 7A (also known as CD108), produced by either monocytes or lymphocytes (or some combination thereof), promotes the monocyte to fibrocyte transition in a β1 integrin-dependent manner32. In vitro studies indicate that the intracellular signalling pathways implicated in murine fibrocyte differentiation include extracellular signal-regulated kinase (ERK)11, mammalian target of rapamycin (mTOR)–phosphoinositide 3-kinase (PI3K)33, RHO-associated protein kinase 1 (ROCK1)34 and the angiotensin 2 receptor35. When viewed in combination, these data indicate that fibrocyte differentiation in both culture and disease models decreases under conditions that perpetuate inflammation and tissue injury and increases under conditions that promote wound healing and tissue remodelling.
Mature macrophages also arise from CD14+ monocytic progenitor cells and assume their final phenotype in response to local cues, such as granulocyte–macrophage colony-stimulating factor (GM-CSF) and T helper 1 (TH1) or TH2 cell cytokines, which result in classical (pro-inflammatory) or alternatively activated (repair) characteristics, respectively. Monocytes and macrophages can also adopt certain characteristics of non-haematopoietic cells, such as intestinal epithelial cells, although this plasticity seems to be related to fusion events followed by nuclear reprogramming36. Macrophages are not known to differentiate into mature fibroblasts.
By contrast, mature fibroblasts originate mainly from post-embryonic tissue precursors20, but can also arise through epithelial–mesenchymal transition (as is seen in the kidney37) or endothelial–mesenchymal transition (as is seen in the lungs38), or from circulating progenitor cells, including mesenchymal stem cells39 and fibrocytes7. Fibroblasts have remarkable plasticity in in vitro studies, demonstrating their ability to be reprogrammed into haematopoietic stem cells with multilineage potential40, although such an event has yet to be shown in vivo. It is generally assumed that fibrocytes are a unique and highly plastic lineage. However, based on these cellular repertoires, an alternative (although as yet unsubstantiated) explanation for the phenotypical overlap between fibrocytes, macrophages and fibroblasts could be that fibrocytes are an intermediate population in the conversion of fibroblasts into macrophages (or possibly vice versa) (FIG. 3). This notion would require that fibroblasts and/or macrophages have sufficient in vivo plasticity to dedifferentiate and re-enter the circulation from inflamed and diseased organs. However, as there is currently no evidence in support of this phenomenon and the haematopoietic origin of circulating and tissue fibrocytes has been confirmed in several murine studies15,41, it is most likely that fibrocytes are a discrete population of reparative cells. Careful lineage-tracing approaches will be required to confirm the monocytic origin of fibrocytes in the setting of different chronic inflammatory stimuli. A comparison of the differentiation pathways that distinguish fibrocytes from macrophages and fibroblasts is shown in FIG. 3.
Figure 3. Differentiation pathways of macrophages, fibrocytes and fibroblasts.

Fibrocytes and macrophages originate from a bone marrow-derived CD14+ precursor found within the circulating monocyte pool. The differentiation of macrophages is stimulated by granulocyte–macrophage colony-stimulating factor (GM-CSF), Toll-like receptor (TLR) activation and phagocytosis, T helper 1 (TH1) cell cytokines and transendothelial migration from the blood to tissues. By contrast, the monocyte to fibrocyte transition is augmented by exposure to TH2 cell cytokines, transforming growth factor-β1 (TGFβ1), semaphorin 7A and apoptotic cells. Further stimulation with TGFβ1 and/or endothelin 1 (ET1) can induce fibrocytes to express α-smooth muscle actin and transition to fibroblast-like effector cells. Importantly, fibroblasts are classically considered to arise from organ-specific post-embryonic mesenchymal cells, although under certain pathological conditions there might be a small contribution by epithelial cells, endothelial cells and/or circulating progenitor cells (such as mesenchymal stem cells (MSCs) and fibrocytes). Fibroblasts have remarkable phenotypical plasticity in their ability to be reprogrammed into haematopoetic stem cells and redifferentiated down the myeloid pathway in vitro, under the control of OCT4 and haematopoietic growth factors. So far, bidirectional differentiation between mature macrophages and fibroblasts has not been shown in vivo. Thus, it is most likely that, rather than acting as an intermediate population between fibroblasts and macrophages, fibrocytes are a unique cell type with properties of both macrophages and fibroblasts.
Fibrocyte functions in the normal host
During the early phases of the response to tissue injury or invasion, fibrocytes are involved in the inflammatory process. In response to IL-1β, TH1 cell cytokines and/or viral infection, cultured human and porcine fibrocytes downregulate collagen expression4,22,42 and increase production of IL-6 (REFS 4,22), IL-8, CC-chemokine ligand 3 (CCL3) and CCL4 (REF. 4), which would promote inflammatory cell recruitment. In addition, the fibrocytes increase cell surface expression of leukocyte adhesion molecules, such as intercellular adhesion molecule 1 (ICAM1)4, which would increase leukocyte trafficking. When cultured in the presence of serum, as might occur in acute injury, fibrocytes have a macrophage-like inflammatory gene programme characterized by the expression of genes encoding chemokine receptors, cytokines and molecules involved in antigen presentation and lipid metabolism27. In porcine models, exposure to innate immune stimulation in the form of TLR agonists increases fibrocyte expression of MHC class I and II molecules and the co-stimulatory proteins CD80 and CD86, which enables fibrocytes to function as antigen-presenting cells for the activation of cytotoxic CD8+ T cells42.
However, fibrocytes also respond to IL-1β by increasing IL-10 production4, which could dampen inflammation and begin the transition to repair and remodelling. Similarly, fibrocytes respond to the presence of apoptotic cells by increasing collagen production43, which is required during repair. Fibrocytes have other reparative functions such as the contraction of ex vivo wound chambers2, although the finding that fibrocytes constitute only a small proportion of αSMA+ cells in animal models of fibrosis15,41 indicates that in this setting the contribution of fibrocytes might be dominated by immunomodulation rather than by direct differentiation to an activated collagen-producing phenotype. Thus, it is important to note that fibrocytes secrete paracrine factors such as platelet-derived growth factor (PDGF) and TGFβ1 that induce the differentiation of fibroblasts to myofibroblasts in culture44.
Fibrocytes secrete high levels of matrix metalloproteinases (MMPs)45, vascular endothelial growth factor (VEGF), PDGF subunit A, hepatocyte growth factor (HGF), GM-CSF, basic fibroblast growth factor (bFGF), IL-8 and IL-1β, which might contribute to their ability to promote neo-angiogenesis in ex vivo models46. Fibrocytes might also influence leukocyte trafficking through the expression of semaphorin 7A32, which activates macrophages47 and dendritic cells (DCs)48 and dampens T cell responses49. Normal human fibrocytes secrete chitinase-3-like protein 1 (also known as YKL40 in humans and BRP39 in mice) (E.L.H. and X. Peng, unpublished data). This protein is known to regulate allergic responses, in part through modulation of DC recruitment and alternative macrophage activation50, and functions as a biomarker of inflammatory diseases, including rheumatoid arthritis, cardiovascular disease and asthma51. In addition, fibrocytes have been identified in human malignancies52,53, and fibrocyte-like cells promote tumour metastasis in rodent models through the suppression of IFNγ and TNF production54 (by an unknown mechanism), as well as through overexpression of MMP9 (REF. 55).
These data indicate that fibrocytes can respond to injurious and inflammatory stimuli by adopting the functional characteristics of both classically and alternatively activated macrophages, as well as of fibroblasts. Whether the inflammatory or reparative fibrocyte phenotype dominates probably depends on local factors.
Fibrocytes in chronic inflammation
Increased numbers of local and/or circulating fibrocytes have been identified in diseases characterized by chronic TH1 and TH2 cell-mediated inflammatory responses (autoimmunity), TH1 cell-dominant responses (cardiovascular disease) or TH2 cell-dominant responses (asthma). However, most recent studies of fibrocytes have focused on describing their associations with various inflammatory conditions, so their effector contributions to inflammatory pathologies remain unknown. In order to highlight areas for future study, the potential role of fibrocytes in these disorders compared with the roles of macrophages and fibroblasts is presented below.
Autoimmune disease
Common to all autoimmune disorders is the breakdown of self-tolerance. This results in lymphocyte-mediated immune responses to autoantigens (which activate innate immune effector pathways involving macrophage accumulation), followed by downstream aberrant activation of stromal connective tissue fibroblast-like cells. Emerging evidence indicates that fibrocytes have a role in the autoimmune effector pathways in human forms and mouse models of scleroderma, autoimmune thyroiditis and rheumatoid arthritis.
A role for fibrocytes in autoimmune pathogenesis is suggested by studies of patients with diffuse scleroderma, a TGFβ1-driven multisystem autoimmune disorder characterized by progressive cutaneous and visceral fibrosis. In this disease, ongoing vascular damage and auto-immunity result in uncontrolled collagen production by activated myofibroblasts56, which also secrete the pro-inflammatory cytokines IL-6 and IL-8 (REF. 57), the auto-stimulatory and pro-angiogenic growth factors TGFβ1 and PDGF56 and the chemotactic and lymphocyte-activating protein ICAM1 (REF. 58). Monocytes12, monocyte-derived DCs59 and lung macrophages60 from patients with scleroderma have increased secretion of TH2 cell-related cytokines and chemokines, such as TGFβ1, IL-1 receptor antagonist (IL-1RA) and CCL18, which is indicative of an alternatively activated phenotype. Although it is not yet known whether these factors reflect or promote disease, alternatively activated macrophages have been shown to stimulate myofibroblast transformation and fibrosis in several murine models31,61.
Flow cytometric analysis of circulating12 or cultured monocytes32,43 shows increased numbers of fibrocytes in patients with scleroderma-related interstitial lung disease. As in other forms of lung fibrosis6, the number of these cells is greatest in patients with progressive disease, demonstrating a biological relationship between fibrocytes and disease activity (S. Mathai, R.A.R. and E.L.H., unpublished data). The presence of these cells is accompanied by increased concentrations of TH1 cell cytokines (such as TNF) and TH2 cell cytokines and chemokines (such as IL-13 and CCL2), although whether fibrocytes produce these soluble factors or are present as a result of them is not known12. Furthermore, the presence of circulating fibrocytes is accompanied by that of CD14+ monocytes that are skewed towards alternative activation, as characterized by increased expression of the scavenger receptor CD163 and enhanced secretion of CCL18 in response to in vitro stimulation with TLR4 agonists12. In these studies, the appearance of fibrocytes correlated very closely with increases in total lymphocyte counts, and later analysis found that CD19+ cells (which have been implicated in the regulation of fibrotic responses in murine studies62) accounted for the increased number of lymphocytes32. Fibrocytes from these patients also expressed semaphorin 7A32, which could negatively regulate T cell responses and promote DC activation47–49. Functional analysis of the relationship between macrophages, fibrocytes and fibroblasts was provided by animal modelling showing that lung fibroblasts fail to adopt a myofibroblast phenotype in response to TGFβ1 in the absence of macrophages and/or fibrocytes32,43; this indicates that fibrocytes, macrophages and fibroblasts are distinct populations with separate but undefined effects on the development of fibrosis and tissue remodelling in the lungs. A functional relationship between fibrocytes and lymphocytes has not been explored in this disease but is also an important area for future investigation.
Further support for a role for fibrocytes in autoimmunity stems from the recent finding of increased fibrocyte numbers in individuals with Graves’ disease, an auto-immune thyroid disorder characterized by persistent inflammation, lipogenesis and fibrosis that is mediated by orbital fibroblasts in the retro-orbital space (thyroid-associated ophthalmopathy)63. The role of macrophages in this process remains relatively unexplored. A novel role for fibrocytes in the pathogenesis of this disorder was recently suggested by a study in which fibrocyte precursors (that were identified by co-expression of CD34, CXC-chemokine receptor 4 (CXCR4), CD11b and collagen I) were found with increased frequency in the blood and ocular lesions of patients with thyroid-associated ophthalmopathy compared with the numbers in normal controls. These cells were similar to orbital fibroblasts in that they expressed the thyroid-stimulating hormone (TSH) receptor, which is a potential target of autoimmunity in this disease, and high levels of the insulin-like growth factor 1 (IGF1) receptor. The fibrocytes also expressed high levels of CD90 (also known as THY1), a marker that in some studies identifies fibroblasts with increased inflammatory capacity64,65. Stimulation of the fibrocytes with TSH led to high-level secretion of pro-inflammatory cytokines such as TNF and IL-6, which could promote disease pathogenesis. Thus, the contribution of fibrocytes to Graves’ disease seems to be similar to that of both fibroblasts and macrophages, in terms of their ECM-producing and pro-inflammatory properties, respectively.
Fibrocytes are also associated with the development of rheumatoid arthritis, an autoimmune polyarthritis characterized by inflammatory infiltration of the joints and proliferative activation of fibroblast-like synovial cells66. Although they are not found with increased frequency in the blood of patients with rheumatoid arthritis, fibrocytes from these patients do have a state of enhanced activation, characterized by the phosphorylation of components of the mitogen-activated protein kinase (MAPK) signalling cascade and of signal transducer and activator of transcription 3 (STAT3) and STAT5 (REF. 13). This activation profile would be expected to augment secretion of the pro-inflammatory cytokines TNF, IL-1 and IL-6 and is remarkably similar to that of activated fibroblast-like synovial cells. This study did not include a direct assessment of the ability of fibrocytes to execute the activities of fibroblast-like synovial cells, such as the synthesis and secretion of pro-inflammatory mediators and MMPs. However, a role for fibrocytes in the inflammatory pathogenesis of chronic synovitis is supported by studies documenting the appearance of activated fibrocytes before the development of arthropathy in the collagen-induced arthritis murine model of rheumatoid arthritis13. Macrophages are also centrally involved in rheumatoid arthritis owing to their production of pro-inflammatory mediators (such as TNF, IL-1β, IL-6 and IL-8) and their roles in phagocytosis, immune complex clearance, antigen presentation and cartilage destruction67. However, these functions of fibrocytes were not tested in this study, thus limiting comparison of these populations.
Given the profound immune dysregulation that occurs in autoimmunity, it is probable that the contribution of fibrocytes to these disorders includes both ECM production and immunomodulation. A better understanding of how fibrocytes interact with other cell types such as lymphocytes, macrophages and fibroblasts, as well as the assessment of fibrocyte functions such as antigen presentation, cytokine production and angiogenesis, might provide new insight into these diseases.
Cardiovascular disease
The development of cardiovascular disease follows a paradigm of macrophage-driven inflammatory changes in blood vessels that ultimately result in the formation of an atherosclerotic plaque (also known as an atheroma) — an accumulation of lipid, inflammatory cells and connective tissue in the arterial vessel wall that results in stenosis of the vessel. When these changes occur in the coronary arteries, myocardial infarction and fibrosis can occur. Fibrocytes are implicated in the development of both vascular plaques and myocardial fibrosis, although their precise contributions to disease pathology may vary.
The driving force in atherosclerosis is provided by classically activated macrophages, which initiate plaque formation early during the course of disease. These macrophages respond to IFNγ and oxidized low-density lipoprotein by transitioning into ‘foam cells’ (which produce pro-inflammatory cytokines, ROS and MMPs) and by enhancing cholesterol uptake68, perhaps in response to defective phagocytic clearance of apoptotic cells (a process known as efferocytosis)69. Late in the course of disease, alternatively activated macrophages might have protective effects characterized by the production of IL-10 and arginase, as well as by enhanced cholesterol efflux68,70. These effects are augmented by fibroblasts, which respond to TGFβ1 that is produced locally by a wide variety of cells (including foam cells and endothelial cells) to form a fibrous cap over the top of the atheroma to prevent rupture and potentially fatal vascular occlusion.
Fibrocytes have been identified in the fibrous caps of human atheromas10 and in several animal models of peripheral vascular disease71,72. In humans, fibrocytes colocalize with lesional TGFβ1 (which would be expected to promote protective responses10) but not with regions associated with the efferocytosis of apoptotic cells (which would be expected to promote classical macrophage activation and thereby contribute to disease). A recent study found that the overexpression of TGFβ1 in apolipoprotein E (Apoe)-null mice (a model of human atherosclerosis) resulted in increased fibrocyte accumulation in atherosclerotic plaques, suggesting that fibrocytes mediate TGFβ1-induced events in this model71. Further studies in Apoe-null mice indicate that fibrocytes do not adopt the fully differentiated phenotype of either macrophages or fibroblasts in atherosclerotic plaques but remain relatively undifferentiated, as evidenced by their persistent expression of CD45 and collagen Iα72,73. This might reflect the fact that fibrocytes are unable to form a discrete effector population given the ongoing injury and impeded repair seen in this disease.
Atherosclerosis affecting the coronary arteries frequently results in injury to and fibrosis of cardiac tissue. In this setting, ischaemic cell death leads to the recruitment of classically activated macrophages that perpetuate tissue injury, in part through expression of TNF74. As the injury phase concludes and healing begins, the alternatively activated macrophage phenotype (characterized by TGFβ1, IL-10 and CD206 expression) dominates, and during this stage cardiac fibroblasts adopt ECM-producing and tissue-remodelling properties74,75. Ischaemic injury also causes these fibroblasts to secrete pro-inflammatory cytokines76, MMPs and tissue inhibitors of metalloproteinases (TIMPs)77, which would be expected to limit the extent of injury, as well as pro-angiogenic substances such as FGFs and VEGF78. In the setting of profound initial injury or persistent damage, or in the presence of profibrotic inflammatory stimuli, cardiac fibrosis and remodelling ensue.
Fibrocytes could contribute to this pathology in various ways. Although fibrocytes have been found in myxomatous human heart valves (which have an abnormal connective tissue composition)79, they have yet to be identified in the normal human myocardium. However, murine modelling indicates that fibrocytes might contribute to the pathogenesis of ischaemic cardiomyopathy. In this model, fibrocytes are recruited to chronically injured myocardium, where they constitute up to 3% of the total number of heart cells. Treatment of these animals with SAP decreases fibrocyte accumulation and fibrosis in an FcγR-dependent manner80 that involves ROCK1 (REF. 34). In all of these studies, the functions attributed to fibrocytes involve ECM production, although it is possible that other activities that are more typically associated with both macrophages and fibroblasts — such as cytokine production, immune cell activation and angiogenesis — are equally important. When viewed in light of the atherosclerosis data described above, these data indicate that fibrocytes might have a protective or reparative effect in the setting of uncontrolled or persistent in vivo TH1 cell cytokine exposure, whereas in response to persistent TH2 cell cytokine exposure (as occurs in the remodelled heart) this phenotype becomes profibrotic.
Asthma
Asthma is a TH2 cell-driven disease characterized by reversible airway obstruction in response to inhaled allergens. A subgroup of patients with asthma have continual lung inflammation and remodelling, which causes persistent airway obstruction. In this setting, subepithelial fibrosis and airway obstruction result from the accumulation of highly proliferative activated myofibroblasts in response to allergen sensitization81, innate immune stimulation82 and pro-angiogenic states (such as VEGF overexpression83 and exposure to chitinase-3-like protein 1 (REF. 50)). By contrast, macrophage function has generally been considered to reflect rather than initiate remodelling responses in the asthmatic lung. However, recent data indicate that alternatively activated macrophages promote the fibroblast to myofibroblast transition and, subsequently, airway remodelling in a mouse model of asthma84 and that modulation of this phenotype can decrease disease85. Furthermore, the finding that chitinase-3-like protein 1, a major product of alternatively activated macrophages, affects allergic inflammation and disease phenotype in murine models of asthma49 supports the contention that alternatively activated macrophages might have a more important role in asthma than previously thought.
Compelling evidence of a role for fibrocytes in asthma was provided by a study in which bronchoscopic lung biopsies obtained from patients with asthma after allergen inhalation showed localization of fibrocytes in the airway submucosa23. Furthermore, murine studies of asthma using the ovalbumin model have found that after allergen inhalation fibrocytes localize to the airway mucosa, where they acquire a myofibroblast phenotype23. The numbers of circulating fibrocytes in patients with asthma are increased only in those individuals with chronic airway obstruction (as compared with mild asthmatics and normal controls)8,86, and cultured fibrocytes from these patients show a TGFβ1-dependent increase in proliferation and the production of αSMA. Although the largest numbers of fibrocytes were observed in patients with severe airway remodelling, fibrocytes were also found in the lungs of patients with mild asthma, indicating that they might have an important role in inflammatory progression86.
In all of these studies, the primary focus has been on the contribution of fibrocytes to the tissue content of myofibroblasts. The paucity of data regarding the immunological function(s) of fibrocytes in asthma prevents a full comparison of fibrocytes and macrophages in this setting. Given the roles of fibrocytes in antigen presentation, cytokine production, angiogenesis and chitinase production, all of which are important components of the asthmatic response, it is likely that fibrocytes participate in these processes to promote ongoing airway inflammation.
Conclusions and perspectives
The association of fibrocytes with heterogeneous chronic inflammatory conditions indicates that they have a role extending far beyond ECM production. As shown in FIG. 4, a paradigm is thus suggested in which fibrocytes recruited early during an inflammatory response have a pro-inflammatory phenotype that is induced by innate immune signals and augmented by IL-1. During this state, the overall effector profile of fibrocytes might more closely resemble that of macrophages than that of fibroblasts. Later, as inflammation regresses and repair and remodelling begin, a programme of matrix production and in some circumstances myofibroblast transformation by fibrocytes ensues. This model contrasts with the restricted differentiation of macrophages and fibroblasts and is consistent with the recruitment of fibrocytes under different pathological conditions. Although it is plausible that fibrocytes function alone to promote the pathologies seen in many forms of chronic inflammation, data supporting this claim are currently lacking and it is more likely that macrophages, fibroblasts and fibrocytes orchestrate repair and remodelling together. The dearth of functional information limiting our ability to draw conclusions about the role of fibrocytes in these disorders represents the great opportunities that exist for further study of these novel cells (BOX 1).
Figure 4. Potential roles of fibrocytes in chronic inflammatory disease.

Using autoimmunity as a model, the possible roles of fibrocytes are proposed. In the setting of autoantigen exposure or acute injury, or following stimulation by interleukin-1β (IL-1β), serum factors and innate immune stimuli, fibrocytes adopt a pro-inflammatory phenotype characterized by the secretion of interferon-γ (IFNγ), IL-6, IL-8, CC-chemokine ligand 3 (CCL3) and CCL4. Leukocyte trafficking is enhanced through expression of intercellular adhesion molecule 1 (ICAM1). Production of extracellular matrix (ECM) components is decreased and antigen-presenting capabilities are increased by the expression of CD80, CD86 and MHC class I and II molecules. Tissue destruction may be increased by expression of matrix metalloproteinases (MMPs). As the local milieu begins to favour repair and remodelling (or perhaps concurrently with ongoing injury in the right biological context), fibrocytes adopt a more reparative phenotype. In this setting, transforming growth factor-β1 (TGFβ1) stimulates fibrocyte development through non-canonical pathways mediated by semaphorin 7A (SEMA7A) and β1 integrin, although other TGFβ1-mediated signalling pathways may also be involved. SEMA7A could activate monocytes and dendritic cells (DCs) while dampening T cell responses. ECM production is also stimulated by T helper 2 (TH2) cell cytokines (such as IL-4 and IL-13), as well as by exposure to apoptotic cells and cellular debris. Myofibroblast transformation is promoted by TGFβ1. Platelet-derived growth factor-α (PDGFα), IL-10, vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF) and basic fibroblast growth factor (bFGF) support neoangiogenesis, and recruitment to sites of injury is promoted by the expression of chemokine receptors such as CXC-chemokine receptor 4 (CXCR4). αSMA, α-smooth muscle actin; CXCL, CXC-chemokine ligand; ERK, extracellular signal-regulated kinase; TLR, Toll-like receptor.
Box 1. Future directions.
The strong association of fibrocytes with diverse forms of chronic inflammation indicates many areas for future study. The continued investigation of differentiation pathways and disease associations will be useful, as will a greater understanding of the role of fibrocytes in inflammatory disorders. In addition, definite clarification of the contribution of fibrocytes relative to those of macrophages and fibroblasts is sorely needed. Thus, future studies should focus on defining the functional phenotype of these cells in terms of cytokine and chemokine production, antigen presentation, angiogenesis, wound repair and tissue remodelling. Furthermore, lineage tracing and chimaera studies should be carried out to determine whether fibrocytes are a truly unique cell population or are an intermediate cell type in the differentiation of macrophages and fibroblasts. These studies will require the development of specific assays and genetic models that enable the isolation of these cells for ex vivo studies and in vivo deletion.
Acknowledgments
We wish to thank E. Tarquino for excellent help with manuscript preparation. We gratefully acknowledge funding from the following sources: the US National Institutes of Health (NIH grant UL1RR024139), the Scleroderma Foundation, the American Thoracic Society and a TRI award from Yale Department of Medicine (all to E.L.H.).
Footnotes
Competing interests statement
The authors declare competing financial interests: see Web version for details.
Contributor Information
Ronald A. Reilkoff, Yale University School of Medicine, Section of Pulmonary and Critical Care Medicine, 300 Cedar Street, New Haven, Connecticut 06520, USA
Richard Bucala, Yale University School of Medicine, Section of Rheumatology, 300 Cedar Street, New Haven, Connecticut 06520, USA.
Erica L. Herzog, Yale University School of Medicine, Section of Pulmonary and Critical Care Medicine, 300 Cedar Street, New Haven, Connecticut 06520, USA
References
- 1.Cohnheim J. Ueber Entzundung und Eiterung (About inflammation and suppuration) Path Anat Physiol Klin Med. 1867;40:1–79. [Google Scholar]
- 2.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 3.Chesney J, Bacher M, Bender A, Bucala R. The peripheral blood fibrocyte is a potent antigen-presenting cell capable of priming naive T cells in situ. Proc Natl Acad Sci USA. 1997;94:6307–6312. doi: 10.1073/pnas.94.12.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala R. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J Immunol. 1998;160:419–425. [PubMed] [Google Scholar]
- 5.Pilling D, Fan T, Huang D, Kaul B, Gomer RH. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS ONE. 2009;4:e7475. doi: 10.1371/journal.pone.0007475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moeller A, et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:588–594. doi: 10.1164/rccm.200810-1534OC. [DOI] [PubMed] [Google Scholar]
- 7.Phillips RJ, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang CH, et al. Increased circulating fibrocytes in asthma with chronic airflow obstruction. Am J Respir Crit Care Med. 2008;178:583–591. doi: 10.1164/rccm.200710-1557OC. [DOI] [PubMed] [Google Scholar]
- 9.Vakil V, et al. Gadolinium-containing magnetic resonance image contrast agent promotes fibrocyte differentiation. J Magn Reson Imaging. 2009;30:1284–1288. doi: 10.1002/jmri.21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medbury H, et al. Monocytes contribute to the atherosclerotic cap by transformation into fibrocytes. Int Angiol. 2008;27:114–123. [PubMed] [Google Scholar]
- 11.Nikam VS, et al. Treprostinil inhibits the recruitment of bone marrow-derived circulating fibrocytes in chronic hypoxic pulmonary hypertension. Eur Respir J. 2010;36:1302–1324. doi: 10.1183/09031936.00028009. [DOI] [PubMed] [Google Scholar]
- 12.Mathai SK, et al. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest. 2010;90:812–823. doi: 10.1038/labinvest.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galligan CL, et al. Fibrocyte activation in rheumatoid arthritis. Rheumatology. 2010;49:640–651. doi: 10.1093/rheumatology/kep265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niedermeier M, et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci USA. 2009;106:17892–17897. doi: 10.1073/pnas.0906070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kisseleva T, et al. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45:429–438. doi: 10.1016/j.jhep.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 16.Haudek SB, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci USA. 2006;103:18284–18289. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vannella KM, et al. Cysteinyl leukotrienes are autocrine and paracrine regulators of fibrocyte function. J Immunol. 2007;179:7883–7890. doi: 10.4049/jimmunol.179.11.7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duffield JS, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 20.Homer RJ, Elias JA, Lee CG, Herzog EL. Modern concepts in pulmonary fibrosis. Arch Pathol Lab Med. doi: 10.5858/2010-0296-RA.1. (in the press) [DOI] [PubMed] [Google Scholar]
- 21.Yang L, et al. Identification of fibrocytes in postburn hypertrophic scar. Wound Repair Regen. 2005;13:398–404. doi: 10.1111/j.1067-1927.2005.130407.x. [DOI] [PubMed] [Google Scholar]
- 22.Balmelli C, et al. Responsiveness of fibrocytes to Toll-like receptor danger signals. Immunobiology. 2007;212:693–699. doi: 10.1016/j.imbio.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;171:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 24.Mattoli S, Barcyk M, Bellini A. Fibrocytes in Asthma. Fibrocytes: New Insights into Tissue Repair and Systemic Fibroses. World Scientific Publishing Co; Singapore: (in the press) [Google Scholar]
- 25.Herzog EL, Bucala R. Fibrocytes in health and disease. Exp Hematol. 2010;38:548–556. doi: 10.1016/j.exphem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pilling D, Tucker NM, Gomer RH. Aggregated IgG inhibits the differentiation of human fibrocytes. J Leukoc Biol. 2006;79:1242–1251. doi: 10.1189/jlb.0805456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curnow SJ, et al. Distinct types of fibrocyte can differentiate from mononuclear cells in the presence and absence of serum. PLoS ONE. 2010;5:e9730. doi: 10.1371/journal.pone.0009730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D. Pivotal advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J Leukoc Biol. 2008;83:1323–1333. doi: 10.1189/jlb.1107782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maharjan AS, Pilling D, Gomer RH. Toll-like receptor 2 agonists inhibit human fibrocyte differentiation. Fibrogenesis Tissue Repair. 2010;3:23–30. doi: 10.1186/1755-1536-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pilling D, Buckley CD, Salmon M, Gomer RH. Inhibition of fibrocyte differentiation by serum amyloid P. J Immunol. 2003;171:5537–5546. doi: 10.4049/jimmunol.171.10.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murray LA, et al. TGF-β driven lung fibrosis is macrophage dependent and blocked by serum amyloid P. Int J Biochem Cell Biol. 2011;43:154–162. doi: 10.1016/j.biocel.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 32.Gan Y, et al. Role of semaphorin 7a in TGF-β1 induced lung fibrosis, and scleroderma-related interstitial lung disease. Arthritis Rheum. 2011 Apr 11; doi: 10.1002/art.30386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehrad B, Burdick MD, Strieter RM. Fibrocyte CXCR4 regulation as a therapeutic target in pulmonary fibrosis. Int J Biochem Cell Biol. 2009;41:1708–1718. doi: 10.1016/j.biocel.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haudek SB, et al. Rho kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc Res. 2009;83:511–518. doi: 10.1093/cvr/cvp135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haudek SB, et al. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;49:499–507. doi: 10.1016/j.yjmcc.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Powell AE, et al. Fusion between intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011;71:1497–1505. doi: 10.1158/0008-5472.CAN-10-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. 2006;69:213–217. doi: 10.1038/sj.ki.5000054. [DOI] [PubMed] [Google Scholar]
- 38.Hashimoto N, et al. Endothelial–mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2009;43:161–172. doi: 10.1165/rcmb.2009-0031OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pereira RF, et al. Marrow stromal cells as a source of progenitor cells for nonhematopoietic tissues in transgenic mice with a phenotype of osteogenesis imperfecta. Proc Natl Acad Sci USA. 1998;95:1142–1147. doi: 10.1073/pnas.95.3.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szabo E, et al. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature. 2010;468:521–526. doi: 10.1038/nature09591. [DOI] [PubMed] [Google Scholar]
- 41.Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113:243–252. doi: 10.1172/JCI18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balmelli C, Ruggli N, McCullough K, Summerfield A. Fibrocytes are potent stimulators of anti-virus cytotoxic T cells. J Leukoc Biol. 2005;77:923–933. doi: 10.1189/jlb.1204701. [DOI] [PubMed] [Google Scholar]
- 43.Peng X, et al. Local apoptosis promotes production of collagen in monocyte derived cells. Fibrogenesis Tissue Repair. doi: 10.1186/1755-1536-4-12. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang JF, et al. Fibrocytes from burn patients regulate the activities of fibroblasts. Wound Repair Regen. 2007;15:113–121. doi: 10.1111/j.1524-475X.2006.00192.x. [DOI] [PubMed] [Google Scholar]
- 45.Garcia-de-Alba C, et al. Expression of matrix metalloproteases by fibrocytes: possible role in migration and homing. Am J Respir Crit Care Med. 2010;182:1144–1152. doi: 10.1164/rccm.201001-0028OC. [DOI] [PubMed] [Google Scholar]
- 46.Hartlapp I, et al. Fibrocytes induce an angiogenic phenotype in cultured endothelial cells and promote angiogenesis in vivo. FASEB J. 2001;15:2215–2224. doi: 10.1096/fj.01-0049com. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki K, et al. Semaphorin 7A initiates T-cell-mediated inflammatory responses through α1β1 integrin. Nature. 2007;446:680–684. doi: 10.1038/nature05652. [DOI] [PubMed] [Google Scholar]
- 48.Holmes S, et al. Sema7A is a potent monocyte stimulator. Scand J Immunol. 2002;56:270–275. doi: 10.1046/j.1365-3083.2002.01129.x. [DOI] [PubMed] [Google Scholar]
- 49.Czopik A. Semaphorin 7A is a negative regulator of T cell responses. Immunity. 2006;5:591–600. doi: 10.1016/j.immuni.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 50.Lee CG, et al. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J Exp Med. 2009;206:1149–1166. doi: 10.1084/jem.20081271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee CG, et al. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Ann Rev Physiol. 2011;73:479–501. doi: 10.1146/annurev-physiol-012110-142250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barth PJ, Ebrahimsade S, Hellinger A, Moll R, Ramaswamy A. CD34+ fibrocytes in neoplastic and inflammatory pancreatic lesions. Virchows Arch. 2002;440:128–133. doi: 10.1007/s00428-001-0551-3. [DOI] [PubMed] [Google Scholar]
- 53.Nimphius W, Moll R, Olbert P, Ramaswamy A, Barth PJ. CD34+ fibrocytes in chronic cystitis and noninvasive and invasive urothelial carcinomas of the urinary bladder. Virchows Arch. 2007;450:179–185. doi: 10.1007/s00428-006-0347-6. [DOI] [PubMed] [Google Scholar]
- 54.Kraman M, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-α. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 55.van Deventer HW, et al. C-C chemokine receptor 5 on pulmonary fibrocytes facilitates migration and promotes metastasis via matrix metalloproteinase 9. Am J Pathol. 2008;173:253–264. doi: 10.2353/ajpath.2008.070732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varga J, Pasche B. Transforming growth factor β as a therapeutic target in systemic sclerosis. Nature Rev Rheumatol. 2009;5:200–206. doi: 10.1038/nrrheum.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kadono T, Kikuchi K, Ihn H, Takehara K, Tamaki K. Increased production of interleukin 6 and interleukin 8 in scleroderma fibroblasts. J Rheumatol. 1998;25:296–301. [PubMed] [Google Scholar]
- 58.Needleman BW. Increased expression of intercellular adhesion molecule 1 on the fibroblasts of scleroderma patients. Arthritis Rheum. 1990;33:1847–1851. doi: 10.1002/art.1780331214. [DOI] [PubMed] [Google Scholar]
- 59.van Leishout AW, et al. Enhanced interleukin-10 production by dendritic cells upon stimulation with Toll-like receptor 4 agonists in systemic sclerosis that is possibly implicated in CCL18 secretion. Scand J Rheumatol. 2009;38:282–290. doi: 10.1080/03009740802572467. [DOI] [PubMed] [Google Scholar]
- 60.Luzina IG, et al. Gene expression in bronchoalveolar lavage cells from scleroderma patients. Am J Respir Cell Mol Biol. 2002;26:549–557. doi: 10.1165/ajrcmb.26.5.4683. [DOI] [PubMed] [Google Scholar]
- 61.Luzina IG, et al. Regulation of pulmonary inflammation and fibrosis through expression of integrins αVβ3 and αVβ5 on pulmonary T lymphocytes. Arthritis Rheum. 2009;60:1530–1539. doi: 10.1002/art.24435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yoshizaki A, et al. CD19 regulates skin and lung fibrosis via Toll-like receptor signaling in a model of bleomycin-induced scleroderma. Am J Pathol. 2008;28:639–650. doi: 10.2353/ajpath.2008.071049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Douglas RS, et al. Increased generation of fibrocytes in thyroid-associated ophthalmopathy. J Clin Endocrinol Metab. 2010;95:430–438. doi: 10.1210/jc.2009-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khoo TK, Coenen MJ, Schiefer AR, Kumar S, Bahn RS. Evidence for enhanced Thy-1 (CD90) expression in orbital fibroblasts of patients with Graves’ ophthalmopathy. Thyroid. 2008;18:1291–1296. doi: 10.1089/thy.2008.0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bahn RS. Graves’ ophthalmopathy. N Engl J Med. 2010;362:726–738. doi: 10.1056/NEJMra0905750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muller-Ladner U, Ospelt C, Gay S, Distler O, Pap T. Cells of the synovium in rheumatoid arthritis. Synovial fibroblasts. Arthritis Res Ther. 2007;9:223. doi: 10.1186/ar2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kinne RW, Stuhlmuller B, Burmester GR. Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res Ther. 2007;9:224. doi: 10.1186/ar2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29:1419–1423. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- 69.Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol. 2009;5:1089–1095. doi: 10.1189/jlb.0209115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saha P, et al. The monocyte/macrophage as a therapeutic target in atherosclerosis. Curr Opin Pharmacol. 2009;9:109–118. doi: 10.1016/j.coph.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 71.Buday A, et al. Elevated systemic TGF-β impairs aortic vasomotor function through activation of NADPH oxidase-driven superoxide production and leads to hypertension, myocardial remodeling, and increased plaque formation in apoE−/− mice. Am J Physiol Heart Circ Physiol. 2010;299:H386–H395. doi: 10.1152/ajpheart.01042.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iwata H, et al. Bone marrow-derived cells contribute to vascular inflammation but do not differentiate into smooth muscle cell lineages. Circulation. 2010;122:2048–2057. doi: 10.1161/CIRCULATIONAHA.110.965202. [DOI] [PubMed] [Google Scholar]
- 73.Daniel JM, et al. Time-course analysis on the differentiation of bone marrow-derived progenitor cells into smooth muscle cells during neointima formation. Arterioscler Thromb Vasc Biol. 2010;30:1890–1896. doi: 10.1161/ATVBAHA.110.209692. [DOI] [PubMed] [Google Scholar]
- 74.Harel-Adar T, et al. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc Natl Acad Sci USA. 2011;108:1827–1835. doi: 10.1073/pnas.1015623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Troidl C, et al. Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. J Cell Mol Med. 2009;13:3485–3494. doi: 10.1111/j.1582-4934.2009.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Turner NA, et al. Mechanism of TNFα-induced IL-1α, IL-1β and IL-6 expression in human cardiac fibroblasts: effects of statins and thiazolidinediones. Cardiovasc Res. 2007;76:81–90. doi: 10.1016/j.cardiores.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 77.Vanhoutte D, Heymans S. TIMPs and cardiac remodeling: ‘Embracing the MMP-independent-side of the family’. J Mol Cell Cardiol. 2010;48:445–453. doi: 10.1016/j.yjmcc.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 78.Murakami M, Simons M. Fibroblast growth factor regulation of neovascularization. Curr Opin Hematol. 2008;15:215–220. doi: 10.1097/MOH.0b013e3282f97d98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barth PJ, Koster H, Moosdorf R. CD34+ fibrocytes in normal mitral valves and myxomatous mitral valve degeneration. Pathol Res Pract. 2005;201:301–304. doi: 10.1016/j.prp.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 80.Haudek SB, et al. Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc Natl Acad Sci USA. 2008;105:10179–10184. doi: 10.1073/pnas.0804910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Simoes DC, et al. Osteopontin deficiency protects against airway remodeling and hyperresponsiveness in chronic asthma. Am J Respir Crit Care Med. 2009;179:894–902. doi: 10.1164/rccm.200807-1081OC. [DOI] [PubMed] [Google Scholar]
- 82.Sugiura H, et al. Activation of Toll-like receptor 3 augments myofibroblast differentiation. Am J Respir Cell Mol Biol. 2009;40:654–662. doi: 10.1165/rcmb.2008-0371OC. [DOI] [PubMed] [Google Scholar]
- 83.Lee CG, et al. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nature Med. 2004;10:1095–1103. doi: 10.1038/nm1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dong L, et al. FIZZ1 plays a crucial role in early stage airway remodeling of OVA-induced asthma. J Asthma. 2008;45:648–653. doi: 10.1080/02770900802126941. [DOI] [PubMed] [Google Scholar]
- 85.Moreira AP, et al. Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore-induced allergic airway disease. J Allergy Clin Immunol. 2010;126:712–721. doi: 10.1016/j.jaci.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 86.Nihlberg K, et al. Tissue fibrocytes in patients with mild asthma: a possible link to thickness of reticular basement membrane? Respir Res. 2006;7:50–59. doi: 10.1186/1465-9921-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang L, et al. Peripheral blood fibrocytes from burn patients: identification and quantification of fibrocytes in adherent cells cultured from peripheral blood mononuclear cells. Lab Invest. 2002;82:1183–1192. doi: 10.1097/01.lab.0000027841.50269.61. [DOI] [PubMed] [Google Scholar]
- 88.Andersson-Sjoland A, Erjefalt JS, Bjermer L, Eriksson L, Westergren-Thorsson G. Fibrocytes are associated with vascular and parenchymal remodelling in patients with obliterative bronchiolitis. Respir Res. 2009;10:103. doi: 10.1186/1465-9921-10-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moore BB, et al. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol. 2006;35:175–181. doi: 10.1165/rcmb.2005-0239OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mehrad B, et al. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. 2007;353:104–108. doi: 10.1016/j.bbrc.2006.11.149. [DOI] [PubMed] [Google Scholar]
- 91.Cowper SE, et al. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23:383–393. doi: 10.1097/00000372-200110000-00001. [DOI] [PubMed] [Google Scholar]