

Abstract
Nitrosative stress has been implicated in the pathophysiology of several CNS disorders including multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE). We have recently shown that protein nitrosothiols (PrSNOs) accumulate in the brain of MS patients and there is indirect evidence that PrSNO levels are also increased in EAE. In this study we sought to identify the major PrSNOs in the spinal cord of EAE animals prepared by active immunization of C57/BL6 mice with MOG35-55 peptide. For this purpose, PrSNOs from control and EAE mice at various disease stages were derivatized with HPDP-biotin, and the biotinylated proteins were isolated with streptavidin-agarose. Proteins from total and streptavidin-bound fractions were then analyzed by western blotting using antibodies against the major S-nitrosylated substrates of CNS tissue. Using this approach we found that the proportion of S-nitrosylated neurofilament proteins, NMDA receptors, α/β-tubulin, β-actin and GAPDH is increased in EAE. Other potential substrates were either not S-nitrosylated in vivo (HCN3, HSP-72, CRMP-2, γ-actin, calbindin) or their S-nitrosylation levels were unaltered in EAE (Na/K ATPase, hexokinase, glycogen phosphorylase). We also discovered that neuronal specific enolase is the major S-nitrosylated protein in acute EAE. Given that S-nitrosylation affects protein function it is likely that the observed changes are significant to the pathophysiology of inflammatory demyelination.
Keywords: MS, nitric oxide, protein nitrosothiol, spinal cord, EAE
Introduction
Experimental autoimmune encephalomyelitis (EAE) is an inflammatory demyelinating CNS disorder that serves as a model for the human disease, multiple sclerosis (MS). As many other inflammatory disorders, MS and EAE are characterized by severe nitrosative stress due to an increase in the inducible form of nitric oxide synthetase (iNOS or NOS-2) (Bo et al., 1994; Lin et al., 1993). One of the major consequences of nitrosative stress is the generation of protein (PrSNO) and non-protein nitrosothiols [e.g. S-nitrosoglutathione (GSNO)], which are believed to produce significant alterations in tissue function (Stamler, 1994; Broillet, 1999). The presence of S-nitrosothiols in MS was indirectly suggested by Boullerne and co-workers (1995) who found elevated levels of antibodies to a conjugated S-nitroso-cysteine epitope in serum and cerebrospinal fluid (CSF) from MS patients, a finding later extended to EAE animals (Boullerne et al., 2002). Calabrese et al. (2003) also reported increased concentration of both nitric oxide metabolites and unidentified low-molecular-weight nitrosothiols (probably GSNO) in CSF from patients with active MS. More recently, we discovered that PrSNOs with molecular masses between 45–200 kDa accumulate in the brain white matter of MS patients, which provides direct evidence for the occurrence of protein S-nitrosylation in inflammatory demyelinating disorders (Bizzozero et al., 2005).
To understand the consequences of protein S-nitrosylation it is critical to identify the protein targets and how function is altered upon incorporation of the NO moiety. S-nitrosylation is a non-enzymatic process and there is no evidence for a linear flanking motif that predicts the S-nitrosylated cysteine residue (Hao et al., 2006). Yet, not every cysteine-containing protein and not every thiol group within a particular protein is subjected to modification by NO, which suggests specificity (Hess et al., 2005). Indeed, incubation of rat spinal cord slices with the GSNO leads to S-nitrosylation of a discrete number of proteins (Romero and Bizzozero, 2006, 2009). These species are likely to be the same as to those previously identified in rat cerebellum homogenates incubated with the NO-donors GSNO or DEA-NONOate (Jeffrey et al., 2001). They comprise 4 metabolic enzymes [glyceraldehyde-3-phosphate dehydrogenase (GAPDH), creatine kinase (CK), hexokinase 1 (HK), and glycogen phosphorylase (GP)], 3 ion channel-related proteins [NMDA-glutamate receptors, hyperpolarization-activated cation channel (HCN3) and Na+/K+ ATPase α2 subunit], 5 structural proteins [neurofilament heavy chain (NFH), α/β-tubulin and β/γ–actin] and 4 signaling proteins [retinoblastoma gene product (Rb), heat-shock protein 72 (Hsp72), collapsing response mediator protein-2 (CRMP-2) and calbindin].
The present study was undertaken to determine whether these major S-nitrosylatable proteins are also modified in vivo during the course of inflammatory demyelination. To this end, S-nitrosylated proteins were isolated from spinal cord of mice with acute and chronic EAE using the “biotin switch” method (Jaffrey et al., 2001), and were analyzed on western blots probed with specific antibodies. We chose this method over the classical 2-D gel electrophoresis/mass spectrometry approach, because of the limited reproducibility and known lack of quantification of the latter. The results show that several of the major in vitro NO targets are also modified in the spinal cord of mice with acute EAE. Nonetheless, the proportion of individual S-nitrosoproteins accumulated in the diseased tissue is, with exception of neuronal specific enolase, very small. This is the first study that directly shows accumulation of PrSNOs in EAE and that identifies many of the modified proteins. A preliminary account of this work has been published in abstract form (Bizzozero and Zheng, 2009).
Materials and Methods
Induction of Experimental Autoimmune Encephalomyelitis (EAE)
Housing and handling of the animals as well as the euthanasia procedure were in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee. Eight-week-old female C57BL/6 mice were purchased from Harlan Bioproducts (Indianapolis, IN) and housed in the UNM-animal resource facility. To induce EAE, animals received a subcutaneous injections into the lower back area of 200μl of MOG35-55 peptide (200μg) in saline mixed with complete Freund’s adjuvant (CFA) supplemented with 4 mg/ml of heat killed Mycobacterium tuberculosis H37Ra (Chondrex Inc; Redmond, WA). Control animals were given CFA without MOG peptide. Two-hours and 48h after EAE induction, all animals received an i.p. injection of 0.3 μg of pertussis toxin (List Biological Laboratories; Campbell, CA) in 100 μl of saline. Seven days after disease induction mice received a second immunization with the MOG peptide in CFA. Animals were weighed and examined daily for the presence of neurological signs. At prescribed days post-immunization (DPI) animals were euthanized by decapitation, and spinal cords (T1-L5) were removed and homogenized by probe sonication in 0.8 ml of HEN buffer (50 mM Hepes pH 7.7, 1 mM EDTA and 0.1 mM neocuproine) containing 60 mM methyl methanethiol sulfonate (MMTS) (Sigma, St. Louis, MO) to block free SH groups and prevent the chemical denitrosylation of proteins. Homogenates were kept at −20°C until use. Protein concentration was assessed with the Bio-Rad DC™ protein assay (Bio-Rad Laboratories; Hercules, CA) using bovine serum albumin as standard.
Pull-down of S-nitrosylated proteins
Excess MMTS was removed by acetone precipitation. Proteins were dissolved in SDS-containing HEN buffer and incubated with 3 mM ascorbic acid (Sigma) and 40mM EZ-link® HPDP-biotin (Thermo Scientific, Rockford, IL) at room temperature for 1 h. HPDP-biotin was removed by acetone precipitation and proteins were dissolved in neutralization buffer (100 mM NaCl, 0.05% SDS and 0.5% Triton X-100 in HEN buffer). Proteins were then incubated for 1 h at 20°C with 25 μl of streptavidin-agarose (Novagen, Madison, WI) previously equilibrated in neutralization buffer. The resin was washed 5 times with neutralization buffer, 4 times with neutralization buffer containing 0.5 M NaCl and once with HEN buffer. Bound-proteins were eluted from the resin by incubation for 30 min at 37°C with SDS-sample buffer containing 1% 2-mercaptoethanol (2-ME). Aliquots from the total fraction (also called the starting material) and the 2-ME eluate were separated by SDS-PAGE and blotted against PVDF membranes. Blots were probed with monoclonal (mAb) or polyclonal antibodies (Ab) (1:1000) against α-spectrin (mAb-1622, Chemicon), neurofilament heavy chain (NFH) (mAb-N5389, Sigma), neurofilament medium chain (NFM) (mAb-N5264, Sigma), neurofilament light chain (NFL) (mAb-1615, Chemicon), glutamate receptor NR2A (Ab-1555P, Millipore), retinoblastoma protein (Rb) (mAb-316, Millipore), glycogen phosphorylase (GP) (mAb-HG-GPBB, Advanced Immunochemicals Inc, Long Beach, CA), hyperpolarizing-activated cation 3 (HCN3) (Ab9338, Millipore), hexokinase I (Ab-3543, Millipore), Na+/K+ ATPase α2 (Ab-07674, Millipore), HSP-72 (Ab-61097, Abcam), fructose-6-phosphate kinase (PFK) (Ab-37583, Abcam), collapsin response mediator protein-2 (CRMP-2) (Ab-9218, Millipore), α-tubulin (mAb-7291, Abcam), β-tubulin (mAb-T4026, Sigma), 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) (mAb-326, Chemicon), neuronal specific enolase (NSE) (Ab-N0649, Sigma), glial fibrillary acidic protein (GFAP) (mAb-G3893, Sigma), creatine kinase (Ab-28898, Santa Cruz), β-actin (Ab-26276, Genetex Inc, San Antonio, TX), γ-actin (Ab-3265, Millipore), GAPDH (mAb-1D4, EnCor Biotechnology, Gainesville, FL), calbindin-D (Ab-CB38a; Swant, Bellizona, Switzerland), triosephosphate isomerase (TPI) (Ab-28760, Abcam), myelin proteolipid protein (PLP) (mAb, a gift from Dr. V.J. Kuchroo, Harvard Medical School) and myelin basic protein (MBP) (mAb-381, Chemicon), followed by incubation with the appropriate HRP-conjugated secondary antibody. Blots were developed by enhanced chemioluminescence (ECL). The developed films were scanned in a Hewlett Packard Scanjet 4890 and the images were quantified using the NIH Image 1.63 imaging analysis program.
Statistical Analysis
Results were analyzed for statistical significance with ANOVA utilizing GraphPad Prism® program (GraphPad Software Inc., San Diego, CA).
Results and Discussion
Characteristics of EAE mice
EAE in female C57BL/6 mice was induced by active immunization with MOG35-55 peptide as described under Materials and Methods. Symptoms were graded according to the following scale: 0, no symptoms; 1, tail weakness; 2, hind limb paresis; 3, hind limb paralysis; 4, complete paralysis; 5, moribund. In this chronic EAE model, neurological symptoms begin at 14 DPI (7 days after the boost with MOG peptide) reaching a peak at 30 DPI, and most animals remain ill (score 3.0–3.5) throughout the entire experimental period (60 DPI) (Fig. 1). Control animals, which received both CFA and pertussis toxin, showed no neurological symptoms. Acute disease was defined as having clinical signs of EAE without any signs of improvement for at least three consecutive days. At this stage the spinal cord pathology is characterized perivenular infiltration of inflammatory cells (Raine et al., 1990). Chronic EAE was defined arbitrarily as animals that remain in the stationary phase of the disease for 20 days (50 DPI). At this stage there is reduced perivascular and parenchymal inflammation, and no evidence of active transmigration of inflammatory cells into the spinal cord (Raine et al., 1990). Biochemical analysis was performed on homogenates of spinal cord sections from vertebrae T1-L5. Since none of the biochemical parameters corresponding to control animals measured in this study changed during the course of the experiment, they were combined to obtain the average control values. Also, since changes in S-nitrosylation levels between control and EAE animals with a clinical score of 4 is just 2–3 fold (see below), we did not attempt to established whether there is a correlation between the amount of S-nitrosylated protein and disease activity at a particular DPI.
Fig. 1.

Clinical course of EAE in C57BL/6 female mice. EAE was induced by active immunization MOG35-55 peptide as described under Materials and Methods. Animals were monitored daily for signs of clinical disease and scored as indicated in the text. Clinical scores represent the mean of 6 control and 10 EAE mice. Three animals with the acute disease (scores 3.5–4.0), three at the chronic phase (score 3–3.5) and the corresponding controls were used for pull-down assays (Figs. 3–8).
Validation of the pull-down assay
Identification of S-nitrosylated proteins and quantification of the extent of modification was performed using a pull-down/western blot method (Jaffrey et al., 2001). In this procedure, protein thiols are first blocked by reaction with MMTS (Fig. 2, step 1), then PrSNOs are decomposed with ascorbate (step 2) and the newly created thiols are biotinylated by reaction with HPDP-biotin (step 3). A small aliquot of this protein homogenate is saved for western blotting and the rest is processed to isolate the biotinylated proteins using streptavidin-agarose. Proteins are eluted from the beads with SDS-sample buffer containing 2-ME, which cleaves the disulfide bond between the protein and the biotin moiety, and aliquots are also analyzed by western blotting. Blots are either stained with Commassie blue to determine the proportion of total proteins that are S-nitrosylated (Fig. 3) or developed with specific antibodies (Figs. 4–8). A number of preliminary studies were carried out to ascertain (1) the concentration of MMTS and time necessary for complete blockage of free thiols groups, (2) the amount of streptavidin-agarose necessary for complete binding of biotinylated proteins, and (3) the composition and number of washes that ensure the proper removal of non-biotinylated proteins from the agarose beads before elution with 2-ME. Also, no material was recovered from the streptavidin agarose when either the HPDP-biotin was omitted from the derivatization step (lane 2 in Figs. 4–8) or when the nitrosothiols were eliminated with dithiothreitol prior to the treatment with MMTS (lane 1 in Figs. 4–8), indicating that the procedure for isolating S-nitroso-proteins is indeed specific. Since quantification was carried out by scanning the western blots developed by ECL, films were exposed for different periods to ascertain the linearity of the response. Yet the technique should be regarded as semi-quantitative since it is difficult to correct for efficiency of protein transfer to the PVDF membrane during electroblotting step, particularly from one run to the next. To minimize this problem, a set of samples (both total and 2-ME eluate fractions) from a control, an acute EAE and a chronic EAE animal were always processed in parallel and ran on the same gel.
Fig. 2.

Schematic representation of biotin-switch/pull-down assay used to identify S-nitrosylated proteins.
Fig. 3.

Levels of S-nitrosoprotein in acute and chronic EAE. Spinal cord proteins were derivatized with HPDP-biotin and biotinylated proteins were isolated with streptavidin-agarose as described under “Materials and Methods”. Aliquots from the starting material (1/600 of the total used) and the 2-ME eluate (1/10 of the total recovered) were analyzed by SDS-PAGE, blotted against PVDF membranes and stained with Commassie blue (panel A). Densitometric scans were obtained to calculate the proportion of S-nitrosylated protein (panel B). Values represent the mean ± SEM of three animals in each group. *Significantly different (p<0.05) from control values. # Significantly different (p<0.05) from acute EAE.
Fig. 4.
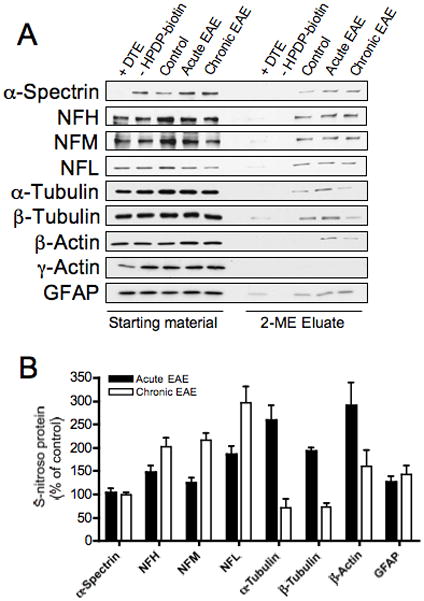
S-Nitrosylation of cytoskeletal proteins in EAE. S-nitrosylated proteins were isolated, separated on SDS-gels and transferred to PVDF membranes as indicated in Fig. 3. Blots were probed with antibodies against the major cytoskeletal proteins and were developed by ECL (panel A). Densitometric scans were obtained to calculate the proportion of each S-nitroso-protein (panel B). Values are expressed as the percentage of control and represent the mean ± SEM of 3 separate experiments.
Fig. 8.

S-Nitrosylation of major myelin proteins in EAE. S-nitrosylated proteins were isolated, separated on SDS-gels and transferred to PVDF membranes as indicated in Fig. 3. Blots were probed with antibodies against the major myelin proteins and developed by ECL (panel A). Densitometric scans were obtained to calculate the proportion of each S-nitroso-protein (panel B). Values are expressed as the percentage of control and represent the mean ± SEM of 3 separate experiments.
Levels of S-nitrosoproteins in EAE spinal cords
Densitometric scanning of the Coomassie blue stained PVDF membranes revealed that approximately 0.2% of the proteins from control spinal cords are S-nitrosylated (Fig. 3). Assuming an average molecular weight of 50,000 and one -SNO moiety per protein molecule that value represents ~ 40 pmol of S-nitrosylated thiols/mg total protein. Considering that there is 60 nmol of protein SH groups/mg spinal cord protein (Romero and Bizzozero, 2006), less 1 in 1000 thiols in the spinal cord is nitrosylated under physiological conditions. This very low value of protein modification explains why direct detection of biotin-derivatized proteins on western blots (Nitroglo assay) and analytical determination of RSNOs by spectrophotometry (Saville, 1958) and spectrofluorometry (Park and Costa, 1997), all failed to detect PrSNOs in the mouse spinal cord (data not shown). Only the pull-down method, which concentrates S-nitrosylated proteins from large amounts of tissue, is capable of detecting basal levels of PrSNOs in murine CNS tissue. The proportion of modified protein increased to 0.6% and 0.3% in the spinal cord of acute and chronic EAE mice, respectively (Fig. 3). The pattern of protein S-nitrosylation during the course of the disease resembles that of iNOS activity (Cross et al., 1996), suggesting that most of the NO utilized for protein S-nitrosylation derives from that enzyme. However, the contribution of the calcium-dependent nNOS and eNOS cannot be ruled out, particularly since there is considerable dysregulation of calcium homeostasis in this disorder (Brand-Schieber and Werner, 2004; Kurnellas et al., 2005). Moreover, there is some evidence that the levels of these two other enzymes are also increased in acute EAE (Kim et al., 2000). Thus, it would be interesting to carry out experiments similar as those in the present study using iNOS- and nNOS-deficient mice, both of which are available in EAE susceptible mouse strains (Fenyk-Melody et al., 1998; Liñares et al., 2006).
Identification of major S-nitrosylated proteins in acute and chronic EAE
Initially, our study focused on the 16 proteins that were identified by Jaffrey et al (2001) as major targets of S-nitrosylation in the CNS, which include several structural proteins, metabolic enzymes, ion channels and signaling proteins. However, because of our interest in myelin/cytoskeletal instability during inflammatory demyelination, several proteins related to those structures were added to the original list.
A- Cytoskeletal proteins
As shown in Fig. 4, not only NFH but all three neurofilament proteins (NFPs) are S-nitrosylated in control spinal cords. The proportion of all three S-nitroso-NFPs augmented in both acute and chronic EAE. In contrast, S-nitrosylation of both α-tubulin and β-tubulin was increased only during acute EAE. These changes may have functional implications since S-nitrosylation and thiolation of tubulins are known to affect their polymerization rate (Landino et al., 2007). The S-nitrosylation of the major microfilament protein β-actin was also elevated only in animals with acute EAE. Like S-nitroso-tubulin, S-nitroso-β-actin has been shown to polymerize less efficiently than unmodified actin as a consequence of reduced annealing rate (Dalle-Donne et al., 2000). α-Spectrin and the astrocyte-specific protein GFAP were S-nitrosylated to the same extent in control and EAE spinal cords. Other major cytoskeletal proteins including γ-actin (Fig. 4A), dynein, ankyrin and tropomyosin (not shown) were not modified by S-nitrosylation in either control or EAE tissues. In sum, it is likely that the abnormal S-nitrosylation of several structural proteins such as NFPs, tubulin and β-actin in EAE may contribute to the pathophysiology of this disorder, particularly if the modified molecules are concentrated in a limited number of cells.
B- Metabolic enzymes
Among the four metabolic enzymes that are S-nitrosylated in vitro (Jaffrey et al., 2001) only GAPDH was S-nitrosylated to a higher level in EAE (Fig. 5A). The activity of this glycolytic enzyme is greatly attenuated after nitrosylation of its critical thiol (cys-149) at the active site (Padgett and Whorton, 1995; Mohr et al., 1996), and this may be relevant to the disease process. In addition, it has been recently shown that GAPDH S-nitros(yl)ation induces its binding to the E3 ubiquitin ligase Siah1 to cause nuclear translocation and to promote apoptosis (Hara et al., 2006). S-nitrosylation of hexokinase (HK) was the same in control and diseased spinal cords. It is noteworthy that incubation of HK with GSNO leads to enzyme inactivation. However, the effect is apparently caused by nitration of several tyrosine residues rather than to nitrosylation of cysteine thiols (Miller et al., 2007). Creatine kinase, whose activity is inhibited by S-nitrosylation (Arstall et al., 1998; Konorev et al., 2000), was barely modified in both control and EAE spinal cord. GP, whose activity is augmented in the presence of NO (Borgs et al., 1996), was S-nitrosylated to the same extent in control and EAE tissues.
Fig. 5.

S-Nitrosylation of metabolic enzymes in EAE. S-nitrosylated proteins were isolated, separated on SDS-gels and transferred to PVDF membranes as indicated in Fig. 3. Blots were probed with antibodies against various metabolic enzymes and developed by ECL (panel A and B). Densitometric scans obtained to calculate the proportion of each S-nitroso-GAPDH and S-nitroso-NSE (panel C). Values are expressed as the percentage of control and represent the mean ± SEM of 3 separate experiments.
We also investigated the possible modification of three additional metabolic enzymes (TPI, PFK, NSE) that are known to be S-nitrosylated by GSNO in cell free systems (Hao et al, 2006). As shown in Fig 5B, the proportion of S-nitroso-PFK and S-nitroso-TPI was the same in control and EAE spinal cord. Surprisingly, NSE, which is minimally S-nitrosylated in control spinal cords, was heavily modified in acute EAE. The proportion of S-nitroso-NSE at the peak of disease is approximately 1.8%, a nearly 30-fold increase relative to control and chronic EAE animals (Fig. 5C).
C- Ion-channels-related proteins
The hyperpolarization-activated cation channel (HCN3) was not S-nitrosylated either in control or EAE tissues (Fig. 6). Note that the HCN3 reactivity in the pull-down lanes corresponding to EAE and controls animals was also present in the DTE-treated and biotin negative controls. Na/K ATPase α-2 subunit was modified equally in control and EAE spinal cords. In contrast, the proportion of S-nitroso-NR2A increased in both acute and chronic EAE. These changes may be of pathological significance since S-nitrosylation of a single cysteine residue in NR2A modulates its channel activity (Choi et al., 2000). However, just like in the case of NFPs (Fig. 1), the elevated proportion of S-nitroso-NR2A in EAE is caused by a decrease in the levels of this protein rather than to a real increase in the amount S-nitrosylated molecules per spinal cord tissue. This could be interpreted as increased S-nitrosylation of a reduced number of protein molecules or that the pool of S-nitroso-NR2A is not the same as the one that gets degraded during the neurodegenerative phase of the disease.
Fig. 6.

S-Nitrosylation of ion channels-related proteins in EAE. S-nitrosylated proteins were isolated, separated on SDS-gels and transferred to PVDF membranes as indicated in Fig. 3. Blots were probed with antibodies against various ion-channel proteins and developed by ECL (panel A). Densitometric scans were obtained to calculate the proportion of each S-nitroso-protein (panel B). Values are expressed as the percentage of control and represent the mean ± SEM of 3 separate experiments.
D- Signal transduction proteins
The retinoblastoma (Rb) protein was not detected in either in the total homogenate or the 2-ME eluates even using an antibody that recognizes both the underphosphorylated and hyperphosphorylated forms of this protein (not shown). The other three signal transduction-related proteins described in the study of Jeffrey et al (2001) (HSP-72, CRMP-2, calbindin) were detected in the total homogenates but they were absent in the 2-ME eluates of control as well as EAE spinal cords (Fig. 7). The barely discernible calbindin reactivity seen in the 2-ME eluates from control and EAE specimens was also observed in the samples pretreated with DTE and in those incubated in the absence of HPDP-biotin, and thus was considered nonspecific. The above findings indicate that while some proteins are susceptible to S-nitrosylation in vitro with various NO-donors, they may not be modified in vivo to any appreciable extent even under severe nitrosative stress conditions.
Fig. 7.

S-Nitrosylation of signaling proteins in EAE. S-nitrosylated proteins were isolated, separated on SDS-gels and transferred to PVDF membranes as indicated in Fig. 3. Blots were probed with antibodies against several signaling proteins and developed by ECL.
E- Myelin proteins
We finally investigated the S-nitrosylation of the three major myelin proteins in EAE (Fig. 8). As expected from its lack of cysteine residues, MBP is not S-nitrosylated. The S-nitrosylation of PLP, the major protein of the myelin sheath, was low and unchanged in the diseased state. It should be noted that PLP is S-nitrosylated in MBP-induced EAE in Lewis rats (Romero and Bizzozero, unpublished results), suggesting that the processes underlying the pathology of these two animal models of MS is somewhat different. S-nitrosylation of PLP has been linked to decompaction of CNS myelin at the level of the intraperiod line where this protein plays an adhesive role (Bizzozero et al., 2004). In contrast to PLP, CNPase was S-nitrosylated in control animals and the extent of modification was slightly increased in acute EAE.
Conclusions
This is the first study to identify S-nitrosylated proteins in an inflammatory demyelinating disorder. The results clearly show that there are several proteins whose S-nitrosylation states increase in EAE. However, the amount of S-nitrosylated proteins is generally low, which is consistent with previous studies showing that under physiological conditions PrSNO levels in brain are < 1 pmol/mg protein, a value that increases by just 2-fold during severe hypoxia (Bryan et al. 2004). We have recently shown that protein S-denitrosylation in spinal cord sections depends almost exclusively on the intracellular concentration of GSH (Romero and Bizzozero, 2009). Thus, not only increased NO synthesis but also reduced GSH levels are needed to cause an accumulation of PrSNOs. In this regard, we have found that the concentration of GSH in the spinal cord diminishes by 30%–40% at the peak of EAE (Smerjac and Bizzozero, 2008). This, along with an increase in iNOS, explains the build-up of S-nitrosoproteins in this phase of the disease. Our data also show that not every cysteine-containing protein and not every protein that is modified in vitro by incubation with NO-donors (e.g. calbindin, γ-actin, CRMP-2, HCN3) is S-nitrosylated in vivo even in pathological conditions characterized by severe nitrosative stress like EAE. In addition, we found proteins that are physiologically S-nitrosylated and yet the extent of the modification does not rise in the course of the disease (e.g. Na/K ATPase, HK, GP). We also discovered other proteins whose S-nitrosylation state increases as a result of a reduction in protein levels (e.g. NR2A, NFPs). Certainly the most remarkable finding was the extensive modification of NSE in acute EAE, and to a lesser extent that of GAPDH in both acute and chronic EAE. While the effect of S-nitrosylation on GAPDH activity has been investigated extensively, there are no studies showing that S-nitroso-NSE is indeed not functional. Work is underway in our laboratory to address this issue. Also, since the identification of the S-nitrosylated proteins was accomplished using only one method, the present findings will have to be validated with alternative approaches as they become available.
At this time, there are a number of reasons why it is difficult to ascertain the pathological consequences of increased protein S-nitrosylation in EAE. First, the proteins whose S-nitrosylation are augmented participate in different biochemical pathways, and are present in different cell types (e.g. NSE and NFPs in neurons, CNPase in oligodendrocytes). Second, for many of these proteins nothing is known regarding the effect of S-nitrosylation on their function (e.g. NSE, NFPs). Third, it is still unclear if increased S-nitrosylation in the course of the disease is indeed deleterious. For instance, administration of GSNO to EAE rats causes S-nitrosylation of p65 in endothelial cells thereby inhibiting the infiltration of immune cells into the CNS and reducing disease activity (Prasad et al., 2007). Also, S-nitrosylation of MyD88 and mitochondrial caspases retards Toll-like receptor signaling (Into et al., 2007) and this could suppress T-cell mediated autoimmunity (Lampropoulou et al., 2008). On the other hand, it is conceivable that S-nitrosylation (and likely inhibition) of important metabolic enzymes like GAPDH and NSE could lead to neuronal death later in the disease process. Fourth, levels of protein modification in the whole EAE spinal cord are low, though S-nitrosylated proteins may be concentrated in specific areas. Indeed, preliminary studies in our laboratory using PLP-induced EAE mice suggest that most S-nitrosylated substrates are present in the vicinity of inflammatory lesions, which is also in agreement with the distribution of iNOS (Parkinson et al., 1997).
Acknowledgments
This work was supported by PHHS grant NS 47448 from the National Institutes of Health.
References
- Arstall MA, Bailey C, Gross WL, Bak M, Balligand JL, Kelly RA. Reversible S-nitrosation of creatine kinase by nitric oxide in adult rat ventricular myocytes. J Mol Cell Cardiol. 1998;30:979–988. doi: 10.1006/jmcc.1998.0662. [DOI] [PubMed] [Google Scholar]
- Bizzozero OA, DeJesus G, Howard T. Exposure of rat optic nerves to nitric oxide causes protein S-nitrosation and myelin decompaction. Neurochem Res. 2004;29:1675–1685. doi: 10.1023/b:nere.0000035802.27087.16. [DOI] [PubMed] [Google Scholar]
- Bizzozero OA, DeJesus G, Bixler HA, Pastuszyn A. Evidence of nitrosative damage in the brain white matter of patients with multiple sclerosis. Neurochem Res. 2005;30:139–149. doi: 10.1007/s11064-004-9695-2. [DOI] [PubMed] [Google Scholar]
- Bizzozero OA, Zheng J. Identification of S-nitrosylated proteins in acute and chronic EAE. J Neurochem. 2009;108(Suppl 1) PTW03–18. [Google Scholar]
- Bo L, Dawson T, Wasswlingh S, Mork S, Choi S, Kong PA, Hanley D, Trapp BD. Induction of nitric oxide synthetase in demyelinating regions of MS brains. Ann Neurol. 1994;36:778–786. doi: 10.1002/ana.410360515. [DOI] [PubMed] [Google Scholar]
- Borgs M, Bollen M, Keppens S, Yap SH, Stalmans W, Vanstapel F. Modulation of basal hepatic glycogenolysis by nitric oxide. Hepatology. 1996;23:1564–1571. doi: 10.1002/hep.510230637. [DOI] [PubMed] [Google Scholar]
- Boullerne AI, Petry K, Meynard M, Geffard M. Indirect evidence for nitric oxide involvement in multiple sclerosis by characterization of circulating antibodies directed against conjugated S-nitrosocysteine. J Neuroimmunol. 1995;60:117–124. doi: 10.1016/0165-5728(95)00061-6. [DOI] [PubMed] [Google Scholar]
- Boullerne AI, Rodriguez JJ, Touil T, Brochet B, Schmidt S, Abrous ND, Le Moal M, Pua JR, Jensen MA, Mayo W, Arnason BG, Petry KG. Anti-S-nitrosocysteine antibodies are a predictive marker for demyelination in experimental autoimmune encephalomyelitis: implications for multiple sclerosis. J Neurosci. 2002;22:123–132. doi: 10.1523/JNEUROSCI.22-01-00123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand-Schieber E, Werner P. Calcium channel blockers ameliorate disease in a mouse model of multiple sclerosis. Exp Neurol. 2004;189:5–9. doi: 10.1016/j.expneurol.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Broillet MC. S-nitrosylation of proteins. Cell Mol Life Sci. 1999;55:1036–1042. doi: 10.1007/s000180050354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan NS, Rassaf T, Maloney RE, Rodriguez CM, Saijo F, Rodriguez JR, Feelisch M. Cellular targets and mechanisms of nitros(yl)ation: an insight into their nature and kinetics in vivo. Proc Natl Acad Sci USA. 2004;101:4308–4313. doi: 10.1073/pnas.0306706101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese V, Scapagnini G, Ravagna A, Bella R, Butterfield DA, Calvani M, Pennisi G, Giuffrida-Stella AM. Disruption of thiol homeostasis and nitrosative stress in the cerebrospinal fluid of patients with active multiple sclerosis: evidence for a protective role of acetylcarnitine. Neurochem Res. 2003;28:1321–1328. doi: 10.1023/a:1024984013069. [DOI] [PubMed] [Google Scholar]
- Choi Y-B, Tenneti L, Le DA, Ortiz J, Bai G, Chen V, Lipton SA. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nature Neurosci. 2000;3:15–21. doi: 10.1038/71090. [DOI] [PubMed] [Google Scholar]
- Cross A, Keeling RM, Goorha S, San M, Rodi C, Wyatt PS, Manning PT, Misko TP. J Neuroimmunol. 1996;71:143–153. doi: 10.1016/s0165-5728(96)00147-6. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Milzani A, Giustarini D, Di Simplicio P, Colombo R, Rossi R. S-NO-actin: S-nitrosylation kinetics and the effect on isolated vascular smooth muscle. J Muscle Res Cell Motil. 2000;21:171–81. doi: 10.1023/a:1005671319604. [DOI] [PubMed] [Google Scholar]
- Fenyk-Melody JE, Garrison AE, Brunnert SR, Weidner JR, Shen F, Shelton BA, Mudgett JS. Experimental autoimmune encephalomyelitis is exacerbated in mice lacking the NOS2 gene. J Immunol. 1998;160:2940–2946. [PubMed] [Google Scholar]
- Hao G, Derekhshan B, Shi L, Campagne F, Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci USA. 2006;103:1012–1017. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara MR, Snyder SH. Nitric oxide-GAPDH-Siah: a novel cell death cascade. Cell Mol Neurobiol. 2006;26:527–538. doi: 10.1007/s10571-006-9011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nature Review of Molecular Cell Biology. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- Into T, Inomata M, Nakashima M, Shibata K, Häcker H, Matsushita K. Regulation of MyD88-dependent signaling events by S nitrosylation retards toll-like receptor signal transduction and initiation of acute-phase immune responses. Mol Cell Biol. 2008;28:1338–4713. doi: 10.1128/MCB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris H, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Kim S, Moon C, Wie MB, Kim H, Tanuma N, Matsumoto Y, Shin T. Enhanced expression of constitutive and inducible forms of nitric oxide synthetase in autoimmune encephalomyelitis. J Vet Sci. 2000;1:11–17. [PubMed] [Google Scholar]
- Konorev EA, Kalyanrsman B, Hogg N. Modification of creatine kinase by S-nitrosothiols: S-nitrosylation vs S-thiolation. Free Rad Biol Med. 2000;28:1671–1678. doi: 10.1016/s0891-5849(00)00281-1. [DOI] [PubMed] [Google Scholar]
- Kurnellas MP, Nicot A, Shull GE, Elkabes S. Plasma membrane calcium ATPase deficiency causes neuronal pathology in the spinal cord: a potential mechanism for neurodegeneration in multiple sclerosis and spinal cord injury. FASEB J. 2005;19:298–300. doi: 10.1096/fj.04-2549fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampropoulou V, Hoehlig K, Roch T, Neves P, Calderón-Gómez E, Sweenie CH, Hao Y, Freitas AA, Steinhoff U, Anderton SM, Fillatreau S. TLR-Activated B cells suppress T cell-mediated autoimmunity. J Immunol. 2008;180:4763–4773. doi: 10.4049/jimmunol.180.7.4763. [DOI] [PubMed] [Google Scholar]
- Landino LM, Koumas MT, Mason CE, Alston JA. Modification of tubulin cysteines by nitric oxide and nitroxyl donors alters tubulin polymerization activity. Chem Res Toxicol. 2007;20:1693–1700. doi: 10.1021/tx7001492. [DOI] [PubMed] [Google Scholar]
- Lin R, Lin T, Tilton RD, Cross A. Nitric oxide localized to spinal cords of mice with experimental allergic encephalomyelitis: an electron paramagnetic resonance study. J Exp Med. 1993;178:643–648. doi: 10.1084/jem.178.2.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liñares D, Taconis M, Maña P, Correcha M, Fordham S, Staykova M, Willenborg DO. Neuronal nitric oxide synthetase plays a key role in CNS demyelination. J Neurosci. 2006;26:12672–12681. doi: 10.1523/JNEUROSCI.0294-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S, Ross-Inta C, Giulivi C. Kinetic and proteomic analyses of S-nitrosoglutathione-treated hexokinase: consequences for cancer energy metabolism. Amino Acids. 2007;32:593–602. doi: 10.1007/s00726-006-0424-9. [DOI] [PubMed] [Google Scholar]
- Mohr S, Stamler JS, Brüne B. Posttranslational modification of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosylation and subsequent NADH attachment. J Biol Chem. 1996;271:4209–4214. doi: 10.1074/jbc.271.8.4209. [DOI] [PubMed] [Google Scholar]
- Padgett CM, Whorton AR. S-nitrosoglutathione reversibly inhibits GAPDH by S-nitrosylation. Am J Physiol. 1995;269:C739–C749. doi: 10.1152/ajpcell.1995.269.3.C739. [DOI] [PubMed] [Google Scholar]
- Park JK, Kostka P. Fluorometric detection of biological S-nitrosothiols. Anal Biochem. 1997;249:61–66. doi: 10.1006/abio.1997.2159. [DOI] [PubMed] [Google Scholar]
- Parkinson JF, Mitrovic B, Merrill JE. The role of nitric oxide in multiple sclerosis. J Mol Med. 1997;75:174–186. doi: 10.1007/s001090050102. [DOI] [PubMed] [Google Scholar]
- Prasad R, Giri S, Nath N, Singh I, Singh AK. GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions. Glia. 2007;55:65–77. doi: 10.1002/glia.20436. [DOI] [PubMed] [Google Scholar]
- Raine CS, Cannella B, Duijvestijin AM, Cross AH. Homing to central nervous system vasculature by antigen specific lymphocytes. Lab Invest. 1990;63:476–489. [PubMed] [Google Scholar]
- Romero JM, Bizzozero OA. Extracellular S-nitrosoglutathione, but not S-nitrosocysteine or N2O3, mediates protein S-nitrosation in rat spinal cord slices. J Neurochem. 2006;99:1299–1310. doi: 10.1111/j.1471-4159.2006.04180.x. [DOI] [PubMed] [Google Scholar]
- Romero JM, Bizzozero OA. Reduced glutathione is required for denitrosylation of CNS proteins. J Neurosci Res. 2009;87:701–709. doi: 10.1002/jnr.21897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saville B. A scheme for the colorimetric determination of microgram amounts of thiols. Analyst (London) 1958;83:670–672. [Google Scholar]
- Smerjac S, Bizzozero OA. Cytoskeletal protein carbonylation and degradation in experimental autoimmune encephalomyelitis. J Neurochem. 2008;105:763–772. doi: 10.1111/j.1471-4159.2007.05178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]