

Abstract
Background
Tuberculosis (TB) continues to cause a high toll of disease and death among children worldwide. The diagnosis of childhood TB is challenged by the paucibacillary nature of the disease and the difficulties in obtaining specimens. Whereas scientific and clinical research efforts to develop novel diagnostic tools have focused on TB in adults, childhood TB has been relatively neglected. Blood transcriptional profiling has improved our understanding of disease pathogenesis of adult TB and may offer future leads for diagnosis and treatment. No studies applying gene expression profiling of children with TB have been published so far.
Results
We identified a 116-gene signature set that showed an average prediction error of 11% for TB vs. latent TB infection (LTBI) and for TB vs. LTBI vs. healthy controls (HC) in our dataset. A minimal gene set of only 9 genes showed the same prediction error of 11% for TB vs. LTBI in our dataset. Furthermore, this minimal set showed a significant discriminatory value for TB vs. LTBI for all previously published adult studies using whole blood gene expression, with average prediction errors between 17% and 23%. In order to identify a robust representative gene set that would perform well in populations of different genetic backgrounds, we selected ten genes that were highly discriminative between TB, LTBI and HC in all literature datasets as well as in our dataset. Functional annotation of these genes highlights a possible role for genes involved in calcium signaling and calcium metabolism as biomarkers for active TB. These ten genes were validated by quantitative real-time polymerase chain reaction in an additional cohort of 54 Warao Amerindian children with LTBI, HC and non-TB pneumonia. Decision tree analysis indicated that five of the ten genes were sufficient to classify 78% of the TB cases correctly with no LTBI subjects wrongly classified as TB (100% specificity).
Conclusions
Our data justify the further exploration of our signature set as biomarkers for potential childhood TB diagnosis. We show that, as the identification of different biomarkers in ethnically distinct cohorts is apparent, it is important to cross-validate newly identified markers in all available cohorts.
Keywords: Biomarker, Children, Mycobacterium tuberculosis, Transcriptomics
Background
It is estimated that one third of the world’s population is infected with Mycobacterium tuberculosis and that each year about nine million people develop tuberculosis (TB), one million (11%) of whom are children under 15 years of age [1]. A unique aspect of TB in children is the rapid progression to disease, typically within the first year following infection, unlike in adults, where TB infection can persist for decades without progression into an active infection [2]. Bacteriological confirmation in the diagnosis of childhood TB is the exception rather than the rule with culture remaining negative in around 70% of cases with probable TB [3]. Using blood transcriptional profiling, several signature gene sets have been identified in adult cohorts from South Africa, The Gambia and The United Kingdom [4-6]. However, a significant overlap was shown with a biomarker set for sarcoidosis, suggesting the need for more specific biomarker sets [7]. To statistically verify differential expression between active TB, latent TB infection (LTBI) and healthy controls (HC) different methods have been used, varying from statistical tests [4,6] to prediction models using the k-nearest neighbours algorithm [4]. Correlation analysis, a method selecting genes that are correlated with a single differentially expressed gene, was used to identify a biomarker set in a Gambian cohort [5]. No studies applying gene expression profiling of children with TB have been published, and it is unknown whether the existing signature gene sets are applicable to childhood cohorts.
In Venezuela, a high TB incidence rate (3190 per 100,000) has been reported in Warao Amerindian children living in the Orinoco Delta in northeastern Venezuela [8]. In this study, we identified new gene signatures in childhood TB by comparing gene expression profiles of Warao Amerindian children with TB, LTBI and HC. We validated the identified gene signatures from this study in an independent cohort of children with LTBI, HC or non-TB pneumonia. Furthermore, we estimated the predictive value of our gene signatures in previously performed adult studies and we compared the discriminatory power of the literature signature gene sets with our gene set.
Results
Identification of signature genes
Genome-wide transcription profiles in whole blood from 9 TB patients, 9 LTBI and 9 HC were generated using Affymetrix exon arrays comprising approximately one million probes, which are mapped to 22011 unique features (Affymetrix core gene set). General characteristics of the study subjects are given in Table 1. Detailed information of the study subjects is given in Additional file 1: Table S1. Random forest analyses were performed to find the signature gene sets used to interrogate whether donors within this study could be divided into distinct groups based on their gene expression profiles. Irrelevant genes were removed from the signature set using the random forest-based local importance measure as described in PhenoLink [9]. A total of 21798 genes were removed in the initial step and the classification or out of bag (OOB) error decreased substantially from 70% to 22%. Next, genes contributing to the correct classification of at least three samples of the same class were selected resulting in a removal of a total of 97 genes and a decrease of the OOB error to approximately 11%. The reduced dataset consisting of 116 genes allowed separation of the three classes with class errors of 11%, 22%, and 0% for the respective classes of TB, LTBI and HC (Table 2). Unsupervised hierarchical cluster analysis of this 116 gene profile showed that all 27 individuals could be successfully clustered into three groups, and each group matched to the corresponding grouping of TB, LTBI and HC (Figure 1). Functional annotation of the 116 genes revealed that genes in the categories of cell proliferation, cell death, phosphorylation and calcium binding were enriched. A full list of enriched gene sets analyzed by the online DAVID tool [10] is provided as supplemental information (Additional file 2: Table S2).
Table 1.
Characteristics of children with TB, LTBI and HC in which microarray analyses were performed
| |
|
TB |
LTBI |
HC |
|---|---|---|---|---|
| Number of donors | 9 | 9 | 9 | |
| Characteristics | ||||
| Age |
Mean (SD) |
7.8 (5.0) |
8.9 (4.5) |
7.2 (3.5) |
| |
Range |
1.1 – 14.5 |
2.2 – 14.6 |
1.3 – 11.5 |
| Gender |
Female |
7 |
3 |
7 |
| Male | 2 | 6 | 2 | |
Table 2.
Class errors of the 116 signature gene set
| Active TB | Latent TB | Healthy controls | Class error (%) | OOB | |
|---|---|---|---|---|---|
| Active |
8 |
0 |
1 |
11.1 |
11.1 |
| Latent |
0 |
7 |
2 |
22.2 |
22.2 |
| Healthy | 0 | 0 | 9 | 0.0 | 0.0 |
Figure 1.
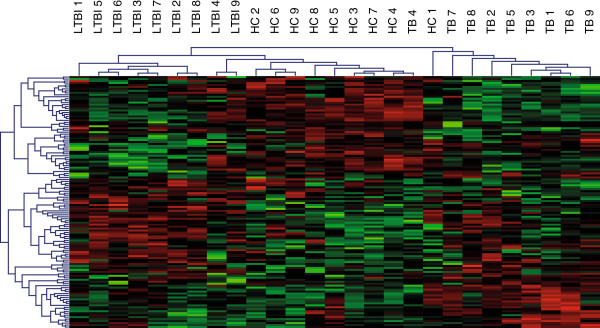
Unsupervised hierarchical cluster analysis of the 116 gene profile.
Validation of signature gene sets in independent sample sets
In order to confirm the value of our identified gene set in comparison with existing signature gene sets in all available cohorts that used whole blood gene expression [4-6], we performed random forest classification of the datasets with all signature gene sets to distinguish TB from LTBI and to distinguish TB, LTBI and HC. The average prediction error for the classification of TB and LTBI patients identified by Berry et al.[4] using our 116-gene signature set was 20.1% (Table 3). The average error for the two sets of Maertzdorf et al.[5,6] was respectively 26.5% and 19.1% (Table 3). Comparison of the other literature sets with each other showed similar average prediction errors when one set was used to discriminate individuals in the other set. However, while our 116-gene set showed a good predictive value for both the childhood cohort described in this study as well as for the adult cohorts identified in other studies, the gene sets identified in those adult studies did not discriminate the children included in our study adequately (average prediction error for TB vs. LTBI 33-50% (Table 3)). As Berry et al. provided demographic characteristics of the subjects included in their study [4], we were able to examine whether these demographic characteristics were related to the chance that a subject with TB was wrongly classified as LTBI using our 116 gene set. Multivariable linear regression analysis showed that age and gender were not significantly associated with wrong classification of TB subjects. In contrast, TB patients from South Africa were significantly more often wrongly classified as LTBI than TB patients from London (beta coefficient corrected for age and gender = 0.339, 95% CI 0.213 – 0.465).
Table 3.
Performance of signature gene sets: cross prediction matrix showing prediction errors
|
Class comparison |
Signature gene set |
This study |
Berry et al. [[4]] |
Maertzdorf et al. [[6]] |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Study | 116 gene set | Minimal TB-LTBI set | Minimal TB-LTBI-HC set | Robust 10 gene q-PCR set | 86 gene set | 393 gene set | 11 gene set | 5 gene set | |
| TB vs. LTBI |
Berry et al.[4] |
20.1 |
19.4 |
nda |
nd |
12.5 |
11.1 |
13.0 |
16.3 |
| Maertzdorf et al.[5] |
26.5 |
22.5 |
nd |
nd |
19.4 |
24.5 |
22.5 |
21.4 |
|
| Maertzdorf et al.[6] |
19.1 |
16.9 |
nd |
nd |
11.3 |
10.1 |
10.2 |
10.1 |
|
| This study |
11.1 |
11.1 |
nd |
nd |
50.0 |
50.0 |
33.0 |
50.0 |
|
|
average prediction error |
19.2 |
17.5 |
nd |
nd |
23.3 |
23.9 |
19.7 |
24.5 |
|
| TB vs. LTBI vs. HC |
Berry et al.[4] |
27.6 |
nd |
23.9 |
34.9 |
20.1 |
14.1 |
27.3 |
30.4 |
| Maertzdorf et al.[5] |
48.6 |
nd |
50.3 |
52.5 |
47.4 |
48.6 |
50.0 |
41.8 |
|
| Maertzdorf et al.[6] |
25.6 |
nd |
25.6 |
48.9 |
17.8 |
21.3 |
26.2 |
32.8 |
|
| This study |
11.1 |
nd |
14.8 |
14.8 |
66.7 |
74.1 |
74.1 |
70.4 |
|
|
28.2 |
nd |
28.7 |
37.8 |
38.0 |
39.5 |
44.4 |
43.8 |
||
| TB vs. LTBI vs. HC vs. other disease | Berry et al.[4] | 37.4 | nd | nd | nd | 27.6 | 18.7 | 28.5 | 37.2 |
Columns represent selected gene biomarker sets in the literature sets as well as in our dataset. Rows represent the studies on which the gene biomarker sets displayed in the columns were tested.
a = not determined.
Identification of the minimal discriminatory signature gene sets
A minimal discriminatory gene set to discriminate between TB and LTBI was identified by variable selection random forest (VarSelRF) [11]. This procedure produced four genes for the set described in this manuscript, three genes for the dataset described by Berry et al.[4], three genes for the dataset described by Maertzdorf et al.[5] and two genes for the other dataset described by Maertzdorf et al.[6] that are required to distinguish TB from LTBI samples (Additional file 3: Table S3). After removal of the overlaps, i.e. the genes that were selected in more than one dataset, nine genes remained (Additional file 3: Table S3). Similarly, a minimal gene set to distinguish TB from LTBI and from HC was identified. In this comparison, six genes were identified for the set described in this manuscript and respectively 35, three, and 25 genes were identified for the three other datasets [4-6] (Additional file 3: Table S3). After removal of the overlaps, 42 genes remained (Additional file 3: Table S3). Performance of these minimal sets was comparable to performance of the 116 gene set (Table 3). However, as only a few genes included in the minimal sets were present in more than one of the datasets, the selected minimal sets seem to be a summary of four small sets that perform well on each of the included datasets rather than a robust representative set that would perform well in ethnically different populations.
Identification and validation of the optimal signature gene set
Because of potential problems with the extrapolation of the minimal gene signature datasets to other populations due to overfitting of these gene sets on the source datasets, we selected ten genes from the random forest signature 116-gene set that were highly discriminative between the three groups of TB, LTBI and HC in our set of Warao Amerindian children. Furthermore, the ten selected genes were consistently selected in the bootstrapping procedure implemented in VarSelfRF from the set of Berry et al. (individuals from The United Kingdom and South Africa, including the individuals suffering from other inflammatory diseases [4]) and from the two sets of Maertzdorf et al. (individuals from The Gambia [5] and South Africa [6]). Additional file 3: Table S3 shows the variable frequencies estimated by the bootstrapping procedure per probe per dataset. The selection of ten genes out of these 116 genes consisted of CHRM2, AMPH, SNX17, PIGC, TAS2R46 (downregulated in TB vs. LTBI) and HBD, GLDC, ACOT7, S100P and STYXL1 (upregulated in TB vs. LTBI). These ten selected genes had the highest variable frequency in .632+ bootstrapped runs of the 116 discriminatory gene set in all cohorts, meaning that they were most frequently present in the trees of the random forest analyses performed (Additional file 3: Table S3). Their possible role in TB, lung disease or inflammatory processes is displayed in Table 4. While the minimal set was a mere combination of genes that had a good predictive value for active TB in each of the source databases, the robust ten gene set represents a set of genes that perform well in the discrimination of active TB from LTBI and HC in all datasets used for its composition and probably also as in future cohorts from other study sites. The overlap between the minimal nine gene set for TB vs. LTBI and the robust ten gene set consisted of three genes (HBD, CHRM2 and GLDC) and the overlap between the minimal 42 gene set for TB vs. LTBI vs. HC and the robust ten gene set consisted of seven genes (HBD, CHRM2, GLDC, ACOT7, SNX17, TAS2R46 and PIGC).
Table 4.
Set of 10 signature genes with their role in TB, lung disease or inflammatory processes
| Gene symbol | Gene name | Function | Possible role in TB, lung disease or inflammatory processes |
|---|---|---|---|
| CHRM2 |
Cholinergic muscarine 2 receptor |
cAMP regulation on airway smooth muscle. |
· Loss of muscarine receptor function is associated with airway hyperreactivity [12]. |
| AMPH |
Amphiphysin |
Phagocytosis, clathrin-mediated endocytosis in alveolar macrophages [13]. |
· Clathrin-mediated endocytosis in the lungs plays an important role in mediating the internalization of human rhinovirus and influenza A virus [14,15]. |
| · Inhibition of clathrin-mediated endocytosis led to inhibition of lipopolysaccharide (LPS) internalization and cytokine/chemokine release from macrophages stimulated by LPS [16]. | |||
| SNX17 |
Sorting nexin 17 |
Intracellular binding protein for the adhesion molecule P-selectin [17]. |
· P-selectin is important in the early phase of cell migration in TB infection and increased P-selectin serum levels are found in TB patients [18]. |
| PIGC |
Phosphatidylinositol glycan anchor biosynthesis class C |
Biosynthesis of glycosylphosphatidylinositol [19]. |
· Incorporation of the mycobacterial cell wall component lipoarabinomannan (LAM) into the macrophage cell membrane, a process that is dependent on successful insertion of a glycosylphosphatidylinositol anchor, is one of the key virulence factors for M. tuberculosis[20]. |
| S100P |
S100 calcium binding protein P |
Calcium-binding protein involved in intracellular and extracellular calcium sensing and signal transduction [21]. |
· M. tuberculosis-mediated inhibition of a cytosolic rise in calcium is one of the essential steps in phagosome maturation [22]. |
| TAS2R46 |
Taste receptor type 2 member 46 |
Regulation of ciliary beat frequency through modulation of intracellular calcium concentration [23]. |
· M. tuberculosis-mediated inhibition of a cytosolic rise in calcium is one of the essential steps in phagosome maturation [22]. Decreased expression of TAS2R receptors has been shown to lead to a decrease in intracellular calcium concentration [23]. |
| · TAS2Rs are expressed on human airway smooth muscle where they cause bronchodilation through a localized calcium response [24]. | |||
| STYXL1 |
Serine/threonine/tyrosine interacting-like1 |
Inhibition of formation of stress granules. |
· Stress granules are host RNA cytoplasmic granules formed in response to infections by a pathway involving phosphorylation of the translation initiation factor eIF2α [25]. |
| HBD |
Hemoglobin delta |
Encodes for the delta globin chain of HbA2. |
· Involved in oxygen transport from the lung to the peripheral tissues. |
| GLDC |
Glycine dehydrogenase (decarboxylating) |
Metabolic enzyme promoting cellular transformation. |
· Altered GLDC expression has been correlated with survival time in lung cancer patients [26]. |
| ACOT7 | Acyl-CoA thioesterase 7 | Expressed in macrophages, plays a role in inflammation through production of arachidonic acid. | · The molecular and cellular functions of ACOT7 have identified the enzyme as a candidate drug target in inflammatory diseases [27]. |
For validation of the ten genes included in the robust ten gene set, we carried out quantitative real-time polymerase chain reaction (qRT-PCR) studies using the same samples as used for the microarray experiment. Additionally, we tested our identified set of ten genes in 54 additional samples (20 LTBI, 16 HC, 18 non-TB pneumonia) from Warao Amerindian children aged 1 to 15 years. Furthermore, from three children with TB a recovery sample taken five months after initiation of anti-TB treatment was tested with qRT-PCR. Analysis of the qRT-PCR data showed that S100P (p = 0.004), GLDC (p = 0.016) and HBD (p = 0.027) significantly discriminated TB from LTBI while PIGC (p = 0.007), SNX17 (p = 0.019), TAS2R46 (p = 0.017) and HBD (p = 0.007) significantly discriminated TB cases from HC. TB cases were separated from non-TB pneumonia cases based on expression of SNX17 (p = 0.027) and HBD (p = 0.006). Active TB cases were separated from all other groups (i.e. LTBI, HC and non-TB pneumonia) based on expression of PIGC (p = 0.045), GLDC (p = 0.044) and HBD (p = 0.025). The values of area under receiver operating characteristic (ROC) curve (AUC) of these genes are shown in Table 5. The quantitative results of qRT-PCR analyses are shown in Additional file 4: Figure S1 and Additional file 5: Table S4. Decision tree analysis indicated that five genes (S100P, HBD, PIGC, CHRM2 and ACOT7) were sufficient to classify 78% of the TB cases correctly with no false-positives among the children with LTBI (100% specificity). Among the HC and non-TB pneumonia cases, false positive rates were 4% and 11% respectively (Figure 2). Interestingly, following this decision tree, the follow-up samples that were taken from three of the nine TB patients when they had received five months of anti-TB treatment were no longer classified as TB while the samples taken on inclusion of these patients were correctly classified as TB.
Table 5.
Receiver operating characteristic analysis of selected genes
|
Gene |
Down- or upregulation in TB |
TB (n = 9) vs. LTBI (n = 29) |
TB (n = 9) vs. HC (n = 25) |
TB (n = 9) vs. pneumonia (n = 18) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AUC | p-value | % Sens | % Spec | AUC | p-value | % Spec | AUC | p-value | % Spec | ||
| ACOT7 |
Upregulation |
0.70 |
0.073 |
67 |
86 |
0.63 |
0.24 |
76 |
0.67 |
0.15 |
72 |
| AMPH |
Downregulation |
0.55 |
0.86 |
56 |
52 |
0.60 |
0.91 |
76 |
0.56 |
1.00 |
61 |
| CHRM2 |
Downregulation |
0.63 |
0.17 |
56 |
62 |
0.62 |
0.33 |
46 |
0.52 |
0.87 |
44 |
| GLDC |
Upregulation |
0.78 |
0.016 |
67 |
79 |
0.64 |
0.19 |
73 |
0.66 |
0.13 |
72 |
| HBD |
Upregulation |
0.79 |
0.027 |
67 |
93 |
0.77 |
<0.01 |
78 |
0.79 |
<0.01 |
83 |
| PIGC |
Downregulation |
0.62 |
0.29 |
55 |
76 |
0.80 |
<0.01 |
89 |
0.73 |
0.076 |
94 |
| S100P |
Upregulation |
0.80 |
<0.01 |
89 |
76 |
0.58 |
0.77 |
35 |
0.64 |
0.24 |
39 |
| SNX17 |
Downregulation |
0.54 |
0.67 |
56 |
65 |
0.74 |
0.019 |
86 |
0.71 |
0.027 |
83 |
| STYXL1 |
Upregulation |
0.58 |
0.67 |
56 |
65 |
0.60 |
0.34 |
30 |
0.62 |
0.27 |
61 |
| TAS2R46 | Downregulation | 0.65 | 0.31 | 67 | 72 | 0.76 | 0.017 | 84 | 0.73 | 0.071 | 78 |
Figure 2.
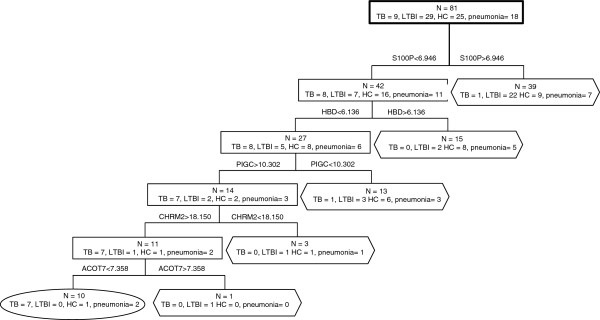
Decision tree analysis to differentiate TB from LTBI, HC and non-TB pneumonia. The combination of S100P, HBD, PIGC, CHRM2 and ACOT7 provides the best discrimination between TB, LTBI, HC and non-TB pneumonia. The sensitivity and specificity of this five-gene panel was 78% and 96% respectively. 94% of the children were correctly classified. Rectangle: internal nodes; Oval and hexagon: terminal nodes showing the number finally determined as TB and non-TB (LTBI, HC or non-TB pneumonia).
Discussion
Although peripheral blood transcriptional signatures discriminating between TB, LTBI and HC subjects have been identified in adult studies [4-6], concerns about the specificity of these signature sets have been raised [7]. Furthermore, the performance of these signature sets in children, that show high rates of progressive tuberculosis due to immaturity of the immune response, has not been investigated so far. In this study, we identified a 116 signature gene set that discriminated TB from LTBI and HC with class errors of 11%, 22%, and 0% for the respective classes of TB, LTBI and HC (Table 2). While this 116 gene signature set also showed a good discriminative value between TB and LTBI in adults from South Africa, The Gambia and The United Kingdom, signature sets that were identified in those adult cohorts were unable to discriminate TB from LTBI in our childhood cohort (Table 3).
Gene clusters that were enriched in our signature set included genes in the categories of (programmed) cell death and calcium binding (Additional file 2: Table S2). Both the Gambian as well as the South-African study of Maertzdorf et al.[5,6] also described enrichment of genes involved in cell death. Other similarities between the functional annotations in the South African study [6] and our study are the enrichment of genes involved in regulation of cell proliferation, regulation of caspase activity and protein kinase activity. Specifically, CD64 was identified as the most powerful discriminating gene seperating TB from LTBI cases in the South African study [6]. As CD64 has also been identified as a marker for general innate immune response activity and sepsis, this marker may not be specific to TB [28]. Berry et al. observed that genes downstream of type I interferon-αβ receptor signaling were over-respresented in patients with active TB [4]. However, type I inferferon signaling is also induced in response to respiratory viruses [29] and Streptococcus pneumoniae[30], questioning the specificity of genes involved in type I interferon receptor signaling as biomarkers for active TB.
The enrichment of genes involved in calcium signaling in our TB biomarker set has not been described before in adult studies using whole-blood gene expression [4-6], nor in studies based on transcriptional profiling of peripheral blood mononuclear cells (PBMCs) [31,32]. A close relation between abnormal calcium metabolism and radiological extent of disease has been described in pulmonary TB patients [33,34]. Alterations in serum calcium, particularly cases of hypercalcemia, have been observed in adult TB patients [33-35]. Hypercalcemia in pediatric TB patients is an infrequently recognized and poorly understood phenomenon [36]. In lung tissue, several processes related to calcium homeostasis are thought to contribute to M. tuberculosis persistence and the aggregation of macrophages in granulomas. Over-production of 1,2-dihydroxyvitamin D3, which plays a traditional role in calcium metabolism, in alveolar macrophages in granulomas has a protective effect against oxidative injuries from the nitric oxide burst from granulomatous macrophages [37-39]. Furthermore, M. tuberculosis inhibits a calcium-dependent phagolysosome formation pathway which leads to the prevention of maturation of M. tuberculosis-containing phagosomes into phagolysosomes. This process, referred to as the M. tuberculosis phagosome maturation arrest, is critical for M. tuberculosis persistence in the human host [22]. S100P, which significantly discriminated TB from LTBI in our study children, and TAS2R46, which significantly distinguished TB cases from HC, are genes involved in calcium signaling [21,23,24]. Possibly, altered expression of these genes in TB patients reflects M. tuberculosis-mediated changes in calcium metabolism in lung tissue that can be measured in peripheral whole blood.
Although the groups of TB, LTBI and HC were reasonably well age-matched (Table 1), we cannot exclude the possibility that age-dependent differences in immune responses have influenced gene expression profiles. Age-related differences in both innate as well as antigen-specific responses to M. tuberculosis are well recognised [40,41]. Alveolar macrophage antimicrobial activity and recruitment of monocytes as well as the production of cytokines and certain aspects of antigen presentation appear to be less efficient in young children. This is particularly true in children younger than one year of age [41]. Therefore, the exclusion of children less than one year of age in our study is likely to have prevented a significant influence of age-related immune differences on gene expression results. Furthermore, the signature set that we identified showed a good discriminative value between TB and LTBI in adults from several regions [4-6]. This is an indication that the genes that were selected in our analysis make up a signature set that performs well in individuals of all ages.
We identified a minimal gene set of 42 genes that was able to separate TB cases from LTBI and HC in all previously described (adult) cohorts [4-6] as well as in our childhood cohort. However, as this minimal set was possibly over-optimized to fit exactly those sets that were used for its composition, this set might not perform well in a newly identified cohort from a different geographic region. As the datasets used for the composition of the minimal set were based on European, African and South American populations the minimal set may not be applicable to individuals from Asia, while this region carries almost two-third of the global TB burden [42]. Furthermore, this signature set could be only indicative of damage to the lung epithelium, similar to what has been described for the overlap of the gene set determined by Berry et al.[4] and the biosignature characteristic for sarcoidosis [7]. Therefore, we used bootstrapping procedures to select a robust set of ten genes that had a high discriminative value in our population, in the two populations described by Maertzdorf et al.[5,6] and in the comparison between TB, LTBI, HC and other inflammatory and infectious diseases in the dataset of Berry et al.[4]. Although this approach probably leads to less overfitting of the selected set towards the source databases used and less overlap with other infectious diseases in comparison with the minimal gene set we identified, the discriminatory power of this ten gene set is less than that of the minimal set (Table 3). Future cohorts can be of help in the reduction of the 116 gene set to a dataset with similar performance in discriminating TB from LTBI, HC and other inflammatory diseases as the minimal gene set without overfitting the dataset to the source datasets.
From the ten gene set, a combination of five (S100P, HBD, PIGC, CHRM2 and ACOT7) could be used in decision tree analysis to differentiate TB from LTBI, HC and non-TB pneumonia with 78% sensitivity and 96% specificity in our dataset (Figure 2). Additionally, the expression profile of children that were treated for TB shifted from an active TB classification (oval in Figure 2) towards a classification as not suffering from active TB (hexagon in Figure 2) at five months post treatment initiation. This indicates that these biomarkers reflect a dynamic response that changes as mycobactericidal activity diminishes.
The discriminatory value of the 116 gene signature set for the classification of cases in the cohort described by Berry and colleagues [4] was significantly better in people from London compared to people from South Africa. An explanation for the greater similarity between our study population with people from London than with people from South Africa comes from population-genetic studies in which a decrease in the level of genetic variation between populations is observed with increasing geographic distance from Africa, consistent with the out-of-Africa spread of human populations [43]. The finding that previously published signature sets based on individuals from South Africa [4,6] do not provide a good discriminatory value between TB, LTBI and HC in The Gambia [5] points towards a high heterozygosity in TB immune response between different African countries. A high-resolution survey of genotype variation based on single-nucleotide polymorphisms, copy-number variants and haplotype analysis of a worldwide sample of 29 populations revealed that the genetic distance between individuals from Asia and Native American or Colombian individuals is significantly less than the genetic distance between Asian and South African populations [44]. Bayesian cluster analysis clustered individuals from East Asia together with Native American or Colombian individuals, indicating their close phylogenetic relationship [44]. Clustering of Native American individuals with Asian individuals based on their genetic similarities was also observed in a recently published quantitative assessment of human genetic variation worldwide [45]. Therefore, we speculate that the applicability of our signature set in Asian populations might be better than the applicability of sets identified in African or European populations.
Conclusions
This study provides a signature gene set that was demonstrated to be instrumental for the diagnosis of childhood TB. As the identification of different biomarkers in ethnically distinct cohorts is apparent, it is important to cross-validate newly identified markers in all available cohorts. Especially, more childhood cohorts should be investigated as TB diagnosis based on traditional methods is less sensitive and specific in children compared to adults.
Methods
Study population
The Warao Amerindians are an indigenous population living in wooden houses raised on stilts along the Orinoco river banks. With a population of approximately 30,000, the Warao people are the second most important Native American group in Venezuela. In this study, 27 HIV-negative children 1 to 15 years of age with TB (n = 9), LTBI (n = 9) and HC (n = 9) were recruited between May 2010 and December 2010. General characteristics of the study subjects are given in Table 1. Detailed information of the study subjects that was recorded on inclusion is given in Additional file 1: Table S1. Tuberculin skin test (TST) and QuantiFERON-TB Gold In-Tube assay (QFT-GIT) were performed in all children. Sputum samples were collected from all children with expectoration and gastric aspirates were taken from all children under 6 years of age. Children with active TB were diagnosed based on culture of M. tuberculosis (n = 2) or on the basis of clinical, epidemiological and radiological features (n = 7). The latter group were children with a TST ≥ 10 mm or a positive QFT-GIT result who presented all of: persistent fever > 38°C recorded daily for at least two weeks, persistent cough for more than three weeks, weight loss (> 5% reduction in weight compared with the highest weight recorded in last three months) or failure to thrive (documented crossing of percentile lines in the preceding three months), persistent lethargy or decrease in playfulness/activity reported by the parent and absence of clinical response on broad-spectrum antibiotics. Standard antero-posterior and lateral chest radiographs (CXRs) were taken from all children. Two independent experts, blinded to all clinical information, evaluated the CXRs and documented their findings on a standard report form. Where the two objective experts disagreed, a third expert was consulted and final consensus was achieved. A diagnosis of TB was only made when the CXR was consistent with TB [46] and the child showed a positive clinical response to anti-TB treatment. Children were followed up clinically, radiologically and, in case of a negative TST at inclusion, by means of TST at six and 12 months after inclusion. LTBI was defined as a TST ≥ 10 mm and a positive QFT-GIT with a negative culture result on inclusion in the absence of radiological and clinical evidence of TB disease on inclusion as well as on six and 12 months after inclusion. HC were children with a TST = 0 mm on inclusion and on six and 12 months after inclusion. The HC had a negative QFT-GIT and a negative culture result on inclusion in the absence of radiological and clinical evidence of TB disease on inclusion as well as on six and 12 months after inclusion. TB patients were sampled before initiation of anti-TB treatment. Of three of the nine TB patients, a follow-up sample was taken when the patient had taken anti-TB treatment for five months.
RNA isolation and microarray procedures
From every child, 2.5 ml of peripheral whole blood was collected in PAXgene RNA tubes (PreAnalytix, Hombrechtikon, Switzerland) and stored at −80°C prior to processing. RNA was isolated using the PAXgene Blood RNA kit (PreAnalytix) following the manufacturer’s protocol. RNA quality was checked using the RNA 6000 Nano Kit on a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). Gene expression data were obtained using GeneChip Human Exon 1.0 ST Arrays (Affymetrix, Santa Clara, CA). The probe preparation and hybridization was done following Affymetrix protocol. Arrays were washed and stained according to the protocol on a GeneChip Fluidics Station 450 (Affymetrix) and scanned on a Genechip Scanner 3000 + autoloader (Affymetrix). Scan results were converted to CEL with the Affymetric scanning software. Spot intensity data were processed with Affymetrix Power Tools (version 1.14-4.1.1) using robust multi-array average (RMA) and plier-gcbg normalization on the core gene set (version HuEx-1 0-st-v2.r2.dt1.hg18). Gene expression values were log2-transformed and differentially expressed genes were identified based on log2 fold changes (M-values). P-values were calculated with a Bayes-regularized one-way ANOVA [47] followed by multiple testing correction of the p-values (q-values) according to the method of Storey and Tibshirani [48]. Microarray data have been deposited in the GEO database under accession GSE41055 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ltmhxwsmskeyyte&acc=GSE41055).
Random forest based identification of TB biomarker genes
A random forest classification was performed to identify signature genes for discrimination of TB, LTBI and HC. This classification model, consisting of 5000 decision trees trained on random subsets of samples and variables was trained based on log2 gene expression data as a function of individuals belonging to either TB, LTBI or HC classes. Irrelevant genes were removed using the random forest-based local importance measure as described in PhenoLink [9] where genes with a negative or neutral contribution of correctly classifying the samples were removed. This process was repeated until fewer than three genes could be removed per iteration. Next, genes were selected provided that they had a contribution to the correct classification of at least three samples of the same class. Again, this process was repeated until fewer than three genes were removed. The resulting set was used for classification and determination of the classification error, both as OOB error and as average error, on all gene expression sets.
Literature datasets
Literature datasets describing gene expression studies of TB, LTBI, HC and other infectious diseases were obtained from GEO (http://nar.oxfordjournals.org/content/39/suppl_1/D1005.full), accession numbers GSE19491 [4], GSE19492 [4], GSE28623 [5] and GSE25534 [6]. For GSE19491, GSE19492 and GSE28623 available normalized data were used, while for GSE25534 the 2 dye array data was normalized using RMA in Arraystar (DNAStar, Madison, WI) to allow inter-slide comparison of gene expression data. First, the TB biomarker set identified in this manuscript was applied to the literature cohorts [4-6] and the biomarker sets identified in the literature cohorts were applied to each other for estimation of the predictive value of each identified set in the other populations. Next, to determine the minimal discriminatory gene sets based on the dataset described in this study as well as on the three previously published datasets [4-6], expression data from probes corresponding with the TB biomarker set determined in this manuscript were subjected to VarSelRF, as described by Diaz-Uriarte et al.[11], which progressively eliminates genes with the lowest random forest-based local importance measure until no further improvements in the OOB error rate are reported. This procedure is designed to identify small, non-redundant sets of genes that have good predictive performance. However, over fitting of the selected minimum discriminatory gene sets on the source databases used could lead to a poor performance of these minimal sets when applying them to classify other patient populations. Therefore, .632+ bootstrapping procedures were performed to select genes with a high discriminative power in all datasets. Bootstrapping repeatedly analyzes subsamples rather than subsets of the data. As each subsample is a random sample with replacement from the full sample, this procedure performs well in the selection of classifiers even when there is over fitting [49].
To test possible associations between the class probability of being wrongly classified as LTBI (dependent variable) and age, gender and geographical region (independent variables) of TB cases included in the GSE19491/GSE19492 dataset, multivariable linear regression analysis was performed.
Functional analysis
Enrichment analysis of signature genes was performed using the web-based DAVID bioinformatics tool (http://david.abcc.ncifcrf.gov) [10]. Q-values for enriched annnotations were determined by a modified Fisher’s exact test (EASE Score [10]) and corrected for multiple testing by the Benjamini-Hochberg approach.
Quantitative RT-PCR on microarray and additional samples
Differential expression of several genes was validated by qRT-PCR. cDNA was generated by reverse transcription using Superscript III Reverse Transcriptase and Random Primers (Invitrogen) following manufacturer’s protocol after DNAse treatment of the RNA using TURBO DNA-free (Ambion). GAPDH was chosen as reference gene. qRT-PCR was performed on the 27 microarray samples as well as on a validation cohort (n = 54). These were RNA samples collected from an additional 54 children during the same period and in the same manner as the microarray samples. Of the 54 children of which RNA was collected for qRT-PCR, 20 were diagnosed with LTBI, 16 were HC and 18 children were diagnosed with a radiologically confirmed pneumonia [50], of which six were LTBI and 12 were HC.
ROC methodology was applied to evaluate the discriminatory ability of signature genes. All expression values were normally distributed (Kolmogorov-Smirnov’s test, p > 0.05). One-way analysis of variance and unpaired Student’s t tests were performed to compare expression values of these genes in children with TB, LTBI, HC and non-TB pneumonia. When the variances across groups were not equal (Levene’s test p < 0.05), Welch correction for nonhomogeneity of variance was applied.
Ethical considerations
The nature and objectives of the study were explained to the parents of children in Spanish or were simultaneously translated to their native language by Spanish-Warao bilingual native interpreters. The study was approved by the ethical committee of the Instituto de Biomedicina, the Regional Health Services, and the Delta Amacuro Indigenous Health Office (Servicio de Atención y Orientación al Indígena). Children were enrolled if their parents or primary caregivers provided written informed consent. Illiterate parents or caregivers signed by means of a thumb print.
Competing interests
All authors declare that they have no competing interests.
Authors’ contributions
LV, MM and JV: patient recruitment and sampling; LV and ME: RNA isolation, microarray and qRT-PCR experiments; AZ and SvanH: bioinformatic analyses; JdeW: coordination and funding of field work and sampling logistics; BdelN: advise on patient recruitment and sampling; PH: supervision and funding of microarray, qRT-PCR and bioinformatic analyses; LV, AZ and PH wrote the paper. All authors commented on the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Detailed characteristics of 27 children with TB, LTBI and HC in which microarray analyses were performed.
Enriched gene sets in the 116 gene set.
Sheet 1. Minimal discriminatory gene set to discriminate between TB and LTBI. Sheet 2. Minimal discriminatory gene set to discriminate between TB, LTBI and HC. Sheet 3. Variable frequencies estimated by the bootstrapping procedure implemented in VarSelRF (1). Sheet 4. Variable frequencies estimated by the bootstrapping procedure implemented in VarSelRF (2).
qRT-PCR cycle threshold (delta Ct) values in TB, LTBI, HC and non-TB pneumonia subjects for each of the ten signature genes. The delta Ct was calculated as Ct value (number of cycles required for the fluorescent signal to exceed the background level) of the target gene – Ct value of the reference gene (GAPDH). The boxplots show the median delta Ct values and the interquartile ranges. The whiskers represent the highest and lowest values that are not outliers. Dots represent outliers and squares in the boxes indicate mean delta Ct values. The p-values for each gene (in parentheses) are the outcomes of the one-way analyses of variance comparing TB, LTBI, HC and non-TB pneumonia. The asterisks indicate statistically significant differences (p < 0.05) between TB and other groups as found by an unpaired Student’s t test.
qRT-PCR cycle threshold (delta Ct) values in TB, LTBI, HC and non-TB pneumonia subjects for each of the ten signature genes. The delta Ct was calculated as Ct value (number of cycles required for the fluorescent signal to exceed the background level, a lower Delta Ct value indicates a higher expression) of the target gene – Ct value of the reference gene (GAPDH).
Contributor Information
Lilly M Verhagen, Email: lillyverhagen@hotmail.com.
Aldert Zomer, Email: A.zomer@cukz.umcn.nl.
Mailis Maes, Email: mailismaes@gmail.com.
Julian A Villalba, Email: jvn1987@gmail.com.
Berenice del Nogal, Email: berenicedelnogal@yahoo.es.
Marc Eleveld, Email: MJ.eleveld@cukz.umcn.nl.
Sacha AFT van Hijum, Email: svhijum@cmbi.ru.nl.
Jacobus H de Waard, Email: jacobusdeward@gmail.com.
Peter WM Hermans, Email: P.hermans@cukz.umcn.nl.
Acknowledgements
The authors thank Mercedes España, coordinator of the Venezuelan National TB Control Program, Maria Eugenia Melendez, head of the pediatric pulmonology department of the Caracas Children’s Hospital ´J.M. de los Ríos´, and Heriberto Perez, pulmonologist at the Caracas Hospital ´José Maria Vargas´, for evaluation of CXRs. Furthermore, we thank the participating families and the field workers involved in the recruitment of participants, in particular the medical students of the Escuela de Medicina ‘José Maria Vargas’ of the Universidad Central de Venezuela and the personnel of the Centros de Diagnostico Medico Cubano (CDIs) in Curiapo and Pedernales. We also thank the staff of the Laboratorio de Tuberculosis, Instituto de Biomedicina (Caracas, Venezuela) and the Laboratory of Pediatric Infectious Diseases, Radboud University Medical Centre (Nijmegen, The Netherlands) for their clinical assistance and technical support. The study was supported by FUNDAIM, Fundación para la Investigación en Micobacterias, Caracas, Venezuela. The ‘Stichting VSB Fonds’ (Utrecht, The Netherlands) is greatly acknowledged for providing a stipend to LV. AZ was supported by the European Commission FP7 Marie Curie IEF Action 274586.
References
- World Health Organization (WHO) Guidance for national tuberculosis programmes on the management of tuberculosis in children. Geneva, Switzerland: WHO; 2006. [PubMed] [Google Scholar]
- Newton SM, Brent AJ, Anderson S, Whittaker E, Kampmann B. Paediatric tuberculosis. Lancet Infect Dis. 2008;8:498–510. doi: 10.1016/S1473-3099(08)70182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar HJ, Hanslo D, Apolles P, Swingler G, Hussey G. Induced sputum versus gastric lavage for microbiological confirmation of pulmonary tuberculosis in infants and young children: a prospective study. Lancet. 2005;365:130–134. doi: 10.1016/S0140-6736(05)17702-2. [DOI] [PubMed] [Google Scholar]
- Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertzdorf J, Ota M, Repsilber D, Mollenkopf HJ, Weiner J, Hill PC, Kaufmann SH. Functional correlations of pathogenesis-driven gene expression signatures in tuberculosis. PLoS One. 2011;6:e26938. doi: 10.1371/journal.pone.0026938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertzdorf J, Repsilber D, Parida SK, Stanley K, Roberts T, Black G, Walzl G, Kaufmann SH. Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun. 2011;12:15–22. doi: 10.1038/gene.2010.51. [DOI] [PubMed] [Google Scholar]
- Maertzdorf J, Weiner J III, Mollenkopf HJ, Network T, Bauer T, Prasse A, Muller-Quernheim J, Kaufmann SH. Common patterns and disease-related signatures in tuberculosis and sarcoidosis. Proc Natl Acad Sci USA. 2012;109:7853–7858. doi: 10.1073/pnas.1121072109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández De Larrea C, Fañdino C, López D, Del Nogal B, Rodríguez N, Convit J, Araujo Z, De Waard JH. Tuberculosis en menores de 15 años en la población Warao de Venezuela. Invest Clin. 2002;43:35–48. [PubMed] [Google Scholar]
- Bayjanov JR, Molenaar D, Tzeneva V, Siezen RJ, Van Hijum SA. PhenoLink - a web-tool for linking phenotype to omics data for bacteria: application to gene-trait matching for Lactobacillus plantarum strains. BMC Genomics. 2012;13:170. doi: 10.1186/1471-2164-13-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Diaz-Uriarte R. GeneSrF and varSelRF: a web-based tool and R package for gene selection and classification using random forest. BMC Bioinformatics. 2007;8:328. doi: 10.1186/1471-2105-8-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson FR, Fryer AD. Muscarinic acetylcholine receptors and airway diseases. Pharmacol Ther. 2003;98:59–69. doi: 10.1016/s0163-7258(03)00004-4. [DOI] [PubMed] [Google Scholar]
- Yamada H, Ohashi E, Abe T, Kusumi N, Li SA, Yoshida Y, Watanabe M, Tomizawa K, Kashiwakura Y, Kumon H, Matsui H, Takei K. Amphiphysin 1 is important for actin polymerization during phagocytosis. Mol Biol Cell. 2007;18:4669–4680. doi: 10.1091/mbc.E07-04-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C, Wang X, Song L, North M, Wiehler S, Proud D, Chow CW. Syk associates with clathrin and mediates phosphatidylinositol 3-kinase activation during human rhinovirus internalization. J Immunol. 2008;180:870–880. doi: 10.4049/jimmunol.180.2.870. [DOI] [PubMed] [Google Scholar]
- Wang H, Jiang C. Influenza A virus H5N1 entry into host cells is through clathrin-dependent endocytosis. Sci China C Life Sci. 2009;52:464–469. doi: 10.1007/s11427-009-0061-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang Y, Liu X, Wang N, Cao H, Lu Y, Zhou H, Zheng J. Inhibition of clathrin/dynamin-dependent internalization interferes with LPS-mediated TRAM-TRIF-dependent signaling pathway. Cell Immunol. 2012;274:121–129. doi: 10.1016/j.cellimm.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Florian V, Schluter T, Bohnensack R. A new member of the sorting nexin family interacts with the C-terminus of P-selectin. Biochem Biophys Res Commun. 2001;281:1045–1050. doi: 10.1006/bbrc.2001.4467. [DOI] [PubMed] [Google Scholar]
- Mukae H, Ashitani J, Tokojima M, Ihi T, Kohno S, Matsukura S. Elevated levels of circulating adhesion molecules in patients with active pulmonary tuberculosis. Respirology. 2003;8:326–331. doi: 10.1046/j.1440-1843.2003.00471.x. [DOI] [PubMed] [Google Scholar]
- Watanabe R, Inoue N, Westfall B, Taron CH, Orlean P, Takeda J, Kinoshita T. The first step of glycosylphosphatidylinositol biosynthesis is mediated by a complex of PIG-A, PIG-H, PIG-C and GPI1. EMBO J. 1998;17:877–885. doi: 10.1093/emboj/17.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welin A, Winberg ME, Abdalla H, Sarndahl E, Rasmusson B, Stendahl O, Lerm M. Incorporation of Mycobacterium tuberculosis lipoarabinomannan into macrophage membrane rafts is a prerequisite for the phagosomal maturation block. Infect Immun. 2008;76:2882–2887. doi: 10.1128/IAI.01549-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austermann J, Nazmi AR, Muller-Tidow C, Gerke V. Characterization of the Ca2+ − regulated ezrin-S100P interaction and its role in tumor cell migration. J Biol Chem. 2008;283:29331–293340. doi: 10.1074/jbc.M806145200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergne I, Chua J, Singh SB, Deretic V. Cell biology of mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol. 2004;20:367–394. doi: 10.1146/annurev.cellbio.20.010403.114015. [DOI] [PubMed] [Google Scholar]
- Shah AS, Ben-Shahar Y, Moninger TO, Kline JN, Welsh MJ. Motile cilia of human airway epithelia are chemosensory. Science. 2009;325:1131–1134. doi: 10.1126/science.1173869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande DA, Wang WC, McIlmoyle EL, Robinett KS, Schillinger RM, An SS, Sham JS, Liggett SB. Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat Med. 2010;16:1299–1304. doi: 10.1038/nm.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist ME, Lifland AW, Utley TJ, Santangelo PJ, Crowe JE Jr. Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J Virol. 2010;84:12274–12284. doi: 10.1128/JVI.00260-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S, Soh BS, Sun LL, Tai BC, Nga ME, Bhakoo KK, Jayapal SR, Nichane M, Yu Q, Ahmed DA, Tan C, Sing WP, Tam J, Thirugananam A, Noghabi MS, Pang YH, Ang HS, Mitchell W, Robson P, Kaldis P, Soo RA, Swarup S, Lim EH, Lim B. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 2012;148:259–272. doi: 10.1016/j.cell.2011.11.050. [DOI] [PubMed] [Google Scholar]
- Forwood JK, Thakur AS, Guncar G, Marfori M, Mouradov D, Meng W, Robinson J, Huber T, Kellie S, Martin JL, Hume DA, Kobe B. Structural basis for recruitment of tandem hotdog domains in acyl-CoA thioesterase 7 and its role in inflammation. Proc Natl Acad Sci USA. 2007;104:10382–10387. doi: 10.1073/pnas.0700974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer W, Pickkers P, Scott CS, van der Hoeven JG, Gunnewiek JK. Hematological indices, inflammatory markers and neutrophil CD64 expression: comparative trends during experimental human endotoxemia. J Endotoxin Res. 2007;13:94–100. doi: 10.1177/0968051907079101. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- Parker D, Martin FJ, Soong G, Harfenist BS, Aguilar JL, Ratner AJ, Fitzgerald KA, Schindler C, Prince A. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. MBio. 2011;2:e00016–11. doi: 10.1128/mBio.00016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen M, Repsilber D, Gutschmidt A, Neher A, Feldmann K, Mollenkopf HJ, Ziegler A, Kaufmann SH. Candidate biomarkers for discrimination between infection and disease caused by Mycobacterium tuberculosis. J Mol Med (Berl) 2007;85:613–621. doi: 10.1007/s00109-007-0157-6. [DOI] [PubMed] [Google Scholar]
- Lu C, Wu J, Wang H, Wang S, Diao N, Wang F, Gao Y, Chen J, Shao L, Weng X, Zhang Y, Zhang W. Novel biomarkers distinguishing active tuberculosis from latent infection identified by gene expression profile of peripheral blood mononuclear cells. PLoS One. 2011;6:e24290. doi: 10.1371/journal.pone.0024290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TY, Chan CH, Shek CC, Davies PD. Hypercalcemia in active pulmonary tuberculosis and its occurrence in relation to the radiographic extent of disease. Southeast Asian J Trop Med Public Health. 1992;23:702–704. [PubMed] [Google Scholar]
- Deniz O, Tozkoparan E, Yonem A, Ciftci F, Bozkanat E, Cakir E, Ozcan O, Narin Y, Bilgic H, Ekiz K, Demirci N. Low parathormone levels and hypercalcaemia in patients with pulmonary tuberculosis: relation to radiological extent of disease and tuberculin skin test. Int J Tuberc Lung Dis. 2005;9:317–321. [PubMed] [Google Scholar]
- Dosumu EA, Momoh JA. Hypercalcemia in patients with newly diagnosed tuberculosis in Abuja. Nigeria. Can Respir J. 2006;13:83–87. doi: 10.1155/2006/407609. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Payne HA, Menson E, Sharland M, Bryant PA. Symptomatic hypercalcaemia in paediatric tuberculosis. Eur Respir Rev. 2011;20:53–56. doi: 10.1183/09059180.00006910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadranel JL, Garabedian M, Milleron B, Guillozzo H, Valeyre D, Paillard F, Akoun G, Hance AJ. Vitamin D metabolism by alveolar immune cells in tuberculosis: correlation with calcium metabolism and clinical manifestations. Eur Respir J. 1994;7:1103–1110. [PubMed] [Google Scholar]
- Chang JM, Kuo MC, Kuo HT, Hwang SJ, Tsai JC, Chen HC, Lai YH. 1-alpha,25-Dihydroxyvitamin D3 regulates inducible nitric oxide synthase messenger RNA expression and nitric oxide release in macrophage-like RAW 264.7 cells. J Lab Clin Med. 2004;143:14–22. doi: 10.1016/j.lab.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Carlberg C, Campbell MJ. Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids. 2012. online publication ahead of print. [DOI] [PMC free article] [PubMed]
- Lewinsohn DA, Gennaro ML, Scholvinck L, Lewinsohn DM. Tuberculosis immunology in children: diagnostic and therapeutic challenges and opportunities. Int J Tuberc Lung Dis. 2004;8:658–674. [PubMed] [Google Scholar]
- Smith S, Jacobs RF, Wilson CB. Immunobiology of childhood tuberculosis: a window on the ontogeny of cellular immunity. J Pediatr. 1997;131:16–26. doi: 10.1016/s0022-3476(97)70120-3. [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) Global Tuberculosis Report 2012. Geneva, Switzerland: WHO; 2012. [Google Scholar]
- DeGiorgio M, Jakobsson M, Rosenberg NA. Out of Africa: modern human origins special feature: explaining worldwide patterns of human genetic variation using a coalescent-based serial founder model of migration outward from Africa. Proc Natl Acad Sci USA. 2009;106:16057–16062. doi: 10.1073/pnas.0903341106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson M, Scholz SW, Scheet P, Gibbs JR, VanLiere JM, Fung HC, Szpiech ZA, Degnan JH, Wang K, Guerreiro R, Bras JM, Schymick JC, Hernandez DG, Traynor BJ, Simon-Sanchez J, Matarin M, Britton A, van de Leemput J, Rafferty I, Bucan M, Cann HM, Hardy JA, Rosenberg NA, Singleton AB. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- Wang C, Zollner S, Rosenberg NA. A Quantitative Comparison of the Similarity between Genes and Geography in Worldwide Human Populations. PLoS Genet. 2012;8:e1002886. doi: 10.1371/journal.pgen.1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais BJ, Gie RP, Schaaf HS, Starke JR, Hesseling AC, Donald PR, Beyers N. A proposed radiological classification of childhood intra-thoracic tuberculosis. Pediatr Radiol. 2004;34:886–894. doi: 10.1007/s00247-004-1238-0. [DOI] [PubMed] [Google Scholar]
- Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t -test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B, Tibshirani RJ. Improvements on cross-validation: the .632+ bootstrap method. J American Statistical Association. 1997;92:548–560. [Google Scholar]
- World Health Organization (WHO) Pneumonia Vaccine Trial Investigators Group. Standardization of interpretation of chest radiographs for the diagnosis of pneumonia in children. Geneva, Switzerland: WHO; 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed characteristics of 27 children with TB, LTBI and HC in which microarray analyses were performed.
Enriched gene sets in the 116 gene set.
Sheet 1. Minimal discriminatory gene set to discriminate between TB and LTBI. Sheet 2. Minimal discriminatory gene set to discriminate between TB, LTBI and HC. Sheet 3. Variable frequencies estimated by the bootstrapping procedure implemented in VarSelRF (1). Sheet 4. Variable frequencies estimated by the bootstrapping procedure implemented in VarSelRF (2).
qRT-PCR cycle threshold (delta Ct) values in TB, LTBI, HC and non-TB pneumonia subjects for each of the ten signature genes. The delta Ct was calculated as Ct value (number of cycles required for the fluorescent signal to exceed the background level) of the target gene – Ct value of the reference gene (GAPDH). The boxplots show the median delta Ct values and the interquartile ranges. The whiskers represent the highest and lowest values that are not outliers. Dots represent outliers and squares in the boxes indicate mean delta Ct values. The p-values for each gene (in parentheses) are the outcomes of the one-way analyses of variance comparing TB, LTBI, HC and non-TB pneumonia. The asterisks indicate statistically significant differences (p < 0.05) between TB and other groups as found by an unpaired Student’s t test.
qRT-PCR cycle threshold (delta Ct) values in TB, LTBI, HC and non-TB pneumonia subjects for each of the ten signature genes. The delta Ct was calculated as Ct value (number of cycles required for the fluorescent signal to exceed the background level, a lower Delta Ct value indicates a higher expression) of the target gene – Ct value of the reference gene (GAPDH).