

Abstract
Multiple myeloma is a malignant disorder of differentiated B-cells for which standard care involves the inhibition of the proteasome. All clinically used proteasome inhibitors, including the chemotherapeutic drug bortezomib, target the catalytic active sites of the proteasome and inhibit protein proteolysis by competing with substrate binding. However, nearly all (~97%) patients become intolerant or resistant to treatments within a few years, after which the average survival time is less than one year. We describe herein the inhibition of the human proteasome via a non-competitive mechanism by the imidazoline scaffold, TCH-13. Consistent with a mechanism distinct from competitive inhibitors, TCH-013 acts additively with and overcomes resistance to bortezomib. Importantly, TCH-013 induces apoptosis a panel of myeloma and leukemia cell lines, but in contrast, normal lymphocytes, primary bone marrow stromal cells and macrophages are resistant to its cytotoxic effects. TCH-013 was equally effective in blocking MM cell growth in co-cultures of MM cells with hBMSC isolated from CD138 negative BM samples of MM patients. The cellular activity translated well in vivo where TCH-013 delayed tumor growth in an MM xenograft model to a similar extent as bortezomib.
Multiple myeloma (MM) is a malignant disorder of differentiated B-cells that remains largely incurable with nearly all patients relapsing 1. MM cells are predominantly located in the bone marrow where they closely interact with bone marrow stromal cells (BMSCs).2 MM cell growth, survival, migration and drug resistance is dictated by the tumor microenvironment through direct cell-to-cell contact or indirectly via secretion of cytokines and soluble growth factors, such as interleukin 6 (IL-6), VEGF, TNF-α, and others 3–5. Of these cytokines, IL-6 plays a predominant role in the terminal differentiation of B cells and is critical in the pathogenesis of MM 2, 6. The interactions between MM cells and BMSCs augments IL-6 secretion via the nuclear factor-κB (NF-κB)-pathway, resulting in increased proliferation and inhibition of apoptosis of MM cells 6–8. In addition to BMSCs, osteoclasts also strongly enhance contact mediated growth and survival of MM cells. Thus, the microenvironment of the bone marrow plays an integral role in the pathogenesis of MM, stimulating a vicious cycle of cytokine production, tumor growth and bone destruction 9, 10. Disruption of this cycle by therapeutics is accomplished by inhibition of the proteasome, which has subsequently evolved to become the central standard of care for MM treatment11–13.
The 26S proteasome consists of a 20S catalytic core and 19S regulatory particles 14–16. The 19S regulatory particles are responsible for recognition, unfolding and translocation of substrates into the 20S core 17. The 20S catalytic core is a threonine protease that exhibits three distinct proteolytic activities: chymotrypsin-like (CT-L), trypsin-like (T-L) and caspase-like (Casp-L) activity, which are responsible for proteolytic degradation of its substrates 18. All clinically relevant proteasome inhibitors, including the peptide-based drug bortezomib (Fig. 1A), elicit their activity via the same mechanism; formation of a covalent bond to the 1N terminal threonine in the catalytic sites of the enzyme11, 19–22. These competitive inhibitors directly compete with substrate binding to the active site(s) in the proteasome. At the cellular level, drug resistance has been attributed, in part, to overexpression of a mutated form of the catalytic subdomains, abrogating drug-protein binding23,24–26. However, tumors that exhibit resistance via this mechanism will retain sensitivity to drugs that bind outside the active site inhibiting activity of the enzyme via a non-competitive mechanism 27–31. Aside from overcoming resistance,32 it has also been suggested that non-competitive modulation of enzyme activity limits off-target effects, reducing toxicity28, 31, 33. Unfortunately, examples of non-competitive proteasome inhibitors are very rare. PR-39, a 39-amino acid peptide, inhibits the 20S proteasome non-competitively via this type of allosteric mechanism, where binding to the α-ring of the 20S proteasome leads to changes in proteasome structure and prevents degradation of specific substrates, including IκB34–36. As for small molecule inhibitors, chloroquine binds to a region of the α-ring but only at lethal, non-physiologically relevant concentrations (>40 μM)37. Schimmer and co-workers reported that 5AHQ inhibits the proteasome through a non-competitive mechanism; however, it is unclear if this is via a direct proteasome-drug interaction or upstream event32. In addition, the non-promiscuous secondary fungal metabolite gliotoxin has also been shown to bind via disulfide bonds to the proteasome but exhibits a multitude of cellular effects and high general toxicity 38.
Figure 1.
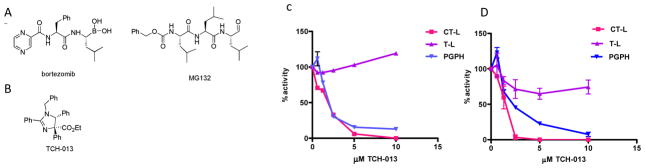
(A) Chemical structures of peptide-based competitive inhibitors bortezomib and MG132, and (B) non-competitive proteasome, small molecule inhibitor, TCH-013. (C) TCH-013 inhibits the CT-L and Casp-L activity of the human proteasome. Fluorogenic substrates Suc-LLVY-AMC, Z-ARR-AMC and Z-LLE-AMC were used to measure CT-L, T-L and Casp-L activities of purified human 20S proteasome particles as described in material and methods. The maximum increase in fluorescence per minute was used to calculate specific activities of each sample. (D) Fluorogenic substrates Suc-LLVY-AMC. Z-ARR-AMC and Z-LLE-AMC were used to measure CT-L, T-L and Casp-L activities of fully assembled proteasomes extracted from RPMI-8226 human multiple myeloma cells with ATP/DTT buffer as described in materials and methods. The maximum increase in fluorescence per minute was used to calculate specific activities of each sample.
Although bortezomib is undoubtedly one of the biggest breakthroughs in this field, additional treatment options are necessary as nearly all (~97%) patients become intolerant or resistant to these competitive inhibitors within a few years, after which the average survival time is less than one year39. We have previously reported that non-peptide based, trans-imidazoline TCH-013 (Fig. 1B) and its R,R enantiomer TCH-013a are orally available anti-arthritic agents that modulate NF-κB-mediated cytokine production via an unknown mechanism40, 41. Considering the important role of cytokine production in the pathogenesis of MM, we subsequently evaluated these agents for their mechanistic details and potential efficacy for MM treatment. Herein, we report that the mechanism of NF-κB-mediated cytokine inhibition by TCH-013 proceeds via a non-competitive modulation of the proteasome. We show that TCH-013 binds to a site other than the substrate binding sites and overcomes resistance to competitive proteasome inhibitors. TCH-013 induces apoptosis a panel of myeloma and leukemia cell lines. In contrast, normal lymphocytes, primary bone marrow stromal cells, and macrophages were resistant to the cytotoxic effects. MM-bone marrow stromal cell co-culture systems indicate that the efficacy of TCH-013 to induce MM cell death is not affected by bone marrow microenvironment. Finally, we show that TCH-013 prevents tumor growth in an MM xenograft mouse model to a similar extent as the maximum tolerated dose of bortezomib.
RESULTS AND DISCUSSION
TCH-013 inhibits the catalytic activity of the proteasome as measured in vitro in purified 20S proteasome and in cell culture
Previously, our lab demonstrated that TCH-013 inhibits the activation of NF-κB mediated IL-6 transcription through an unknown mechanism 41. NF-κB activation is tightly controlled by its inhibitory protein, IκB, which in turn is controlled by the ubiquitin-proteasome system (UPS). Thus, in order to elucidate the mechanism of NF-κB inhibition by TCH-013, the ability of TCH-013 to inhibit the proteasome was investigated in vitro and in cell culture. Purified 20S proteasome and the following fluorogenic substrates at their respective KM values (SFig. 1) were used: Suc-LLVY-AMC (for CT-L activity), Boc-LRR-AMC (for T-L activity) and Z-LLE-AMC (for Casp-L activity) 30. The rates of hydrolysis were monitored by fluorescence increase over time and the linear portions of the curves were used to calculate the IC50 values (Fig. 1C). In vitro analysis indicates that TCH-013 selectively inhibits the CT-L, (IC50 2.80 ±0.66 μM) and Casp-L (IC50 1.60 ±0.26 μM) activities of the 20S catalytic core of the human proteasome. The T-L activity was not inhibited. Consistent with in vitro results, TCH-013 also inhibited the CT-L and Casp-L activity in cell extracts from MM cells RPMI-8226 (Fig. 1D), with no significant activity towards the T-L activity. This data indicates that TCH-013 inhibits the 20S proteasome in purified protein assays as well as in crude MM cell extracts at similar efficacies, thus making it unlikely that activity is controlled by an indirect upstream regulation of the proteasome in the cell.
TCH-013 is not a promiscuous inhibitor and does not form aggregates or micelles
In order to examine whether TCH-013 exhibits protease selectivity, we evaluated the compound against various proteases, including the 20S human immunoproteasome, 20S proteasome isolated from S. cerevisiae, (SFig. 2), calpain and trypsin (data not shown). TCH-013 was found to be equally effective in modulating the different types of purified 20S proteasomes but was devoid of any activity against the other proteases tested. This data indicates that TCH-013 selectively inhibits the proteasome over other proteases tested. False positives in in vitro assays can often be observed when amphipathic molecules form colloidal aggregates in aqueous buffers 42. We tested TCH-013 in various assays to examine the possibility of aggregation or micelle formation 43. TCH-013 did not form micelles or aggregates at relevant concentrations. The various assays and results are described in detail in the supplemental data (S. Fig. 3 and STable1).
TCH-013 induces a modest accumulation of ubiquitinylated proteins in cell culture and prevents the degradation of IκBα
The 26S proteasome is responsible for the proteolytic degradation of proteins in order to maintain biological homeostasis. Protein ubiquitinylation typically precedes proteasome mediated proteolytic degradation, thus inhibition of the proteasome results in an accumulation of ubiquitinylated cellular proteins.44 To investigate whether TCH-013 inhibits the proteasome in cells, we examined the accumulation of ubiquitinylated cellular proteins. Confocal microscopy was performed on HeLa cells following 2 hours of incubation with either vehicle (0.1% DMSO) or TCH-013 (1.0 μM) to detect and localize the accumulation of ubiquitinylated protein (soluble or insoluble) in whole cells. A relatively low dose of TCH-013 was used (1.0 μM) to avoid significant cell death. Treatment of the cells with TCH-013 resulted in an increase in cellular ubiquitin accumulation as measured by anti-ubiquitin immunofluorescence assays (Fig. 2A). Quantification of the fluorescent intensity of the ubiquitin–alexa568 conjugates indicated a statistically significant increase (33% fold increase) in accumulation of ubiquitinylated proteins (p=0.0291), consistent with a mechanism of proteasome inhibition.
Figure 2. TCH-013 causes the accumulation of ubiquitinylated proteins and inhibits the degradation of IκBα.
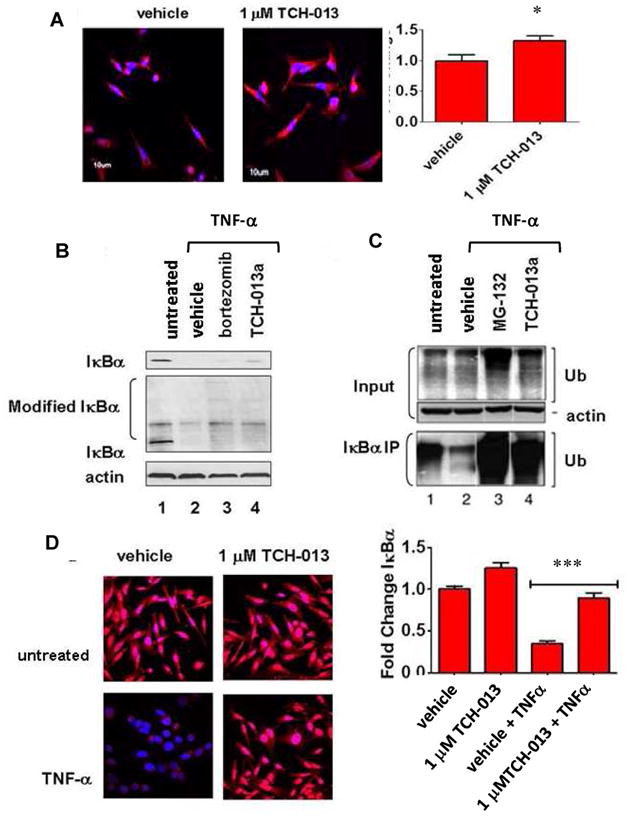
(A) Representative photos showing fluorescent confocal images of HeLa cells treated with either vehicle, or 1 μM TCH-013 for two hours and subsequently stained for the presence of ubiquitinylated proteins (red) and counterstained for DNA with DAPI (blue). Fluorescent intensity of the ubiquitinylated proteins obtained from each treatment were measured using Olympus FV1000 software (n=6, p=0.0291). (B) (top panel) PRMI-8226 cells were left untreated or treated with either vehicle, 1 μM bortezomib or 10 μM TCH-013 for thirty minutes and then stimulated with 10 ng/mL TNF-α. Whole cell extracts were probed for the presence of unmodified IκBα using a monoclonal antibody against the N-terminal of IκBα (L35A5). (middle panel) The same whole cell extracts were probed for the presence of unmodified and modified IκBα using a polyclonal antibody against the C-terminal of IκBα (C-21). The presence of actin was used as a loading control on the same blot. (C) HeLa cells were left untreated or treated with either vehicle, 1 μM MG-132 or 10 μM TCH-013 for thirty minutes and stimulated with 10 ng/mL TNF-α. (input) Whole cell extracts from the samples were probed for the presence of ubiquitin using a monoclonal antibody directed toward ubiquitin (P4D1) that detetects ubiquitin, polyubiquitin and ubiquitinylated proteins. The presence of actin was used as a loading control. (bottom panel) IκBα was immunoprecipitated from the whole cell extracts using a polyclonal antibody directed toward the C-terminal of IκBα (C-21). The IκBα immunoprecipitates were probed for the presence of ubiquitinylated proteins. (D) Representative photos showing fluorescent confocal images of HeLa cells treated with either vehicle or 1 μM TCH-013 and either stimulated with 10 ng/mL TNF-α or left untreated. The cells were fixed and subsequently stained for the presence of IκBα (red) and counterstained for DNA with DAPI (blue). (n=6, p=0.0001).
One of the many pathways the proteasome regulates is the pro-inflammatory, anti-apoptotic NF-κB signaling pathway, via the proteolytic degradation of NF-κB endogenous inhibitor, IκB.45 In response to cytokine stimulation, IκB undergoes phosphorylation and ubiquitinylation, followed by its subsequent proteasomal degradation, which liberates NF-κB for nuclear translocation and gene transcription.46 Evaluation of IκB protein levels can thus serve as a specific protein indicator of proteasome activity. Considering the previously reported inhibition of NF-κB by TCH-013,41, 47 we subsequently evaluated the proteolytic degradation of NF-κB’s inhibitory protein, IκBα. Total inhibition of cellular protein proteolysis was evaluated by Western blot using whole cell extracts from RPMI-8226 (constitutively active NF-κB) and HeLa cells treated with either the competitive proteasome inhibitors bortezomib or MG-132, and TCH-013. When extracts were probed with a polyclonal antibody directed toward ubiquitin, Western blot analysis indicated strong accumulation of ubiquitin products when treated with the competitive inhibitors bortezomib and MG-132, respectively (Fig. 2B and C, lane 3). Interestingly, compared to bortezomib or MG-132 only modest levels of total ubiquitinylated products were found in the TCH-013 treated whole cell extracts (Fig. 2B and C, lane 4). In contrast, when IκBα immunoprecipitates from HeLa cells were probed for ubiquitin accumulation, both MG-132 and TCH-013 treatment prevented IκBα degradation (Fig. 2C, lanes 3 and 4, respectively). RPMI-8226 cells that contain constitutively active NF-κB provided similar results when probed for modified forms of IκBα (Fig. 2B, lanes 3 and 4, respectively).
In order to further verify the effect of TCH013 on cellular IκB levels, we utilized confocal microscopy to visualize IκBα in the presence and absence of TCH-013 (1 μM) in HeLa cells with and without stimulation with TNFα. Cells were stained for IκBα with an antibody directed toward the C-terminal of IκBα (red) and DAPI (blue) was used to visualize the DNA. In vehicle treated cells, IκBα is distributed throughout the cytoplasm of the cells (Fig. 2D), whereas upon stimulation with TNFα, IκBα is degraded and the fluorescent signal (red) is significantly decreased. Cells treated with TCH-013 prior to TNF-α stimulation retained a robust red fluorescent signal, clearly indicating that TCH-013 inhibits the degradation of IκBα. Quantification of the fluorescent signal illustrates that the prevention of IκBα degradation by TCH-013 is statistically significant (p= 0.0001), when compared to its respective TNF-α/vehicle control. This experiment was repeated with an antibody directed toward the N-terminal of IκBα, which recognizes endogenous levels of total IκBα with similar results (SFig. 4). These studies indicate that TCH-013 induces the robust accumulation of ubiquitinylated isoforms of IκB, consistent with inhibition of the proteasome.
TCH-013 inhibits the 20S proteasome via a non-competitive mechanism of binding
Intrigued by the modest accumulation of globally ubiquitinylated proteins, but robust accumulation of IκBα in both cell types, we examined in detail the mechanism for proteasome inhibition by TCH-013. For these studies we used the R,R-enantiomer, TCH-013a, to ensure clear kinetic results. The mechanism by which TCH-013a inhibits the proteasome was investigated using Michaelis-Menton analysis to determine of KM and Vmax and then further illustrated using a Lineweaver-Burk double reciprocal plot of the kinetic data. Kinetic analysis of CT-L activity of purified 20S particles indicate that when the substrate (Suc-LLVY-AMC) concentration was increased incrementally and measurements were taken at five different concentrations of TCH-013a or vehicle, the Vmax of the CT-L activity diminished with the increasing concentration of substrate and the KM remained constant (Fig. 3A). This is a pattern that is consistent with non-competitive type inhibition 48. By contrast, the proteasome inhibitor MG-132 (Fig. 1A), which binds the active site of the proteasome, inhibits the human 20S proteasome via a competitive type mechanism (data not shown). The results suggest that the site(s) of TCH-013 binding on the 20S proteasome is not directly in competition with substrate binding and proceeds via a non-competitive mechanism of binding. The Ki values (2.4 μM ±0.4) were determined using a Dixon plot (supplemental data FigS5) and matched the IC50 value (IC50 2.80 ±0.66 μM), which is also consistent with non-competitive kinetics.
Figure 3. TCH-013 inhibits the 20S proteasome non-competitively and binds to a site other than the CT-L active site.

(A) Kinetic analysis of 20S inhibition by TCH-013 was accomplished using purified human 20S proteasome treated with either: vehicle, 10, 5, 2.5, 1.25 or 0.6 μM TCH-013. Km and Vmax values were calculated from Michaelis-Menton analysis using a range of Suc-LLVY-AMC substrate from 1 to 20 μM. (Km values were 5.35 ±0.28, 5.40 ±0.81, 7.37 ±0.59, 5.66 ±0.49, 6.02 ±0.36, and 5.43 ± 0.52 for vehicle, 10, 5, 2.5, 1.25, and 0.6 μM treatment respectively, while Vmax values were 0.99 ±0.03, 0.02 ±.001, 0.24 ±0.01, 0.72 ±0.03, 0.85 ±0.03, and 0.82 ±0.04 U/sec for vehicle, 10, 5, 2.5, 1.25, and 0.6 μM treatment respectively). A representative double reciprocal Lineweaver-Burk plot in which all of the treatments result in intersecting on the same point of the X-axis (1/[S]) illustrates non-competitive inhibition. (B) The biotinylated non-reversible covalent and competitive inhibitor Ada-Lys(biotinyl)-(Ahx)3-(Leu)3-vinyl sulfone is not blocked from binding to the catalytic binding sites of the 20S proteasome. Purified human 20S proteasome particles were pretreated for one hour with vehicle, 10, 5, 2.5, 1.3 or 0.6 μM TCH-013a. Ada-Lys(biotinyl)-(Ahx)3-(Leu)3-vinyl sulfone was then used to probe for catalytic binding sites that were not occupied by TCH-013a. The proteasome particles were then resolved by 12.5% SDS-PAGE and transferred to PVDF. Immunoblotting was performed using an avidin-HRP conjugate. (C) TCH-013 inhibition of the 20S proteasome is not restored after extensive washing. 1 nM 20S proteasome was treated with vehicle, 10 μM TCH-013 or 1 μM MG-132 and tested for CT-L activity. The treated samples were washed extensively 500 volumes of assay buffer and tested for CT-L activity. (D) The effect of TCH-013 on CT-L activity of the 20S proteasome is additive to the effect of bortezomib. Dose response curves of bortezomib in combination with vehicle or varying amounts of TCH-013. (E) TCH-013 and bortezomib retain constant relative potencies as demonstrated by the linear isobole created using the dose of bortezomib necessary to obtain 50% inhibition of CT-L activity with varying amounts of TCH-013.
TCH-013 binds to a site(s) other than the catalytic site
Non-competitive binding kinetics implies that an inhibitor binds to a site on the enzyme that is distinct from the substrate binding site. We examined if TCH-013a binds to a site other than the substrate binding site using a probe directed toward the catalytic subunits of the 20S proteasome (Fig. 3B). Purified 20S enzyme was pretreated for 1 hour with vehicle, an excess of MG-132 (1 μM), or an excess of TCH-013a (10 μM). The drug bound enzyme was then probed with 1 μM biotinylated adamantane-acetyl-(6-aminohexanoyl)3(leucinyl)3-vinyl-(methyl)-sulfone (a potent, covalent, irreversible inhibitor of all three catalytic subunits ) at an amount determined to give 50% the maximum binding to the catalytic center of the β5 subunit (SFig. 6) 49. The samples were electrophoresed, transferred to PVDF and probed for the biotin label. The results shown in Figure 3 indicate that treatment of the 20S proteasome with MG-132 blocked the binding of the biotinylated probe to the β5 catalytic binding site 49. However, pretreatment of the enzyme for 1 hour with TCH-013a at concentrations ranging from 0.6 to 10 μM had no effect on the binding of the probe to any of the catalytic sites (β5, β1, or β2).
A difference in binding affinity between TCH-013a and the biotinylated probe for the active sites of the 20S proteasome could allow the probe to compete with TCH-013a, and bind to the catalytic subunits as seen in Figure 3B. Based on the non-competitive kinetics of TCH-013a this would seem unlikely. However, we further challenged this possibility by evaluating the reversibility of TCH-013a binding to the enzyme, compared to MG-132, using membrane washout experiments. Treatment of the 20S proteasome with TCH-013a or MG-132 completely abrogated the CT-L activity of the enzyme. After extensively washing with 500 volumes of activity buffer on a membrane concentrator, the retentates were analyzed again for 20S CT-L activity (Fig. 3C). Removal of MG-132 by extensive washing with reaction buffer completely restored proteolytic activity. Unlike the reversible competitive inhibitor MG-132, the proteolytic activity of the 20S proteasome was not restored for TCH013a-treated samples (Fig. 3C, >70% inactive), indicating that either TCH-013a is strongly bound to the enzyme or that the enzymatic activity is irreversibly compromised. Thus, consistent with our kinetic results, the catalytic sites are free to bind with the biotinylated probe in the presences of proteasome-bound TCH-013a.
Given that TCH-013 inhibits the proteasome at a site that is distinct from competitive inhibitors such as bortezomib, we determined the effect of the combination of TCH-013 and bortezomib on the CT-L activity of the proteasome. When added to the proteasome in combination, both TCH-013 and bortezomib retained a linear relationship of relative potencies indicating that the agents act additively when added in combination to the 20S proteasome (Fig. 3D and E) in the purified enzyme assay. This additive affect translated in cell culture where the combination treatment of bortezomib and TCH-013 in multiple myeloma cells indicated additive affects of the compounds (SFig. 7). These data further supports our findings that both agents affect proteasome activity via distinct mechanisms.
TCH-013 induces rapid cell death in bortezomib resistant THP-1 cells
Although recent advances in the treatment of MM have made a dramatic impact, nearly all patients relapse 50. Several mechanisms of resistance have been identified, including mutation and overexpression of the β5 catalytic subunit of the proteasome 24. Inhibitors with mechanisms that are distinct from these competitive inhibitors, such as bortezomib (BTZ), could bypass this type of resistance. Given that TCH-013 binds to a site(s) other than the substrate binding sites of the proteasome, we evaluated the cytotoxicity of TCH-013 in bortezomib-resistant human myelomonocytic THP-1 cells. The mechanism of acquired resistance in this cell line was previously determined to be due to the overexpression of a mutated β5 catalytic subunit of the proteasome. Proteasomes assembled with this mutant subunit also exhibited strong cross-resistances across the board of competitive proteasome inhibitors 24. Wild-type THP-1, and THP-1 cells with increasing levels of bortezomib resistance (THP-1/BTZ50, THP-1/BTZ200 and THP-1/BTZ500 cells) were treated with TCH-013 or bortezomib (control) and cell viability was measured after 72 hours using an MTS assay (Table 1A). THP-1/BTZ50, THP-1/BTZ200, and THP-1/BTZ500 were 48, 94 and 247-fold 24 more resistant to bortezomib, respectively. Importantly, TCH-013 retained efficacy against the bortezomib resistant cell lines and displayed similar potency regardless of the extent of bortezomib resistance. These results are consistent with the observed drug-proteasome binding interaction that is distinct from the binding of competitive type inhibitors, such as bortezomib.
TCH-013 induces cell death in MM cells
In order to assess whether non-competitive proteasome inhibition effectively kills MM cells, the cytotoxicity of TCH-013 was evaluated on a panel of myeloma and leukemia cell lines, as well as supportive bone marrow stromal cells, macrophages, and osteoclasts in cell culture. As indicated in Figure 4B, TCH-013 significantly decreased viability of myeloma and leukemia cell lines (GI50 1–4 μM). In contrast, normal lymphocytes, primary bone marrow stromal cells and macrophages were resistant to the cytotoxic effects of TCH-013 with GI50’s of 14μM to >20μM (Fig. 4B). Notably, these levels exceed the maximum serum concentration of TCH-013 that is attained following intraperitoneal injections (SFig. 8).
Figure 4. Table 1.

(A) TCH-013 inhibits cell growth in bortezomib (BTZ) resistant THP-1 cells that overexpress mutated β5 subunits. Parental and BTZ resistant THP-1 cell lines were treated with TCH-013 continuously for 72 hours and viability was measured using a MTS dye conversion assay. Resistance factor is an expression of the GI50 of the BTZ resistant THP-1 cell line divided by the CC50 of the parental THP-1 cell line. (B) In vitro viability testing results for leukemia, myeloma and supportive primary cell lines. (C) Murine bone marrow derived macrophages (mononuclear cells) were cultured under osteogenic differentiation conditions (50ng/ml M-CSF and 50ng/ml RANKL) in the continual presence of vehicle or indicated concentrations of TCH-013. Formation of multinucleated osteoclasts is visualized by tartrate-resistant acid phosphatase (TRAP) staining on day 6. At concentrations similar to those required to exert tumor cells cytotoxicity, TCH-013 inhibited the formation of multinucleated osteoclasts.(D) Survival of RPMI8226 cells alone(left) or in the presence of human BMSCs(right) as measured by Luciferase Activity(D, top panels) and MTT Assay(D, bottom panels), cells were incubated with various concentrations of TCH-013 ranging from 1.25μM to 20μM. The IC50 was determined by Prism(Graphpad).
In addition to its anti-tumor properties, an important mechanism of bortezomib is its ability to prevent pathological bone resorption by inhibiting osteoclast differentiation and resorption.51 Similar to these positive side-effects of bortezomib, treatment of bone marrow macrophages with TCH-013 inhibited their differentiation into multinucleated, bone resorbing osteoclasts at concentrations similar to those required to induce cytotoxicity in tumor cells (Fig. 4C).
The supporting microenvironment of the BMSC induces MM survival and renders many therapeutic options less effective. Given the notable lack of toxicity to the primary bone marrow stromal cells but excellent cytotoxicity to the MM cells, we investigated the efficacy of TCH-013 in co-cultures of MM cells with hBMSC isolated from CD138 negative BM samples of MM patients, as a model for this natural microenviroment. In order to differentiate the MM cells from primary hBMSC, we used RPMI-8226-luc cells in order to visualize cytotoxicity of MM cells by a decrease in luciferase activity. We found that TCH-013 induced cell death in both isolated MM cells and in MM cells co-cultured with hBMSC (Fig. 4D). The reduction of luciferase activity, indicative of survival of the RPMI-8226-luc cells for both MM alone (IC50 2.37 μM) and co-cultured (IC50 2.62 μM) corresponded with our previous cytotoxity data in isolated MM cells (GI50 1.84 μM).
To further examine the time course for apoptosis induced by bortezomib and TCH-013, RPMI-8226 cells were incubated with these compounds and the percentage of cells exhibiting an apoptotic phenotype was determined by propidium iodide staining and fluorescence activated cell sorting (FACS) to measure cellular chromosomal DNA content (SFig. 8A). The DNA content per cell can be used as a diagnostic indicator of cells in the G1 (2N content), G2 (4N content), and S (between 2N-4N content) phases. The labeling of cells with apparent DNA content less than the G1 peak (sub-G1) demonstrates that DNA fragmentation has occurred and is suggestive of apoptosis. TCH-013 (at 5X GI50, 10 μM) induced DNA fragmentation rapidly, indicative of programmed cell death or apoptosis, with a similar percentage of cells exhibiting a sub-G1 staining pattern over time, as compared to the clinically used MM drug bortezomib (at 500X GI50, 1.0 μM) (SFig. 8B). These data indicate that TCH-013 can induce substantial and rapid apoptosis in MM cells.
TCH-013 is well tolerated in non-tumor bearing immunocompetent mice and effective in an RPMI-8226 mouse xenograft model
In order to evaluate whether tumoral cytotoxicity translates in vivo, we determined the activity of TCH-013 against established RPMI-8226 human MM in female NIH-III mice (Fig. 5). The study, using four to six mice per group, evaluated the efficacy of 150 mg/kg TCH-013 administered intraperitoneal on tumor cell growth and host survival. This dose was determined to obtain serum levels of about 5 μM three hours post IP injection with a half-life of 3.3 hours (SFig. 9). The study had a control cohort treated with the vehicle alone (10% propylene glycol in sterile water). The mice were inoculated subcutaneously, high in the right axilla with RPMI-8226 MM cells with 76.4% of the animals developing measurable tumors. Treatment began when the mean estimated tumor mass for all groups was 122 mg (range, 111–124 mg). Mice were then assigned to treatment groups (0.2 mL/20g 10% propylene glycol in sterile water twice a day for 8 days then two days off, 1.25 mg/kg bortezomib intravenously once a day for three days then two days off, and 150 mg/kg TCH-013 twice a day for eight days then two days off). This dosing regimen did not indicate any detectable anemia, gross neurologic toxicity (data not shown), weight loss, or liver toxicity as earlier reported.40 In a log-linear regression model of the mice during treatment (SFig. 10), the mean tumor volume doubling time was 6.3 days for the vehicle treated group and >25 days for TCH-013 (P < 0.0001). 50% of bortezomib treated mice exhibited no tumor growth during the treatment period and 17% of the bortezomib treated mice experienced complete tumor regression after treatment was removed. The remainder of the bortezomib treated mice had a mean tumor doubling time of 20 days. Fifty percent of the TCH-013 treated mice also did not exhibit tumor growth during the treatment period. The other half of the TCH-013 treated mice had a doubling time longer than 25 days (n=4, P < 0.0001 when compared to the vehicle treated group) during the 10 day treatment period. Tumors began to grow after the cease of treatment on day 10 for both the bortezomib and TCH-013 treated groups, at similar doubling rates.
Figure 5.

TCH-013 delays tumor growth in an RPMI-8226 xenograft mouse model. The anti-tumor activity of TCH-013 against established RPMI-8226 human MM in female NIH-III mice was evaluated as described in materials and methods. TCH-013 was administered IP at 150 mg/kg twice daily. Bortezomib was administered intravenously twice weekly at the maximum tolerated dose. Tumor burden (mg) = (L X W2)/2, where L and W are the respective orthogonal tumor length and width measurements (mm). Shaded area included treatment days.
Conclusion
Without doubt, competitive proteasome inhibitors significantly improve the clinical outcome of patients with MM, however inherent toxicity (e.g. neuropathy and cytopenias) and cross-resistance is often observed in patients undergoing treatment. Nearly all patients eventually become resistant and/or intolerant after which the survival average is less than one year12, 13, 39. Resistance to bortezomib has been demonstrated to occur through several different mechanisms, including the overexpression of mutated catalytic subunits of the proteasome, 24, 25 which is the site in the proteasome where bortezomib and all other proteasome inhibitors under clinical evaluation bind 11. In this study, we examined TCH-013, a non-peptide based, small molecule NF-κB inhibitor40, 41. Michaelis-Menton kinetics demonstrates that TCH-013 inhibits the CT-L and Casp-L activities of the 20S proteasome via non-competitively kinetics. Our data verifies that TCH-013 binds to site(s) other than the substrate binding site and consistent with a distinct mechanism from competitive proteasome inhibitors. Importantly, TCH-013 acted additively with and overcame resistance to the competitive proteasome inhibitor bortezomib, consistent with a mode of action distinct from competitive proteasome inhibitors.
In addition, TCH-013 induced cell death in myeloma and leukemia cells preferentially over normal hematopoietic and stromal cells, and indicated activity in MM cells co-cultured with primary human BMSC isolated from CD138 negative BM samples of MM patients. Similar to bortezomib, TCH-013 was found to be effective as a single agent in MM cells albeit at higher concentrations than bortezomib. However, the GI50 of TCH-013 in the RPMI-8226 MM cell lines was comparable to the clinically significant drugs doxorubicin but better than melphalan 13. The concentration at which TCH-013 induced cell death in myeloma cells was similar to concentrations at which it inhibited the proteasome and blocked NF-κB transcription, which suggests that the cytotoxicity of TCH-013 is related to its effects on protein degradation by the proteasome. The efficacy of TCH-013 on RPMI-8226 cells in cell culture translated well into the RPMI-8226 mouse xenograft model. Mice treated with TCH-013 (150mg/Kg) resulted in a significant delay in tumor growth during the treatment period. This markedly delayed tumor growth was comparable to bortezomib treatment at its maximum tolerated dose. These studies indicate that the non-competitive inhibitor TCH-013 was effective in blocking MM tumor growth in vivo.
The binding site(s) at which TCH-013 non-competitively inhibits the proteasome is not known at this time and examples of non-competitive proteasome modulators are scares. One possibility is that TCH-013 binding induces a conformational change that prevents certain substrates from entering the proteolytic chamber of the proteasome complex. Detailed co-crystal structures of the TCH-013-proteasome interaction may to reveal the binding site(s) by which non-competitive inhibitors interact with the proteasome.
In conclusion, the orally available, well tolerated, trans-imidazoline TCH-013 represents a novel class of non-competitive proteasome inhibitor with significant efficacy in the delay of MM tumor growth in mice. Importantly, TCH-013 acts additively with and overcomes resistance to the competitive proteasome inhibitor bortezomib, consistent with a mode of action distinct from competitive proteasome inhibitors.
METHODS
Materials and Methods
Reagents
Ada-Lys(biotinyl)-(Ahx)3-(Leu)3-vinyl sulfone was purchased from Enzo Life Sciences. Bortezomib was purchased from Selleck Biochemicals, MG-132 was purchased from EMD. Fluorogenic substrates and purified proteasome particles were purchased from Boston Biochem. All antibodies were purchased from Santa Cruz Biotechnologies and Cell Signaling. Propidium Iodide, RNase and all cell culture media and supplements were purchased from Invitrogen. Luciferase Assay System was purchased from Promega. Black 96-well-plate were purchased from Corning and 1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformaza (MTT), was purchased from Sigma. All other chemicals were purchased from Sigma.
Cell culture
The human cell lines HeLa and RPMI-8226 were purchased from American Type Culture Collection. THP1, THP1/BTZ50, THP1/BTZ200 and THP1/BTZ500 were a kind gift from Drs. J. Cloos and G. Jansen, Dept. of Hematology, Research coordinator Pediatric Oncology/Hematology VU University Medical Center, Cancer Center Amsterdam, Amsterdam, The Netherlands 24. The hemopoaetic cell lines were maintained in RPMI-1640 supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 mg/mL streptomycin, 1 mM sodium pyruvate, and 0.2 mM L-glutamine. HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 mg/mL streptomycin, 1 mM sodium pyruvate, and 0.2 mM L-glutamine. Cells were cultured at 37 °C, 5% CO2. RPMI8226-Luc cells were grown in RPMI1640 medium with 10%FBS. hBMSC was isolated from CD138 negative BM samples of MM patients, and cultured in MEM Alpha medium with 20%FBS in cell culture coated plate for about 14–21 days(various between patients), change the medium when needed.
Cell viability assays
Human white blood cells were counted manually using a BD Unopette reservoir and a hemacytometer. CCRF-CEM, K-562, MOLT-4, RPMI-8226 and SR cell viability was determined in the NCI Developmental Therapeutics Program 60 Cell Line Screen. All other cell line viability was assayed using the MTS dye conversion assay (Promega) in our lab following the manufacturer’s protocol. The absorbance of formazan was measured at 490 nm on a SpectraMaxM5e microplate reader. Background absorbance was subtracted from each measurement and viability was calculated using the untreated control as 100%. Drug potency was expressed as GI50 (concentrations that inhibit viability by 50%). GI50 was calculated by a non-linear regression analysis of the values.
FACS analysis
Cells were treated with vehicle (0.1% DMSO), bortezomib, or TCH-013 for 0, 8, 16 and 24 hours at 37 °C, 5% CO2. The cells were harvested and resuspended in PBS with 50% FBS. The cells were fixed with 70% ice cold ethanol overnight. After centrifugation of the cells, the pellet was washed two times with 5 mL ice cold PBS containing 10% FBS and resulting cell pellet was resuspended in staining solutions (PBS containing 50 μg propidium iodide, 10 μL 0.1M EDTA, 14 units RNaseA). The DNA content was analyzed by flow cytometry using a FACS Vantage SE analyzer with ModFit LT software.
Proteasome extraction and measurement of proteasome activity
Proteasome was extracted from RPMI-8226 cells with ATP/DTT buffer as previously described by Holyoake 52. The fluorogenic substrates Suc-LLVY-AMC, Boc-ARR-AMC and Z-LLE-AMC were used to measure CT-L, T-L and Casp-L proteasome activities using 1 nM purified 20S proteasome or 10 μg ATP/DTT lysate 53. The rate of cleavage of fluorogenic peptide substrates was determined by monitoring the fluorescence of released aminomethylcoumarin using a SpectraMax M5e multiwell plate reader at an excitation wavelength of 380 nm and emission wavelength of 460 nm. Fluorescence was measured at 37° C every minute over a 30 minute period and the maximum increase in fluorescence per minute was used to calculate specific activities of each sample. KM, Ki, and Vmax values were calculated from Michaelis–Menten analysis using a concentrations range of Suc-LLVY-AMC (1 to 20 μM) and TCH-013a (0.6–10 μM).
Detection of biotinylated Ada-Lys(biotinyl)-(Ahx)3-(Leu)3-vinly sulfone labeled proteasome subunits
Inhibitors were mixed with 200ng of the human 20S proteasome isolated from human erythrocytes in enzyme assay buffer (50 mM Tris-HCl pH 7.5) for 1 hour at 37 °C. Subsequently 100 nM Ada-Lys(biotinyl)-(Ahx)3-(Leu)3-vinly sulfone was added and incubated for hour at 37°C. Reaction mixtures were boiled with Laemmli’s buffer containing 2-mercapto-ethanol for 3 minutes and resolved on 12.5% SDS-PAGE. The proteins were transferred to a polyvinylidiene difluoride membrane. Immunoblotting was performed using avidin-HRP conjugate and developed using ECL detection.
Western blot analysis of whole cell lysates and immunoprecipitates
Harvested cells were washed twice with PBS and suspended in ice cold lysis buffer (20 mM tris-HCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X-100, 1 mM PMSF, 2.5 mM sodium pyrophosphate, 1 mM glycerophosphate, 1 mM Na3VO4, and 1 mg/mL leupeptin. The suspension was kept on ice for 20 minutes and centrifuged at 15,000 X g for 10 minutes at 4°C. The supernatant was recovered as total cell lysate and 20 μg lysate was separated on a 10% Tris-HCl gel. Immunoblotting was performed using 1:1000 dilution of the indicated antibodies described below. Mouse monoclonal antibodies were used against the N-terminal of IκBα and ubiquitin (from Cell Signaling) and rabbit polyclonal antibody against the C-terminal of IκBα (Santa Cruz Biotechnology). The second antibody, HRP conjugated anti-mouse or anti-rabbit IgG antibody, was used at the dilution of 1:3000. For IκBα immunoprecipitation, whole cell lysates were prepared as above. Approximately 1 mg of protein from whole cell lysates was immunoprecipitated using the rabbit polyclonal antibody against the C-terminal of IκBα (Santa Cruz Biotechnology) following standard procedures. One quarter of the immunoprecipitate was separated on a 10% Tris-HCl gel and transferred to PVDF. Immunoblotting was performed using mouse monoclonal antibody against ubiquitin (Cell Signaling).
Detection of ubiquitin and IκBα by confocal microscopy
HeLa cells were grown on glass coverslips and treated with vehicle (0.1 % DMSO) or 1.0 μM TCH-013. After pretreatment with compounds, the cells were stimulated with 20 ng/mL TNF-α (30 minutes for IκBα and 2 hours for ubiquitin experiments) while the control cells were left un-stimulated. Cells were then fixed with 4% paraformaldehyde in PBS, permeabilized in 0.5% Triton in PBS, and pre-incubated for 1 h in blocking buffer (5% BSA, 0.05% Tween-20 in PBS). The cells were stained overnight with primary antibody at 4° C (1:250 of either rabbit anti-ubiquitin (P4D1), mouse anti-IκBα (L35A5) or rabbit anti-IκBα (C21) in blocking buffer) and for 1 h with Alexa568 labeled secondary antibody (1:1000, Invitrogen). Coverslips were mounted with Fluorogel containing DAPI (4,6-diamidino-2-phenylindole; 1 mg/ml). Cells were imaged using an Olympus FV1000 scanning confocal microscope using the one way XY scan mode.
In vitro murine osteoclast differentiation
Total bone marrow was isolated from long bones of 8-week old C56Bl/6 mice. Bone marrow derived macrophages (BMM) were generated by culturing first in αMEM supplemented with 100 ng/mL M-CSF for 3 days. To generate mature OCs, BMMs were then replated at 4x104 cells/mL in αMEM supplemented with 50 ng/mL M-CSF and 50 ng/mL RANKL, refeeding every 48 hours for 6 days as previously described.54 Cells were fixed and stained for TRAP using the leukocyte acid phosphatase kit (Sigma-Aldrich).
MM-hBMSC Co-culture
Human BMSCs were plated in 96-well-plate in 20% FBS MEM Alpha and allowed to attach for overnight and then the wells were gently washed with PBS twice. Medium was then exchanged to MEM Alpha with 0.5%FBS and cells were starved for 12hours. Then medium was exchanged to RPMI1640 with 0.5%FBS when RPMI8226-Luc cells were added to the plate with medium alone or in the presence of various concentrations of TCH-013 and cells were allowed to grow for 48hours. After incubation, the survival of the RPMI8226 cells was assessed by Luciferase Assay System (Promega) per manufacture’s instruction and by 1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan (MTT; Sigma) assay.
Human MM xenograft model
The RPMI-8226 xenograft model was performed at Charles River Laboratories using Female NIH-III mice (Crl: NIH-Lyst bgFoxn1nuBtkxid). Mice were 5–6 weeks old on day 1 of the experiment. A 2.5 X 107 cells/mL suspension (92.35% viable based on trypan blue exclusion) was prepared in 50% serum free media and 50% matrigel. The mice were implanted subcutaneously, high in the right axilla, with 5 X 106 cells in 200 μL using a 27-guage needle attached to a 1 cc syringe. Post-injection viability was determined by trypan blue exclusion to be 76.4%. Tumor burden (mg) was estimated from caliper measurements by the formula for the volume of a prolate ellipsoid assuming unit density as: Tumor burden (mg) = (L X W2)/2, where L and W are the respective orthogonal tumor length and width measurements (mm).
Supplementary Material
Acknowledgments
The authors gratefully acknowledge financial support in part from the MM Research Foundation (MMRF), the National Institutes of Health (CA-142644-01), Bioscience Research and Commercialization Center (BRCC) and the Michigan Initiative for Innovation & Entrepreneurship (MIIE). The authors would like to thank M. Frame for her technical help with confocal microscopy and C. Geiger and C. Hornick for technical help with the bone marrow studies. We would also like to thank MIR Discovery and Imaging Services/Charles River Laboratories for their work on the RPMI-8226 mouse xenograft model. A great deal of appreciation is given to J. Cloos, G. Jansen and J. Meerloo for the kind gift of the bortezomib resistant cell lines.
Footnotes
Supplementary information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mahindra A, Laubach J, Raje N, Munshi N, Richardson PG, Anderson K. Latest advances and current challenges in the treatment of multiple myeloma. Nat Rev Clin Oncol. 2012;9:135–143. doi: 10.1038/nrclinonc.2012.15. [DOI] [PubMed] [Google Scholar]
- 2.Anderson KC, Carrasco RD. Pathogenesis of myeloma. Annu Rev Pathol. 2012;6:249–274. doi: 10.1146/annurev-pathol-011110-130249. [DOI] [PubMed] [Google Scholar]
- 3.Zheng Y, Cai Z, Wang S, Zhang X, Qian J, Hong S, Li H, Wang M, Yang J, Yi Q. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood. 2009;114:3625–3628. doi: 10.1182/blood-2009-05-220285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hideshima T, Chauhan D, Schlossman R, Richardson P, Anderson KC. The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene. 2001;20:4519–4527. doi: 10.1038/sj.onc.1204623. [DOI] [PubMed] [Google Scholar]
- 5.Hideshima T, Podar K, Chauhan D, Anderson KC. Cytokines and signal transduction. Best Pract Res Clin Haematol. 2005;18:509–524. doi: 10.1016/j.beha.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Chiang MY, Stadtmauer EA. NF-kappaB, IL-6 and myeloma cell growth: making the connection. Cancer Biol Ther. 2004;3:1018–1020. doi: 10.4161/cbt.3.10.1326. [DOI] [PubMed] [Google Scholar]
- 7.Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, Anderson KC. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996;87:1104–1112. [PubMed] [Google Scholar]
- 8.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J, Anderson KC. NF-kappa B as a Therapeutic Target in Multiple Myeloma. J Biol Chem. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 9.Huston A, Roodman GD. Role of the microenvironment in multiple myeloma bone disease. Future Oncol. 2006;2:371–378. doi: 10.2217/14796694.2.3.371. [DOI] [PubMed] [Google Scholar]
- 10.Bommert K, Bargou RC, Stuhmer T. Signalling and survival pathways in multiple myeloma. Eur J Cancer. 2006;42:1574–1580. doi: 10.1016/j.ejca.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 11.Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol. 2012;19:99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stewart AK. Novel therapeutics in multiple myeloma. Hematology. 2012;17(Suppl 1):S105–108. doi: 10.1179/102453312X13336169156131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson KC, Alsina M, Bensinger W, Biermann JS, Chanan-Khan A, Cohen AD, Devine S, Djulbegovic B, Faber EA, Jr, Gasparetto C, Huff CA, Kassim A, Medeiros BC, Meredith R, Raje N, Schriber J, Singhal S, Somlo G, Stockerl-Goldstein K, Treon SP, Tricot G, Weber DM, Yahalom J, Yunus F. Multiple myeloma. J Natl Compr Canc Netw. 2012;9:1146–1183. doi: 10.6004/jnccn.2011.0095. [DOI] [PubMed] [Google Scholar]
- 14.Groll M, Potts BC. Proteasome structure, function, and lessons learned from beta-lactone inhibitors. Curr Top Med Chem. 2011;11:2850–2878. doi: 10.2174/156802611798281320. [DOI] [PubMed] [Google Scholar]
- 15.Groll M, Huber R, Moroder L. The persisting challenge of selective and specific proteasome inhibition. J Pept Sci. 2009;15:58–66. doi: 10.1002/psc.1107. [DOI] [PubMed] [Google Scholar]
- 16.Groll M, Clausen T. Molecular shredders: how proteasomes fulfill their role. Curr Opin Struct Biol. 2003;13:665–673. doi: 10.1016/j.sbi.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 18.Groll M, Heinemeyer W, Jager S, Ullrich T, Bochtler M, Wolf DH, Huber R. The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc Natl Acad Sci U S A. 1999;96:10976–10983. doi: 10.1073/pnas.96.20.10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kisselev AF. Joining the army of proteasome inhibitors. Chem Biol. 2008;15:419–421. doi: 10.1016/j.chembiol.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 20.Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 21.Shah JJ, Orlowski RZ. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia. 2009;23:1964–1979. doi: 10.1038/leu.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navon A, Ciechanover A. The 26 S proteasome: from basic mechanisms to drug targeting. J Biol Chem. 2009;284:33713–33718. doi: 10.1074/jbc.R109.018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franke NE, Niewerth D, Assaraf YG, van Meerloo J, Vojtekova K, van Zantwijk CH, Zweegman S, Chan ET, Kirk CJ, Geerke DP, Schimmer AD, Kaspers GJ, Jansen G, Cloos J. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia. 2012;26:757–768. doi: 10.1038/leu.2011.256. [DOI] [PubMed] [Google Scholar]
- 24.Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, van der Heijden JW, Ylstra B, Peters GJ, Kaspers GL, Dijkmans BA, Scheper RJ, Jansen G. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112:2489–2499. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- 25.Verbrugge SE, Assaraf YG, Dijkmans BA, Scheffer GL, Al M, den Uyl D, Oerlemans R, Chan ET, Kirk CJ, Peters GJ, van der Heijden JW, de Gruijl TD, Scheper RJ, Jansen G. Inactivating PSMB5 mutations and P-glycoprotein (MDR1/ ABCB1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno) proteasome inhibitors in mononuclear blood cells from rheumatoid arthritis patients. J Pharmacol Exp Ther. 2012;341:174–182. doi: 10.1124/jpet.111.187542. [DOI] [PubMed] [Google Scholar]
- 26.de Wilt LH, Jansen G, Assaraf YG, van Meerloo J, Cloos J, Schimmer AD, Chan ET, Kirk CJ, Peters GJ, Kruyt FA. Proteasome-based mechanisms of intrinsic and acquired bortezomib resistance in non-small cell lung cancer. Biochem Pharmacol. 2012;83:207–217. doi: 10.1016/j.bcp.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Jankowska E, Gaczynska M, Osmulski P, Sikorska E, Rostankowski R, Madabhushi S, Tokmina-Lukaszewska M, Kasprzykowski F. Potential allosteric modulators of the proteasome activity. Biopolymers. 2010;93:481–495. doi: 10.1002/bip.21381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wenner M. A new kind of drug target. Sci Am. 2009;301:70–74. 76. doi: 10.1038/scientificamerican0809-70. [DOI] [PubMed] [Google Scholar]
- 29.Hauske P, Ottmann C, Meltzer M, Ehrmann M, Kaiser M. Allosteric regulation of proteases. Chembiochem. 2008;9:2920–2928. doi: 10.1002/cbic.200800528. [DOI] [PubMed] [Google Scholar]
- 30.Gaczynska M, Osmulski PA. Characterization of noncompetitive regulators of proteasome activity. Methods Enzymol. 2005;398:425–438. doi: 10.1016/S0076-6879(05)98035-X. [DOI] [PubMed] [Google Scholar]
- 31.DeDecker BS. Allosteric drugs: thinking outside the active-site box. Chem Biol. 2000;7:R103–107. doi: 10.1016/s1074-5521(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 32.Li X, Wood TE, Sprangers R, Jansen G, Franke NE, Mao X, Wang X, Zhang Y, Verbrugge SE, Adomat H, Li ZH, Trudel S, Chen C, Religa TL, Jamal N, Messner H, Cloos J, Rose DR, Navon A, Guns E, Batey RA, Kay LE, Schimmer AD. Effect of noncompetitive proteasome inhibition on bortezomib resistance. J Natl Cancer Inst. 2010;102:1069–1082. doi: 10.1093/jnci/djq198. [DOI] [PubMed] [Google Scholar]
- 33.Groebe DR. In search of negative allosteric modulators of biological targets. Drug Discov Today. 2009;14:41–49. doi: 10.1016/j.drudis.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 34.Anbanandam A, Albarado DC, Tirziu DC, Simons M, Veeraraghavan S. Molecular basis for proline- and arginine-rich peptide inhibition of proteasome. J Mol Biol. 2008;384:219–227. doi: 10.1016/j.jmb.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaczynska M, Osmulski PA, Gao Y, Post MJ, Simons M. Proline- and arginine-rich peptides constitute a novel class of allosteric inhibitors of proteasome activity. Biochemistry. 2003;42:8663–8670. doi: 10.1021/bi034784f. [DOI] [PubMed] [Google Scholar]
- 36.Gao Y, Lecker S, Post MJ, Hietaranta AJ, Li J, Volk R, Li M, Sato K, Saluja AK, Steer ML, Goldberg AL, Simons M. Inhibition of ubiquitin-proteasome pathway-mediated I kappa B alpha degradation by a naturally occurring antibacterial peptide. J Clin Invest. 2000;106:439–448. doi: 10.1172/JCI9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sprangers R, Li X, Mao X, Rubinstein JL, Schimmer AD, Kay LE. TROSY-based NMR evidence for a novel class of 20S proteasome inhibitors. Biochemistry. 2008;47:6727–6734. doi: 10.1021/bi8005913. [DOI] [PubMed] [Google Scholar]
- 38.Kroll M, Arenzana-Seisdedos F, Bachelerie F, Thomas D, Friguet B, Conconi M. The secondary fungal metabolite gliotoxin targets proteolytic activities of the proteasome. Chem Biol. 1999;6:689–698. doi: 10.1016/s1074-5521(00)80016-2. [DOI] [PubMed] [Google Scholar]
- 39.Alexanian R, Delasalle K, Wang M, Thomas S, Weber D. Curability of multiple myeloma. Bone Marrow Res. 2012;2012:916479. doi: 10.1155/2012/916479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lansdell TA, O’Reilly S, Woolliscroft T, Kahlon DK, Hovde S, McCormick JJ, Henry RW, Cornicelli JA, Tepe JJ. Attenuation of collagen-induced arthritis by orally available imidazoline-based NF-kB inhibitors. Bioorg Med Chem Lett. 2012;22:4816–4819. doi: 10.1016/j.bmcl.2012.05.056. [DOI] [PubMed] [Google Scholar]
- 41.Kahlon DK, Lansdell TA, Fisk JS, Hupp CD, Friebe TL, Hovde S, Jones AD, Dyer RD, Henry RW, Tepe JJ. Nuclear Factor-kappaB Mediated Inhibition of Cytokine Production by Imidazoline Scaffolds. J Med Chem. 2009;52:1302–1309. doi: 10.1021/jm8013162. [DOI] [PubMed] [Google Scholar]
- 42.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 43.Dominquez A, Fernandez A, Gonzalez N, Iglesias E, Montenegro L. Determination of critical micelle concentration of some surfactants by three techniques. J Chem Ed. 1997;74:1227–1231. [Google Scholar]
- 44.Thompson SJ, Loftus LT, Ashley MD, Meller R. Ubiquitin-proteasome system as a modulator of cell fate. Curr Opin Pharmacol. 2008;8:90–95. doi: 10.1016/j.coph.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma V, Lansdell TA, Peddibhotla S, Tepe JJ. Sensitization of tumor cells towards chemotherapy: Enhancing the efficacy of camptothecin by novel imidazolines. Chem Biol. 2004;11:1689–1699. doi: 10.1016/j.chembiol.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 48.Segel IH. Enzyme Kinetics. John Wiley & Sons, Inc; New York: 1975. [Google Scholar]
- 49.Borodovsky A, Ovaa H, Meester WJ, Venanzi ES, Bogyo MS, Hekking BG, Ploegh HL, Kessler BM, Overkleeft HS. Small-molecule inhibitors and probes for ubiquitin- and ubiquitin-like-specific proteases. Chembiochem. 2005;6:287–291. doi: 10.1002/cbic.200400236. [DOI] [PubMed] [Google Scholar]
- 50.Richards T, Weber D. Advances in treatment for relapses and refractory multiple myeloma. Med Oncol. 2010;27(Suppl 1):S25–42. doi: 10.1007/s12032-009-9407-5. [DOI] [PubMed] [Google Scholar]
- 51.Rossia M, Di Martino MT, Morelli E, Leotta M, Rizzo A, Grimaldi A, Misso G, Caraglia M. Molecular Targets for the Treatment of Multiple Myeloma. Curr Cancer Drug Targets. 2012 doi: 10.2174/156800912802429300. [DOI] [PubMed] [Google Scholar]
- 52.Heaney NB, Pellicano F, Zhang B, Crawford L, Chu S, Kazmi SM, Allan EK, Jorgensen HG, Irvine AE, Bhatia R, Holyoake TL. Bortezomib induces apoptosis in primitive chronic myeloid leukemia cells including LTC-IC and NOD/SCID repopulating cells. Blood. 2010;115:2241–2250. doi: 10.1182/blood-2008-06-164582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci U S A. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uluckan O, Becker SN, Deng H, Zou W, Prior JL, Piwnica-Worms D, Frazier WA, Weilbaecher KN. CD47 regulates bone mass and tumor metastasis to bone. Cancer Res. 2009;69:3196–3204. doi: 10.1158/0008-5472.CAN-08-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.