

Abstract
Protein carbonylation, the non-enzymatic addition of aldehydes or ketones to specific amino acid residues, has been implicated in the pathophysiology of multiple sclerosis (MS). In this study we investigated whether protein carbonyls (PCOs) also accumulate in the spinal cord of Lewis rats with acute experimental autoimmune encephalomyelitis (EAE). Western blots analysis after derivatization with dinitrophenyl hydrazine (oxyblot) showed elevated protein carbonylation at the time of maximal clinical disability. During the same period glutathione levels were substantially reduced, suggesting a causal relationship between these two markers. In contrast, lipid peroxidation products accumulated in EAE spinal cord well before the appearance of neurological symptoms. Carbonyl staining was not restricted to inflammatory lesions but present throughout the spinal cord particularly in neuronal cell bodies and axons. By 2-dimensional-oxyblot we identified several cytoskeletal proteins, including β-actin, GFAP and the neurofilament proteins as the major targets of carbonylation. These findings were confirmed by pull-down experiments, which also showed an increase in the number of carbonylated β-actin molecules and a decrease in that of oxidized neurofilament proteins in EAE. These data suggest the possibility that oxidation targets neurofilament proteins for degradation, which may contribute to axonal pathology observed in MS and EAE.
Keywords: protein carbonylation, oxidative stress, cytoskeletal degradation, experimental autoimmune encephalomyelitis, multiple sclerosis
Experimental autoimmune encephalomyelitis (EAE) shares a number of clinical and pathological features with MS, and is routinely employed to study the mechanistic bases of disease and to test therapeutic approaches (Gold et al. 2000). This animal model of MS can be induced in a variety of mammalian species by active immunization with myelin-specific antigens (e.g. MBP, PLP, MOG) or by adoptive transfer of T-cells from immunized animals into naïve recipients (Day 2005). MBP-induced EAE in the Lewis rat, the animal model used in this study, is a n acute monophasic clinical disease that resolves spontaneously within 5 days of onset. At the peak of disease there are perivenular infiltrates of inflammatory cells within the spinal cord. There is, however, no demyelination of CNS axons, making this an ideal model to study the neuroinflammatory aspects of MS.
In recent years a significant body of experimental evidence has accumulated demonstrating that oxidative stress is a major player in the pathogenesis of both MS and EAE (for reviews, see LeVine 1992, Gilgun-Sherki et al. 2004). While oxidation of lipids and nucleic acids in these disorders has been studied extensively, protein oxidation has received little attention. The non-enzymatic addition of aldehydes or ketones to specific amino acid residues (i.e. carbonylation) constitutes the major and most common oxidative alteration (Berlett & Stadtman 1997, Dalle-Donne et al. 2003, Nystrom 2005). Carbonyl groups can be introduced into proteins either by direct (oxidative) reaction of ROS (e.g. hydrogen peroxide, lipid hydroperoxides) with protein side chains or indirect (non-oxidative) addition of reactive carbonyl species (RCS) to Cys, His and Lys residues. RCS are carbonyl-containing molecules resulting from the oxidation of lipids (e.g. 4-hydroxynonenal, malondialdehyde (MDA), acrolein) or carbohydrates (e.g. glyoxal, methylglyoxal). Protein carbonylation is an irreversible form of damage that results from severe or prolonged oxidative stress conditions (Dalle-Donne et al. 2003), which occurs in many neurodegenerative disorders including Alzheimer's disease (Aksenov et al. 2001), Parkinson's disease (Floor & Wetzel 1998) and amyotrophic lateral sclerosis (Ferrante et al. 1997). Our discovery that protein carbonyls are augmented in MS (Bizzozero et al. 2005) suggests that this type of chemical modification may also play a critical pathophysiological role in inflammatory demyelinating diseases.
There are multiple possible fates for carbonylated proteins; the outcome likely depends on the specific protein being carbonylated as well as the level of oxidative damage it sustains. The concentration of carbonylated proteins within cells is normally extremely low, although even moderate increases in the carbonylation can have tremendous effects on protein function, aggregate formation, or metabolic stability. Maintenance of low carbonylation level is achieved through the action of several proteolytic systems that preferentially digest oxidized proteins. The core 20s proteasome is known to catalyze the degradation of oxidized proteins in an ATP- and ubiquitin-independent pathway, and is thought to be the primary clearance mechanism for carbonylated proteins in cells (Grune et al. 1997). However, other proteases have been implicated in the hydrolysis of oxidized proteins as well. For example, the calcium-dependent cysteine protease calpain has been shown to preferentially degrade oxidized neurofilament over non-oxidized protein (Troncoso et al. 1995). While many proteins become more susceptible to degradation by cellular proteases upon carbonylation of one or more residues (Stadtman 1990), other oxidized proteins become more resistant to proteolysis. Additionally, carbonylated proteins can inhibit the proteases that degrade them, and carbonylation of the proteases themselves impairs their function (Friguet et al. 1994, Shacter 2000). Ultimately it is this phenomenon what likely causes the accumulation of carbonylated species in diseased tissues. As their concentration within cells build up, carbonylated proteins tend to crosslink forming high-molecular-weight aggregates, which are both cytotoxic and no longer susceptible to proteolysis (Nie et al. 2007, Smith et al. 1995).
The present study was designed to determine if EAE displays oxidative damage to proteins similar to that seen in MS. We sought to determine whether changes in protein carbonylation levels occur during the course of EAE and to identify specific proteins that are carbonylated in this disorder. Our focus was on cytoskeletal proteins because (1) their abundance in cells makes them common targets of various ROS and RCS (Dalle-Donne et al. 2006), and (2) they are major targets of oxidation in MS (Bizzozero 2007). The results clearly show that the oxidative stress conditions generated during the course of MBP-induced EAE in the Lewis rat are sufficient to cause protein carbonylation and that this process may trigger proteolytic degradation of oxidized cytoskeletal proteins. To the best of our knowledge this is the first study demonstrating protein carbonylation in EAE. A preliminary account of these findings has been presented in abstract form (Smerjac & Bizzozero 2006, Smerjac & Bizzozero 2007).
Materials and Methods
Induction of Experimental Autoimmune Encephalomyelitis (EAE)
Housing and handling of the animals as well as the euthanasia procedure were in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee. Seven-week-old male Lewis rats were purchased from Harlan Bioproducts (Indianapolis, IN) and housed in the UNM-animal resource facility. EAE was induced in these animals as described by Schaecher et al (2002) with slight modifications. Briefly, rats received two subcutaneous injections into the lower back area of 100μl each containing 200 mg/ml of guinea pig spinal cord homogenate plus 280 μg/ml of guinea pig myelin basic protein (Sigma, St. Louis, MO) in saline mixed with complete Freund's adjuvant (CFA) supplemented with 1 mg/ml of heat killed Mycobacterium tuberculosis H37Ra (Chondrex Inc; Redmond, WA). Control animals were given CFA without myelin antigens. Two hours after EAE induction, all animals received an i.p. injection of 2 μg of pertussis toxin (List Biological Laboratories; Campbell, CA) in 100 μl of saline. Animals were weighed and examined daily for the presence of neurological signs. At prescribed days post-injection (DPI) animals were euthanized by decapitation. Spinal cords were dissected and either fixed with methacarn or homogenized in PEN buffer (20 mM sodium phosphate pH 7.5, 1 mM EDTA and 0.1 mM neocuproine) containing 2 mM 4,5 dihydroxy-1,3 benzene disulfonic acid and 1 mM dithiothreitol (DTT). Protein homogenates were stored at −80°C until use. Protein concentration was assessed with the Bio-Rad DCT protein assay (Bio-Rad Laboratories; Hercules, CA) using bovine serum albumin as standard.
Determination of glutathione (GSH)
The amount of non-protein thiols, of which > 90% is GSH (Vitvitsky et al. 2006), was determined with 5,5'-dithiobis-(2-nitrobenzoic acid) (DTNB; Sigma). Briefly, an aliquot of the spinal cord homogenate prepared in PEN buffer without DTT was mixed with an equal volume of 2% sulfosalicylic acid to precipitate the proteins. After centrifugation at 10,000g for 15 min, the supernatants were mixed with 0.2M sodium phosphate buffer pH 7.5 containing 0.3 mM DTNB, 10 mM EDTA and 1% SDS and incubated for 15 min at room temperature. Absorbance was measured at 412 nm using a Hewlett-Packard 8452-A Diode Array Spectrophotometer. The amount of thiol groups was calculated using a molar extinction coefficient of 13,600 cm−1 for the thionitrobenzoate anion (Riddles et al. 1979).
Measurement of lipid peroxidation
Lipid peroxidation was assessed by measuring the amount of thiobarbituric acid reactive substances (TBARS), which are byproducts of lipid peroxidation, in the tissue homogenates (Ohkawa et al. 1979). One-hundred-μl aliquots (~500μg of protein) were mixed with 25μl of 2% (w/v) butylated hydroxytoluene and 875μl of 1% (w/v) thiobarbituric acid (Aldrich; Milwaukee, WI) prepared in 10% (w/v) trichloroacetic acid. Samples were incubated for 20 min at 90°C. Aggregated material was removed by centrifugation at 10,000g for 15 min and the absorbance of the supernatant was measured at 532 nm. The amount of TBARS was calculated using a standard curve prepared with 1,1,3,3-tetraethoxypropane (Aldrich).
Assessment of protein carbonylation by western blotting
Protein carbonyl groups were measured with the OxyBlot™ protein oxidation detection kit (Intergen Co., Purchase, NY), following the protocol provided by the manufacturer. In brief, proteins (5μg) were incubated with 2,4-dinitrophenylhydrazine to form the 2,4-dinitrophenyl (DNP) hydrazone derivatives. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted to polyvinylidene difluoride (PVDF) membranes. DNP-containing proteins were then immunostained using rabbit anti-DNP antiserum (1:500) and goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP) (1:2000). Blots were developed by enhanced chemioluminescence (ECL) using the Western Lightning ECL™ kit from Perkin-Elmer (Boston, MA). The developed films were scanned in a Hewlett Packard Scanjet 4890 and the images quantified using the Scion Image for Windows, version Alpha 4.0.3.2 (Scion Corporation, Frederick, MD).
Two-dimensional gel electrophoresis of carbonylated spinal cord proteins
DNP-labeled proteins were also dissolved in isoelectric focusing (IEF) buffer (8M urea, 2% CHAPS, 50mM DTT and 0.2% (w/v) Bio-Lyte® 3/10 ampholytes). Proteins were absorbed on 11cm-long ReadyStrip IPG® strips (pH range 3–10 non-linear; BioRad) and IEF was carried out at 30,000 V-hr. IEF strips were equilibrated for 10 min in 375mM Tris-HCl buffer pH 8.8 containing 6M urea, 2% SDS, 20% glycerol with 130mM DTT, followed by 10 min the same buffer containing 135mM iodoacetamide instead of DTT. Proteins were separated on 10% Criterion™ pre-cast gels (BioRad) followed by western blotting carried out as described above. Blots were probed with anti-DNP antibodies, then stripped and probed with antibodies against specific cytoskeletal proteins. Spot matching between blots was performed using The Discovery Series PDQuest 2-D Analysis Software Version 7.0.1 (BioRad).
Identification of carbonylated proteins
Tissue proteins (0.5mg), dissolved in 200 μl 2% SDS, were incubated at room temperature with 5 mM biotin-hydrazide (Sigma). After 60 min, proteins were precipitated with 1 ml of acetone at −20°C and collected by centrifugation at 10,000g for 10 min. Pellets were washed three times with 1 ml of acetone-water (3:1, v/v) and dissolved in 100 μl of 100 mM Tris-HCl buffer pH 7 containing 1% SDS and 100 mM NaCl. The solutions were diluted 20-fold with 100 mM NaCl and centrifuged at 10,000g for 10 min to remove any aggregated material. The supernatants were then incubated for 1 h at 20°C with 50μl of streptavidin-agarose (Sigma) previously equilibrated in 100 mM Tris-HCl pH 7.6 containing 0.05% SDS and 100mM NaCl (buffer A). The resin was washed 5-times with 500 μl of buffer A, 4-times with buffer A containing 1M NaCl, and once with buffer A without NaCl. Bound-proteins were eluted from the resin with 100 μl of SDS-sample buffer containing 1% 2-mercaptoethanol. Aliquots from the total and bound fractions were separated by SDS-PAGE on 10% polyacrylamide gels and blotted against PVDF membranes. Blots were probed with antibodies against NFH (1:1000, rabbit polyclonal; Chemicon, Temecula, CA), NFM (1:4000, rabbit polyclonal, Chemicon), NFL (1:1000, mouse monoclonal, Chemicon), β-tubulin (1:1000, mouse monoclonal; Sigma), GFAP (1:5000, rabbit polyclonal; Dako, Carpinteria, CA) and β-actin (1:1000, mouse monoclonal; Abcam Inc., Cambridge, MA), followed by incubation with the appropriate HRP-conjugated secondary antibody. Blots were developed by ECL as described above.
Immunohistochemical detection of protein carbonyls
Spinal cord segments were fixed overnight in methacarn (methanol : chloroform : acetic acid, 60:30:10 by vol) and then mounted in paraffin. Tissue was sectioned in the horizontal plane (6 μm-thick) and mounted on Vectabond™-treated slides (Vector Laboratories, Burlingame, CA). Sections were deparafinized with xylenes and a graded alcohol series, and then rinsed with phosphate buffered saline (PBS) solution for 10 min. Carbonyl groups were converted into DNP-hydrazones by reaction with 1mg/ml DNPH (Sigma) prepared in 2N HCl for 30 min. Sections were rinsed 3 times with PBS, blocked with 10% (v/v) normal goat serum and incubated overnight with rabbit anti-DNP (1:1000) (Sigma). After removing the primary antibody with PBS containing 0.1% triton X-100 (PBS-T), the sections were incubated for 3h with TRITC (Tetramethyl Rhodamine IsoThioCyanate)-conjugated goat anti-rabbit antibody (1:100) (Jackson ImmunoResearch laboratories; West Grove, PA). Sections were rinsed twice with PBS-T, once with PBS, and then mounted in a buffered glycerol solution containing p-phenylenediamine as anti-fade reagent. Images were captured with a Zeiss 200m microscope equipped with a Hamamatsu C4742-95 digital camera.
Statistical Analysis
Results were analyzed for statistical significance with Student's unpaired t test or ANOVA utilizing the GraphPad Prism® (version 4) program (GraphPad Software Inc., San Diego, CA).
Results
Characteristics of Lewis rat EAE animals
EAE in male Lewis rats was induced as described under “Materials and Methods.” Symptoms were graded according to the following scale: 0, no symptoms; 1, tail weakness; 1.5, clumsy gait; 2, hind limb paresis; 2.5, partial hind limb dragging; 3, hind limb paralysis; 3.5, hind limb paralysis with fore limb paresis; 4, complete paralysis; 5, moribund. Following immunization with MBP, Lewis rats experienced an acute and monophasic inflammation of the spinal cord resulting in clinical disability. Neurological symptoms begin at 9–11 DPI, peaking at 12–13 DPI, with full recovery by 16 DPI (Table 1). CFA-injected control and EAE rats were sacrificed on days 7, 9, 10, 11, 13, and 15 after injection. Animals injected with antigen that did not show symptoms after 12 DPI were excluded from our analysis. Spinal cord sections from vertebrae T6-L2 were removed; portions from each animal were homogenized for biochemical analysis or fixed in methacarn for IHC. Since none of the biochemical parameters corresponding to CFA injected control animals measured in this study changed during the course of the experiment, they were combined to obtain the average control values.
Table 1.
Characteristics of disease in EAE animals
| Number of days post-induction (DPI) of EAE | Number of CFA controls | Number of animals that developed EAE | Clinical Score at sacrifice |
|---|---|---|---|
| 7 | 1 | 0/3 | 3 = Grade 0 |
| 9 | 1 | 1/3 | 2 = Grade 0 |
| 1 = Grade 1.5 | |||
| 10 | 1 | 4/4 | 1 = Grade 3 |
| 1 = Grade 3.5 | |||
| 2 = Grade 4 | |||
| 11 | 5 | 9/9 | 3 = Grade 3 |
| 5 = Grade 3.5 | |||
| 1 = Grade 4 | |||
| 13 | 4 | 6/6 | 1 = Grade 2 |
| 4 = Grade 3 | |||
| 1 = Grade 3.5 | |||
| 15 | 3 | 4/4 | 1 = Grade 0 |
| 1 = Grade 1 | |||
| 1 = Grade 1.5 | |||
| 1 = Grade 3 |
Oxidative stress occurs in the spinal cord of EAE rats: oxidation of glutathione and lipids
To establish the occurrence of oxidative damage in EAE, we measured the levels of GSH in the spinal cords of the affected animals throughout the disease course. As shown in Figure 1A, GSH levels in EAE spinal cords was 58% and 72% of control values at 10 and 11DPI, respectively, indicating that the CNS of the affected animals is subjected to considerable oxidative stress. Normal GSH levels were observed prior to 10 DPI and after 11 DPI.
Figure 1.
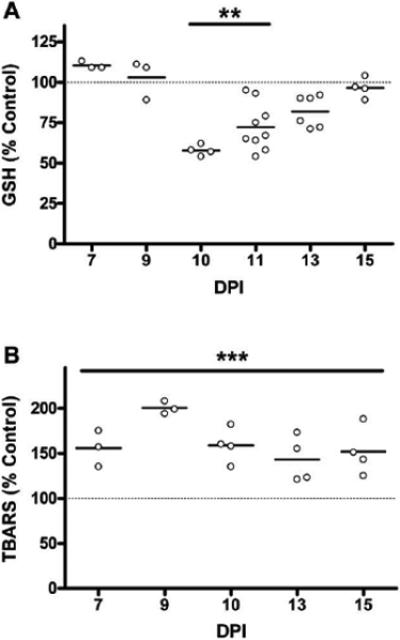
Temporal profile of glutathione oxidation and lipid peroxidation in EAE spinal cord. (A) Free glutathione (GSH) levels in EAE spinal cord homogenates through disease course measured using DTNB (see Materials and Methods). Data is represented as a percent of average control GSH levels (8.8 ± 1.3 nmol GSH/mg protein). (B) Lipid peroxidation was assessed by measuring TBARS as described in Materials and Methods in spinal cord homogenates through disease course. EAE values are expressed as a percentage of the average control value (0.48 ± 0.07 nmol TBARS/mg protein). GSH and TBARS data were analyzed for statistical significance using a one-way ANOVA with Dunnett's multiple comparison post-test to determine statistical significance from control, (**) p<0.01, (***) p<0.001. Lower glutathione at 10 –11 DPI points to considerable oxidative stress in the CNS during onset of neurological symptoms. Increase in TBARS at 7–15 DPI indicates carbonyl stress throughout the disease course.
Lipid oxidation, another commonly used marker of oxidative stress conditions, was determined by measuring the amount of thiobarbituric acid reactive substances (TBARS) in spinal cords. As depicted in Figure 1B, the level of TBARS augmented in EAE at all time points examined (i.e. 7–15 DPI), demonstrating carbonyl stress conditions in the EAE spinal cord before and after the period of clinical disability.
Oxidative stress damage increases protein carbonylation in EAE
We recently reported that protein carbonylation in brain slices can be stimulated by depleting GSH and/or inducing lipid peroxidation (Bizzozero et al. 2006). Because both of these changes take place during the course of EAE, we thought that protein carbonyls might also accumulate in this disorder. The presence of protein carbonyls in the spinal cord tissue of CFA-injected control animals and EAE rats throughout the disease course was investigated using the oxyblot technique. A representative western blot detecting protein carbonyls (i.e. oxyblot) of one control and 5 EAE spinal cords from different DPI is shown in Figure 2A. At 11 and 13 DPI, there is a clear increase in the carbonyl content of several protein bands from EAE rat spinal cords relative to control. Quantitative analysis of the oxyblots revealed that at 11 and 13 DPI, total protein carbonyl levels in EAE spinal cords increase by 70% relative to control spinal cords (Figure 2B) returning to control values at 15 DPI. Since carbonylation is an irreversible protein modification, the above results indicate that a clearance mechanism to eliminate these damaged proteins must operate in the disease tissue.
Figure 2.

Protein carbonyls in the spinal cord of EAE rats increase during disease course. Spinal cord homogenates from control and EAE animals were derivatized with 2,4 dinitrophenylhydrazine then separated by SDS-PAGE. Carbonylated proteins were determined using western blotting as described in Materials and Methods. (A) Representative blot showing one control and 5 EAE animals sacrificed at a range of days post injection (DPI). (B) Protein carbonyls (PCO) were tracked throughout the disease course and are expressed as a percentage of the average carbonylation in control spinal cords. Mean carbonyl levels (line) and the value for individual animals are shown at various DPI. Data was analyzed for statistical significance using one-way ANOVA with Dunnett's multiple comparison post-test to determine statistical significance from control, (**) p<0.01. Carbonylated protein species accumulate in the spinal cord of animals with acute EAE at 11 and 13 DPI and return to control levels at 15 DPI indicating a clearance mechanism for removal of oxidized species.
The accumulation of carbonyls in EAE spinal cords was also assessed using the DNPH-based IHC procedure that we have recently developed to determine the anatomical distribution of PCOs in brain sections (Bizzozero et al. 2006). In this case, however, detection of PCOs was performed by immunofluorescence utilizing a TRITC-labeled secondary antibody. Figure 3 shows the IHC distribution of carbonyls in the spinal cord of a control and an EAE animal at 11 DPI. The amount of carbonyls in both the gray matter (GM) and white matter (WM) from the CFA-injected control is relatively low. There is an overall increase in protein carbonyls in both WM and GM from EAE spinal cords, as opposed to localization around inflammatory lesions. Motor neurons and axons (arrows) were the most intensely stained structures in the diseased tissue, and this distribution is reminiscent to that of cytoskeletal elements. Validation of this IHC technique was performed by omitting the DNPH-treatment, the anti-DNP antibody or the secondary anti-rabbit IgG antibody (not shown). In addition, we carried out a positive control in which carbonyls are generated by incubating spinal cords sections with FeSO4/H2O2 and a specificity control in which endogenous carbonyls are removed by incubation with NaBH4.
Figure 3.
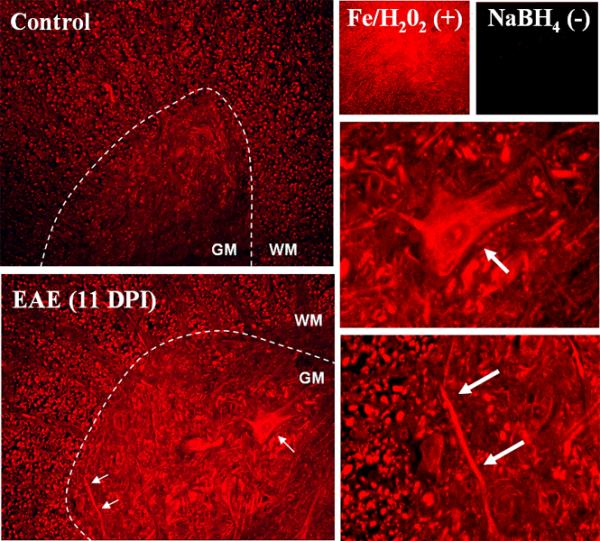
Immunohistochemical localization of protein carbonyls in the rat spinal cord of EAE animals. Carbonyls in thoracic spinal cord sections of a control and an EAE rat (score = 3.0) were detected by immunohistochemistry as described in Materials and Methods. Images show carbonyls the spinal cord ventral horn. Both neuronal cell bodies and axons are heavily stained in the EAE sample (arrows and insets). The carbonyls increase is seen throughout the spinal cord tissue, not localized around lesions. Tissue sections were treated with Fe/H2O2 to generate carbonyls (positive control) or NaBH4 to block endogenous carbonyls (negative control).
Oxidation levels of β-actin and NFL change during disease
The particular staining pattern for carbonyls prompted us to investigate whether the major cytoskeletal proteins are subjected to carbonylation. Identification of carbonyl-containing proteins was initially carried out by spot matching DNP-labeled proteins on a 2-D gel with those of specific cytoskeletal elements. As shown in Figure 4, β-actin, GFAP and the neurofilament proteins are among the major carbonylated targets in EAE. Quantification of the extent of oxidation of these species was performed by the pull-down/western blot procedure recently used in our laboratory to identify carbonylated cytoskeletal proteins during GSH depletion (Bizzozero et al. 2006, Bizzozero et al. 2007). To this end, protein carbonyls from control and 13 DPI EAE spinal cord homogenates were first converted into biotinylated residues by reaction with biotin-hydrazide. Biotin-containing proteins were then isolated with streptavidin-agarose and analyzed by western blotting employing antibodies against the three neurofilament chains, β-tubulin, β-actin, and GFAP. The results show that all six of these proteins are targets of carbonylation, with significant changes in the amount of oxidized NFL and β-actin (Error! Reference source not found.Table 2). It is important to note that, while total protein oxidation increases in EAE (Figure 2), the number of molecules that are carbonylated for specific proteins may increase or decrease depending on whether or not the modified species is selectively degraded during the course of the disease.
Figure 4.

Two-dimensional Oxyblot analysis of EAE spinal cord proteins. Spinal cord homogenates from EAE animals were derivatized with 2, 4-dinitrophenylhydrazine. Proteins were separated by 2-D-gel electrophoresis and blotted to PVDF membranes. Membranes were immunostained with anti-DNP, then stripped and re-probed with antibodies against the major cytoskeletal proteins. Cytoskeletal proteins on anti-DNP blots were identified by spot matching using BioRad PD-Quest software. The major carbonylated targets in EAE include β-actin, GFAP and the neurofilament proteins.
Table 2.
Specific oxidation of cytoskeletal proteins in control and EAE spinal cord.
| Control (% oxidized) | EAE (13 DPI) (% oxidized) | |
|---|---|---|
| NFH | 0.87 ± 0.09 | 1.71 ± 0.80 |
| NFM | 0.24 ± 0.04 | 0.32 ± 0.04 |
| NFL | 0.19 ± 0.04 | 0.08 ± 0.04 * |
| β-Tubulin | 0.08 ± 0.04 | 0.17 ± 0.06 |
| β-Actin | 0.39 ± 0.09 | 0.76 ± 0.06 * |
| GFAP | 1.87 ± 0.49 | 0.63 ± 0.38 |
Spinal cord homogenates from control and EAE (13 DPI) animals were derivatized with biotin hydrazide and carbonylated proteins were isolated with streptavidin-agarose as described in Materials and Methods. Total and bound fractions were analyzed by SDS-PAGE followed by western blotting for six cytoskeletal proteins (3 neurofilament chains, β-tubulin, β-actin, and GFAP). Specific oxidation (percentage of total protein modified by carbonylation) is reported. Data was analyzed for statistical significance using t-test,
p<0.05.
β-actin showed increased carbonylation, whereas NFL had less oxidation indicating the oxidized NFL may be degraded.
Neurofilaments and β-tubulin are degraded in EAE spinal cord
The integrity of CNS cytoskeletal network was investigated by western blotting of control and EAE spinal cord proteins. Figure 5A shows representative western blots of the three neurofilament chains, β-tubulin, β-actin and GFAP from control and EAE spinal cord (9 and 13 DPI). Figure 5B depicts the relative level of these proteins (normalized for coomassie blue staining) compared to average control values throughout the disease course. There are significantly lower levels of NFH, NFM, NFL, and β-tubulin in EAE compared to CFA-treated control. Since cytoskeletal proteins are very stable (Nixon & Logvinenko 1986, Li & Black 1996), this finding clearly indicates that their disappearance from the EAE spinal cord is due to increased degradation rather than diminished synthesis. A decrease in NFL protein at 10–12 DPI has been reported previously (Shields & Banik 1998) and is believed to be indicative of increased calpain activity in EAE spinal cord. GFAP and β-actin are not degraded, and the increase in GFAP protein at 15 DPI corroborates previous reports of gliosis beginning at 14 DPI in this model (Aquino et al. 1988). The specific oxidation (percentage of total protein modified by carbonylation) of β-actin (a protein that is not degraded) is higher in EAE, whereas that of NFL decreases in EAE. This finding suggests that, while β-actin is resistant to proteolysis, the oxidized neurofilament may be preferentially degraded in this disorder.
Figure 5.

Levels of cytoskeletal proteins in EAE spinal cord. Spinal cord homogenates from control and EAE animals (5 μg protein) were separated by SDS-PAGE followed by western blotting. Membranes were immunostained with antibodies against six cytoskeletal proteins (3 neurofilament chains, β-tubulin, β-actin, and GFAP). (A) Representative blot with 3 control and 3 EAE samples prepared from animals at 9 and 13 DPI are shown. (B) Levels of each cytoskeletal protein were tracked throughout the disease course and values are expressed as a percentage of the average protein level in control spinal cords. Values for individual animals as well as the mean value (line) are shown at each DPI. Data were analyzed for statistical significance using one-way ANOVA with Dunnett's multiple comparison post-test to determine statistical significance from control, (*) p<0.05, (**) p<0.01. Neurofilaments and β-tubulin are degraded in the disease, which is possibly a consequence of oxidative damage to these proteins.
Discussion
In this study we have characterized oxidative stress conditions that occur in MBP-induced EAE in the Lewis rat by measuring established markers for oxidative damage throughout the disease course. We discovered that the concentration of free GSH in the spinal cord of EAE rats is greatly decreased at 10-11 DPI, returning to normal values at 13 DPI. There are several, and not mutually exclusive, reasons underlying the reduction of free GSH in the diseased spinal cord. These include (1) oxidation of GSH to GSSG with hydrogen peroxide or lipid hydroperoxides via a reaction catalyzed by glutathione peroxidase; (2) conjugation of GSH with reactive α,β-unsaturated aldehydes derived form lipid peroxidation (e.g. acrolein, 4-hydroxynonenal) by a reaction catalyzed by gluthathione S-transferase; (3) reduced cellular uptake of cysteine, required for GSH synthesis, due to high levels of glutamate (Sagara & Schubert 1998); (4) decreased activity of enzymes involved in the GSH synthesis (e.g. glutathione synthetase) and/or recycling of GSSG (e.g. glutathione reductase); and (5) diminished amount of NADPH, which is needed for GSSG reduction. Preliminary results in our laboratory have shown that the amount of total glutathione (i.e. GSH + GSSG) in EAE spinal cord is also reduced, suggesting that the disappearance of GSH cannot be simply attributed to its oxidation. However, regardless of the mechanism responsible for the decrease in low-molecular-weight thiols, GSH levels return to normal values as inflammation subsides and animals recover.
TBARS, the most commonly used measurement for lipid peroxidation, detects dialdehydes (mostly MDA) resulting from breakdown of lipid hydroperoxides and endoperoxides (Esterbauer et al. 1991). In contrast to the reduction of GSH levels, which parallels disease activity, the concentration of TBARS in the EAE spinal cords is elevated at every time point measured (i.e. 7–15 DPI). This result indicates that oxidative stress takes place prior to the appearance of inflammatory lesions in the spinal cord of EAE animals, a pathological process that correlates temporally with neurological symptoms (Schaecher et al. 2002). The presence of TBARS in 15 DPI animals, which are recovering neurologically, may be due to continued ROS generation or more likely to the persistence of dialdehydes in the tissue (MDA t1/2= 2 days) (Aldini et al. 2006).
The most important finding in this study is that several carbonylated protein species accumulate in the spinal cord of EAE rats at 11–13 DPI. The degree and distribution of protein carbonylation present during autoimmune neuroinflammation was unexpected and indicates that the oxidative stress conditions in this model are very severe. Nonetheless, in this acute animal model of MS there appears to be a mechanism that eliminates these damaged proteins and bring carbonylation to control levels as the inflammatory process subsides. Indeed, it is believed that the accumulation of carbonylated proteins is a multifaceted process that takes into account both the rate of oxidation (dependent on both ROS formation and antioxidant levels) and the rate of proteolysis of the damaged proteins (Sayre et al. 2005). It will be important to know if repeated tissue injury would result in the long-term accumulation of oxidized proteins as seen in multiple sclerosis (Bizzozero et al. 2005), and ongoing studies are addressing this issue in chronic animal models of MS.
Since ROS have variable but still relatively short half-lives (10−9 s for hydroxyl radicals, 10−6 s for superoxide, 10−3 s for peroxynitrite, 100 s peroxyl radicals and 102 s for hydrogen peroxide) (Reth 2002), they normally damage targets that are in close proximity to their source. Thus, if ROS in EAE spinal cords were generated mostly by inflammatory cells, one would expect to find accumulation of protein carbonyls near the inflammatory foci. Instead, we discovered that protein carbonyls are not circumscribed to inflammatory lesions but accumulate throughout the spinal cord. This more generalized carbonyl increase, staining multiple cell types including neurons, suggests that the origin of ROS is likely metabolic. We have recently shown that total or even partial depletion of GSH with diethyl maleate or 1,2-bis(2-chloroethyl)-1-nitrosourea results in abundant protein carbonylation by a process involving mitochondrial production of ROS (Bizzozero et al. 2006). While other cellular/subcellular sources of ROS cannot be excluded, it is tempting to speculate that a similar mechanism takes place in EAE. Supporting this idea are the facts that (1) there is mitochondrial damage and augmented superoxide production in EAE and MS (Kalman et al. 2007) and (2) protein carbonyls do not increase until after this antioxidant is depleted. The lack of protein carbonylation in the presence of sustained RCS production is noteworthy and suggests that carbonylation of proteins in EAE takes place via a direct mechanism and not indirectly by reaction with dialdehydes (MDA, glyoxal, methyl glyoxal). In this regard, we have recently found that, while dependent on lipid peroxidation, the carbonylation of cytoskeletal proteins during depletion of glutathione probably occurs via lipid hydroperoxides rather than RCS (Bizzozero et al. 2007).
Another major finding in this study is the identification of several prominent cytoskeletal proteins as targets of carbonylation in EAE. Cytoskeletal proteins are established targets of protein carbonylation in many diseases including several CNS disorders (Aksenov et al. 2001, Muntane et al. 2006). More importantly, carbonylation of cytoskeletal proteins is known to have deleterious effects on the macromolecule structures. For instance, actin filaments and microtubules both destabilize and disassemble upon oxidation of their protein components (Dalle-Donne et al. 2001, Ozeki et al. 2005, Banan et al. 2001, Banan et al. 2004, Neely et al. 2005). The secondary structure of neurofilaments is also altered when the individual proteins become carbonylated (Gelinas et al. 2000), often leading to the formation of dense aggregates (Smith et al. 1995).
In the present study, we have demonstrated that neurofilament proteins and β-tubulin are extensively degraded in the EAE spinal cord, which likely causes alterations in the cytoskeletal network. We have also observed that the specific oxidation of NFL protein decreases in EAE, suggesting the possibility that oxidized neurofilament may be preferentially degraded in this disease. Although the heavy and medium chain neurofilament proteins do not show changes in the specific oxidation at the 13 DPI time point analyzed, variations in the susceptibility of these oxidized high molecular weight neurofilament chains to degradation or aggregate formation may reconcile these differences. Additionally, the more rapidly formation of newly synthesized, and therefore unoxidized, light chain neurofilament could contribute to the lower specific oxidation at this time point. It is well known that proteolysis of carbonylated proteins in cells under conditions of oxidative stress aids in preventing formation of large aggregates that could be cytotoxic (Davies & Shringarpure 2006, Shringarpure et al. 2001). The 20s proteasome has been shown to selectively recognize and degrade oxidized proteins (Grune et al. 1995, Pacifici et al. 1993, Rivett 1985), and upon its inhibition carbonylated proteins do indeed accumulate (Lee et al. 2001). Calpain is also believed to be responsible for neurofilament degradation in EAE (Shields & Banik 1998), but it is unclear if this calcium-activated protease has preference for oxidized over native polypeptides. Studies using proteasome and calpain inhibitors in EAE have primarily focused on abatement of neurological symptoms without looking at specific proteins that are protected from proteolysis (Hosseini et al. 2001, Vanderlugt et al. 2000, Hassen et al. 2006, Guyton et al. 2005). Whether calpain, the proteasome, or a combination of these proteolysis pathways is responsible for the degradation of carbonylated neurofilament proteins in EAE is currently under investigation through the use of specific protease inhibitors in vivo. Recognizing the triggers for neurofilament degradation may help us to understand the mechanisms of axonal degeneration during neuroinflammation.
Acknowledgements
This work was supported by PHHS grant NS 47448 from the National Institutes of Health.
Abbreviations
- CFA
complete Freund's adjuvant
- DPI
days post-injection
- EAE
experimental autoimmune encephalomyelitis
- GFAP
glial acidic fibrillary protein
- GSH
glutathione
- IHC
immunohistochemistry
- MDA
malondialdehyde
- MS
multiple sclerosis
- NFH
neurofilament heavy (200kDa) protein
- NFL
neurofilament light (69kDa) protein
- NFM
neurofilament medium (150kDa) protein
- PAGE
polyacrylamide gel electrophoresis
- PCOs
protein carbonyls
- RCS
reactive carbonyl species
- ROS
reactive oxygen species
- SDS
sodium dodecyl sulfate
- TBARS
thiobarbituric acid reactive substances
References
- Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2001;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- Aldini G, Dalle-Donne I, Facino RM, Milzani A, Carini M. Intervention strategies to inhibit protein carbonylation by lipoxidation-derived reactive carbonyls. Med. Res. Rev. 2006;27:817–868. doi: 10.1002/med.20073. [DOI] [PubMed] [Google Scholar]
- Aquino DA, Chiu FC, Brosnan CF, Norton WT. Glial fibrillary acidic protein increases in the spinal cord of Lewis rats with acute experimental autoimmune encephalomyelitis. J. Neurochem. 1988;51:1085–1096. doi: 10.1111/j.1471-4159.1988.tb03072.x. [DOI] [PubMed] [Google Scholar]
- Banan A, Fitzpatrick L, Zhang Y, Keshavarzian A. OPC-compounds prevent oxidant-induced carbonylation and depolymerization of the F-actin cytoskeleton and intestinal barrier hyperpermeability. Free Radic. Biol. Med. 2001;30:287–298. doi: 10.1016/s0891-5849(00)00471-8. [DOI] [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Shaikh M, Fields JZ, Farhadi A, Keshavarzian A. Novel effect of NF-kappaB activation: carbonylation and nitration injury to cytoskeleton and disruption of monolayer barrier in intestinal epithelium. Am. J. Physiol. Cell Physiol. 2004;287:C1139–1151. doi: 10.1152/ajpcell.00146.2004. [DOI] [PubMed] [Google Scholar]
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol. Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- Bizzozero OA. ISN-satellite meeting on Myelin development and function. Chichen Itza; Mexico: 2007. Major cytoskeletal proteins are carbonylated in multiple sclerosis; p. 33. [Google Scholar]
- Bizzozero OA, DeJesus G, Callahan K, Pastuszyn A. Elevated protein carbonylation in the brain white matter and gray matter of patients with multiple sclerosis. J. Neurosci. Res. 2005;81:687–695. doi: 10.1002/jnr.20587. [DOI] [PubMed] [Google Scholar]
- Bizzozero OA, Reyes S, Ziegler J, Smerjac S. Lipid peroxidation scavengers prevent the carbonylation of cytoskeletal brain proteins induced by glutathione depletion. Neurochem. Res. 2007 Jun 6; doi: 10.1007/s11064-007-9377-y. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Bizzozero OA, Ziegler JL, De Jesus G, Bolognani F. Acute depletion of reduced glutathione causes extensive carbonylation of rat brain proteins. J. Neurosci. Res. 2006;83:656–667. doi: 10.1002/jnr.20771. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Aldini G, Carini M, Colombo R, Rossi R, Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends Mol. Med. 2003;9:169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Lusini L, Milzani A, Di Simplicio P, Colombo R. Actin carbonylation: from a simple marker of protein oxidation to relevant signs of severe functional impairment. Free Radic. Biol. Med. 2001;31:1075–1083. doi: 10.1016/s0891-5849(01)00690-6. [DOI] [PubMed] [Google Scholar]
- Davies KJ, Shringarpure R. Preferential degradation of oxidized proteins by the 20S proteasome may be inhibited in aging and in inflammatory neuromuscular diseases. Neurology. 2006;66:S93–96. doi: 10.1212/01.wnl.0000192308.43151.63. [DOI] [PubMed] [Google Scholar]
- Day M. Histopathology of EAE. In: Lavi E, Constantinescu C, editors. Experimental models of multiple sclerosis. Springer; NY: 2005. pp. 25–43. [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, Kowall NW, Brown RH, Beal MF. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997;69:2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- Floor E, Wetzel MG. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J. Neurochem. 1998;70:268–275. doi: 10.1046/j.1471-4159.1998.70010268.x. [DOI] [PubMed] [Google Scholar]
- Friguet B, Szweda LI, Stadtman ER. Susceptibility of glucose-6-phosphate dehydrogenase modified by 4-hydroxy-2-nonenal and metal-catalyzed oxidation to proteolysis by the multicatalytic protease. Arch. Biochem. Biophys. 1994;311:168–173. doi: 10.1006/abbi.1994.1222. [DOI] [PubMed] [Google Scholar]
- Gelinas S, Chapados C, Beauregard M, Gosselin I, Martinoli MG. Effect of oxidative stress on stability and structure of neurofilament proteins. Biochem. Cell Biol. 2000;78:667–674. [PubMed] [Google Scholar]
- Gilgun-Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J. Neurol. 2004;251:261–268. doi: 10.1007/s00415-004-0348-9. [DOI] [PubMed] [Google Scholar]
- Gold R, Hartung HP, Toyka KV. Animal models for autoimmune demyelinating disorders of the nervous system. Mol. Med. Today. 2000;6:88–91. doi: 10.1016/s1357-4310(99)01639-1. [DOI] [PubMed] [Google Scholar]
- Grune T, Reinheckel T, Davies KJ. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997;11:526–534. [PubMed] [Google Scholar]
- Grune T, Reinheckel T, Joshi M, Davies KJ. Proteolysis in cultured liver epithelial cells during oxidative stress. Role of the multicatalytic proteinase complex, proteasome. J. Biol. Chem. 1995;270:2344–2351. doi: 10.1074/jbc.270.5.2344. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Sribnick EA, Wingrave JM, Ray SK, Banik NL. Axonal damage and neuronal death in multiple sclerosis and experimental autoimmune encephalomyelitis: the role of calpain. In: Waxman S, editor. Multiple sclerosis as a neuronal disease. Elsevier; NY: 2005. pp. 293–303. [Google Scholar]
- Hassen GW, Feliberti J, Kesner L, Stracher A, Mokhtarian F. A novel calpain inhibitor for the treatment of acute experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2006;180:135–146. doi: 10.1016/j.jneuroim.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Hosseini H, Andre P, Lefevre N, Viala L, Walzer T, Peschanski M, Lotteau V. Protection against experimental autoimmune encephalomyelitis by a proteasome modulator. J. Neuroimmunol. 2001;118:233–244. doi: 10.1016/s0165-5728(01)00352-6. [DOI] [PubMed] [Google Scholar]
- Kalman B, Laitinen K, Komoly S. The involvement of mitochondria in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2007;188:1–12. doi: 10.1016/j.jneuroim.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Lee MH, Hyun DH, Jenner P, Halliwell B. Effect of proteasome inhibition on cellular oxidative damage, antioxidant defences and nitric oxide production. J. Neurochem. 2001;78:32–41. doi: 10.1046/j.1471-4159.2001.00416.x. [DOI] [PubMed] [Google Scholar]
- LeVine SM. The role of reactive oxygen species in the pathogenesis of multiple sclerosis. Med. Hypotheses. 1992;39:271–274. doi: 10.1016/0306-9877(92)90121-r. [DOI] [PubMed] [Google Scholar]
- Li Y, Black MM. Microtubule assembly and turnover in growing axons. J. Neurosci. 1996;16:531–544. doi: 10.1523/JNEUROSCI.16-02-00531.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntane G, Daldo E, Martinez A, Rey MJ, Avila J, Perez M, Portero M, Pamplona R, Ayala V, Ferrer I. Glial fibrillary acidic protein is a major target of glycoxidative and lipoxidative damage in Pick's disease. J. Neurochem. 2006;99:177–185. doi: 10.1111/j.1471-4159.2006.04032.x. [DOI] [PubMed] [Google Scholar]
- Neely MD, Boutte A, Milatovic D, Montine TJ. Mechanisms of 4-hydroxynonenal-induced neuronal microtubule dysfunction. Brain Res. 2005;1037:90–98. doi: 10.1016/j.brainres.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Nie CL, Wei Y, Chen X, Liu YY, Dui W, Liu Y, Davies MC, Tendler SJ, He RG. Formaldehyde at low concentration induces protein tau into globular amyloid-like aggregates in vitro and in vivo. PLoS ONE. 2007;2:e629. doi: 10.1371/journal.pone.0000629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Logvinenko KB. Multiple fates of newly synthesized neurofilament proteins: evidence for a stationary neurofilament network distributed nonuniformly along axons of retinal ganglion cell neurons. J. Cell Biol. 1986;102:647–659. doi: 10.1083/jcb.102.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystrom T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005;24:1311–1317. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- Ozeki M, Miyagawa-Hayashino A, Akatsuka S, Shirase T, Lee WH, Uchida K, Toyokuni S. Susceptibility of actin to modification by 4-hydroxy-2-nonenal. J. Chromatogr. 2005;827:119–126. doi: 10.1016/j.jchromb.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Pacifici RE, Kono Y, Davies KJ. Hydrophobicity as the signal for selective degradation of hydroxyl radical-modified hemoglobin by the multicatalytic proteinase complex, proteasome. J. Biol. Chem. 1993;268:15405–15411. [PubMed] [Google Scholar]
- Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 2002;3:1129–1134. doi: 10.1038/ni1202-1129. [DOI] [PubMed] [Google Scholar]
- Riddles PW, Blakeley RL, Zerner B. Ellman's reagent: 5,5'-dithiobis(2-nitrobenzoic acid)--a reexamination. Anal. Biochem. 1979;94:75–81. doi: 10.1016/0003-2697(79)90792-9. [DOI] [PubMed] [Google Scholar]
- Rivett AJ. Preferential degradation of the oxidatively modified form of glutamine synthetase by intracellular mammalian proteases. J. Biol. Chem. 1985;260:300–305. [PubMed] [Google Scholar]
- Sagara Y, Schubert D. The activation of metabotropic glutamate receptors protects nerve cells from oxidative stress. J. Neurosci. 1998;18:6662–6671. doi: 10.1523/JNEUROSCI.18-17-06662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayre LM, Moreira PI, Smith MA, Perry G. Metal ions and oxidative protein modification in neurological disease. Ann. Ist Super. Sanita. 2005;41:143–164. [PubMed] [Google Scholar]
- Schaecher K, Rocchini A, Dinkins J, Matzelle DD, Banik NL. Calpain expression and infiltration of activated T cells in experimental allergic encephalomyelitis over time: increased calpain activity begins with onset of disease. J. Neuroimmunol. 2002;129:1–9. doi: 10.1016/s0165-5728(02)00142-x. [DOI] [PubMed] [Google Scholar]
- Shacter E. Quantification and significance of protein oxidation in biological samples. Drug Metab. Rev. 2000;32:307–326. doi: 10.1081/dmr-100102336. [DOI] [PubMed] [Google Scholar]
- Shields DC, Banik NL. Upregulation of calpain activity and expression in experimental allergic encephalomyelitis: a putative role for calpain in demyelination. Brain Res. 1998;794:68–74. doi: 10.1016/s0006-8993(98)00193-0. [DOI] [PubMed] [Google Scholar]
- Shringarpure R, Grune T, Davies KJ. Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cell Mol. Life Sci. 2001;58:1442–1450. doi: 10.1007/PL00000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smerjac S, Bizzozero OA. Evidence for increased protein carbonylation in experimental autoimmune encephalomyelitis. J. Neurochem. 2006;96(suppl. 1):132. doi: 10.1111/j.1471-4159.2007.05178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smerjac S, Bizzozero OA. Cytoskeletal degradation and oxidation in experimental autoimmune encephalomyelits. J. Neurochem. 2007;102(suppl. 1):226. doi: 10.1111/j.1471-4159.2007.05178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Rudnicka-Nawrot M, Richey PL, Praprotnik D, Mulvihill P, Miller CA, Sayre LM, Perry G. Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer's disease. J. Neurochem. 1995;64:2660–2666. doi: 10.1046/j.1471-4159.1995.64062660.x. [DOI] [PubMed] [Google Scholar]
- Stadtman ER. Covalent modification reactions are marking steps in protein turnover. Biochemistry. 1990;29:6323–6331. doi: 10.1021/bi00479a001. [DOI] [PubMed] [Google Scholar]
- Troncoso JC, Costello AC, Kim JH, Johnson GV. Metal-catalyzed oxidation of bovine neurofilaments in vitro. Free Radic. Biol. Med. 1995;18:891–899. doi: 10.1016/0891-5849(94)00224-8. [DOI] [PubMed] [Google Scholar]
- Vanderlugt CL, Rahbe SM, Elliott PJ, Del Canto MC, Miller SD. Treatment of established relapsing experimental autoimmune encephalomyelitis with the proteasome inhibitor PS-519. J. Autoimmun. 2000;14:205–211. doi: 10.1006/jaut.2000.0370. [DOI] [PubMed] [Google Scholar]
- Vitvitsky V, Thomas M, Ghorpade A, Gendelman HE, Banerjee R. A functional trans-sulfuration pathway in the brain links to glutathione homeostasis. J. Biol. Chem. 2006;281:35785–35793. doi: 10.1074/jbc.M602799200. [DOI] [PubMed] [Google Scholar]