

Abstract
Although the affinity optimization of protein binders is straightforward, engineering epitope specificity is more challenging. Targeting a specific surface patch is important because the biological relevance of protein binders depends on how they interact with the target. They are particularly useful to test hypotheses motivated by biochemical and structural studies. We used yeast display to engineer monobodies that bind a defined surface patch on the mitogen activated protein kinase (MAPK), Erk-2. The targeted area (“CD” domain) is known to control the specificity and catalytic efficiency of phosphorylation by the kinase by binding a linear peptide (“D” peptide) on substrates and regulators. An inhibitor of the interaction should thus be useful for regulating Erk-2 signaling in vivo. Although the CD domain constitutes only a small percentage of the surface area of the enzyme (~ 5%), sorting a yeast displayed monobody library with wild type (wt) Erk-2 and a rationally designed mutant led to isolation of high affinity clones with desired epitope specificity. The engineered binders inhibited the activity of Erk-2 in vitro and in mammalian cells. Furthermore, they specifically inhibited the activity of Erk-2 orthologs in yeast and suppressed a mutant phenotype in round worms caused by overactive MAPK signaling. The study therefore shows that positive and negative screening can be used to bias the evolution of epitope specificity and predictably design inhibitors of biologically relevant protein-protein interaction.
Keywords: MAPK, Erk-2, Fn3, protein-protein interaction, yeast display
Introduction
Structural and biochemical studies often lead to hypotheses regarding how the activity of a protein may be inhibited using an epitope specific binder. Although reagents with defined binding activity are useful for hypothesis testing and target validation, engineering epitope specificity is challenging for both rational design and directed evolution. Instead, protein inhibitors are designed by first engineering high affinity binders and then screening the selected clones for relevant function (1, 2). This strategy does not make full use of existing information about the target molecule, e.g. a surface that has been identified to be functionally relevant. Also, engineering an inhibitor in and of itself does not test a hypothesis motivated by biochemical and structural studies, because finding a binder that turns out to be an inhibitor is not the same as testing if targeting a particular surface patch leads to inhibition of a biologically relevant response.
A number of factors will determine whether epitope selectivity can be engineered efficiently, including the target molecule, the protein scaffold, and the method of screen. Although in vitro display platforms, such as phage or ribosome display, can also be used to engineer epitope specificity, cellular display systems, including yeast and bacterial displays are particularly well suited for the task because they allow analysis of displayed molecules by flow cytometry in order to fine tune molecular interaction (3, 4). In this study, we used yeast display to simultaneously engineer target affinity and epitope specificity. The quantitative information collected on the individual clones during the sort was then used to model their in vitro and in vivo activity. We chose the monobody scaffold derived from the tenth type 3 domain of fibronectin (Fn3) for our study because it has a compact structure (~100 amino acids) and can be engineered by mutating flexible loops to interact with an arbitrary target with high affinity and selectivity (5). Fn3 monobodies are ideal for targeting intracellular proteins, since they do not require a disulfide bond for structural stability.
We designed epitope specific Fn3 binders to target a surface patch on Erk-2 known to be important for signaling (Fig. 1a). The targeted area, the CD domain, appears on a solvent exposed surface away from the catalytic site, but plays a critical role in Erk-2 activity by binding a linear D-peptide found on its substrates and regulators (6–9). Since the interaction between the CD domain and a D peptide is used to impose specificity of signaling in all MAPKs, disrupting the interaction at the CD domain should result in selective inhibition of MAPK signaling (Fig. S1). Yet, conclusive evidence that links inhibition of D peptide binding with physiological changes in vivo is lacking due to the lack of a suitable reagent. For example, isolated D peptides typically have limited affinity and specificity (10), and are biologically unstable for in vivo testing. Computationally designed small molecule inhibitors were shown to induce apoptotic responses in cultured cells and change the egg laying behavior of C. elegans (11) but they have low affinity (12) and have not been shown to work through the docking site of Erk-2. Given the challenges of designing small molecule inhibitors (13), epitope specific protein binders are attractive alternatives to small molecule inhibitors for target validation in drug discovery as well as for basic research applications.
Figure 1. Engineering monobodies to target the Erk-2 CD domain.
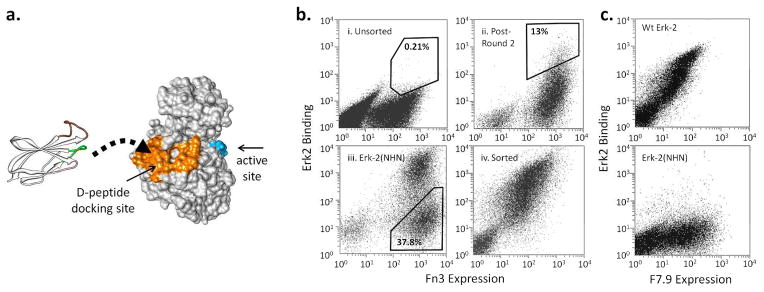
a. The Erk-2 residues within 5 Å of bound D-peptide (2GPH) are colored orange. The activation loop residues are colored in blue to show the separation between the docking domain and the active site. An Fn3 structure is shown to the scale on the left and is depicted as binding the docking surface. The randomized loops are shown in color: BC (green), DE (pink), FG (brown).
b. The yeast displayed Fn3 library was sorted using streptavidin-coated magnetic beads and FACS. During FACS, the cells were labeled with a cMyc antibody and Erk-2 to normalize binding with protein expression. i) Unsorted cells labeled with 1 μM of Erk-2. ii) Cells that have been sorted twice by FACS and then labeled with 250 nM of Erk-2. iii) Cells labeled with Erk-2(NHN) during negative sorting (Round 6). iv) Cells after the final round of FACS labeled with 10 nM of Erk-2. The percentages indicate the fraction of the cells within the indicated gates.
c. A representative selected clone, F7.9, binds 10 nM wt Erk-2 but not Erk-2(NHN). The lack of binding to the mutant indicates that the monobody epitope includes one or more of the mutated residues.
Because the area involved in the binding of a D peptide is only ~ 5% of the total surface area (14), screening of Erk-2 mutants may yield a similarly low percentage of the binders capable of disrupting the docking interaction. We therefore tested the use of a rationally designed Erk-2 mutant to engineer epitope specific binders that specifically target the docking domain. The study yielded a surprising finding that the CD domain may constitute an interaction hot spot by demonstrating that there exists a significant selection bias at the docking site over the rest of the protein surface. The selected binders interfered with Erk-2 activity in vitro and in cultured cells as expected. Importantly, when expressed in yeast and round worms, the designed binders predictably interfered with orthologous Erk-2 signaling in each organism, thus providing for the first time definitive evidence that blocking the CD domain is a valid strategy for selective inhibition of Erk-2 signaling in vivo.
Results and Discussion
Screening of Fn3 yeast library
Existing biochemical and structural data suggest that targeting the CD domain of Erk-2 would lead to inhibition of the kinase activity, but testing of this idea in vivo has been elusive due to the lack of a suitable reagent. To identify monobody binders of the Erk-2 CD domain, we screened an Fn3 library on the yeast surface using recombinant Erk-2 as bait (Fig. 1b, S2). We used both magnetic sorting and fluorescence activated cell sorting (FACS) to cope with the large size of the library (1.5 × 108) (15). The sorting strategy is described in Supplementary Information (SI, Fig. S3). After the fourth round of FACS, most of the selected clones could be labeled intensely using 10 nM of Erk-2. To identify the monobodies that bind Erk-2 at the CD domain, we constructed a rationally designed mutant, Erk-2(NHN), that contains three mutations (H123N, Y126H, and D319N) at the docking site. The mutations prevent the binding of a D-peptide (Fig. S4). We reasoned that these mutations should similarly disrupt the binding of a monobody whose epitope overlaps with the CD domain. Therefore, we labeled the yeast with Erk-2(NHN) and collected the cells that do not bind mutant Erk-2, which presumably correspond to epitope-specific monobodies (Round 6).
A large fraction (~38%) of the cells did not bind the mutant protein (Fig. 1b, iii), which is significantly greater than expected based on the 5% fractional surface area of the docking domain, and suggests that the CD domain may constitute a preferred interaction surface. A recent study described the use of phosphorylated and unphosphorylated forms of Erk-2 to engineer conformation-specific intrabodies (16). The use of a rationally designed mutant should be useful to guide epitope evolution toward an arbitrary surface patch and engineer functionally relevant binders independent of conformational changes. The Fn3 monobody scaffold was previously used to bind intracellular targets, including the SH2 domain of Abl kinase or SUMO (17, 18). Monobody binding was sufficient to inhibit the activity of the target proteins by disrupting protein-protein interaction. However, these studies did not provide quantitative evaluation of the binding preference at the target sites, which makes it difficult to determine if similar success can be expected on other systems. Furthermore, these molecules have smaller surface areas than Erk-2, which likely increased the probability of finding binders at or near functionally relevant sites without explicit negative selection. Introducing negative selection should play a more important role for engineering epitope specificity as the target size increases.
Characterization of individual clones
We characterized twenty clones among those selected after sixth and seventh rounds of sorting. Sixteen clones bound wt Erk-2 only, three clones bound both wt and Erk-2(NHN), and one bound neither (Fig. 1c, S5). Therefore, the majority of the selected clones had desired epitope specificity. The difference between epitope-specific binders and those that lack the specificity was apparent, since each examined clone either exhibited specificity for the CD domain or not. The inclusion of nonselective clones in the final pool suggests that a more stringent counter-selection may be needed. We sequenced eleven clones and obtained five unique sequences (Fig. 2a). Most mutants contained one or more arginines in the FG loop, which may interact with the acidic patch of the CD domain. Interestingly, F6.2, F7.1, F7.8 and F7.9 contained residues within their FG loop and the G strand that recapitulated the canonical D-peptide motif (R-X3-φ-X-φ, where φ is a hydrophobic residue) (19). There was no detectable consensus sequence in the BC or DE loop.
Figure 2. Binding characteristics of selected monobodies.

a. The yeast displaying various Fn3 clones were labeled with purified Erk-2, and the MFI of the displaying population was fitted to a binding curve to extract the apparent binding constant Kd. All measurements were done in triplicates or more to compute the mean and standard deviation. The loop sequences correspond to the Fn3 residues that were randomized in the library. The stability of F6.2 and F7.9 was measured by circular dichroism spectroscopy. The specificity of Erk-2 binding was tested by labeling yeast cells with 1 μM biotinylated Erk-2, p38, or JNK; and SA-PE. The MFI of the displaying population was measured.
We measured the Erk-2 affinity of individual clones on the yeast surface. Affinity measurement on the yeast surface is known to yield values that largely agree with in-solution methods, such as surface Plasmon resonance (20). Most of the clones had a dissociation constant Kd in low nM (Fig. 2a). Two clones, F6.2 and F7.9, that were characterized by circular dichroism spectroscopy had denaturation temperature Tm = 69 – 71 °C, indicating that the mutations in the loops did not destabilize the molecules (Fig. 2a, S6).
Specificity of interaction
We repeated the binding study with purified p38α and JNK1, which share 69% and 60% sequence similarity with Erk-2, respectively. The sequence variation is greater within the docking domain, since of the 28 Erk-2 residues within 5 Å of the bound peptide, only 15 (eight) are conserved in p38α (JNK1). Targeting a divergent epitope should be an effective way to increase the specificity of designed inhibitors and reduce nonspecific binding. Most of the clones did not bind p38α or JNK1 at 1 μM concentration (Fig. 2b). F7.8 bound p38α with Kd = 0.2 μM, which is still 155-fold higher than its affinity for Erk-2 (Kd = 1.3 nM). Therefore, all selected Fn3 mutants bound Erk-2 with high specificity, even though we did not actively sort against other enzymes. A likely explanation is that beneficial mutations are rare on average and therefore targets that are not used in positive selection are implicitly selected against (21). Although our study is limited in scope, the docking site residues are equally or more divergent in other MAPKs, including p38β, p38γ, JNK2, JNK3, and Erk5, which should reduce the likelihood of significant binding to these targets.
Monobodies inhibit Erk-2 activity in vitro
Using purified monobodies, we performed in vitro binding and kinase inhibition assays. To test if the monobodies disrupt the interaction at the CD domain (Fig. 3a), we used a model D-peptide, pepHePTP, fused to GST to pull down Erk-2 in the presence of different Fn3 competitors. All clones reduced the efficiency of precipitation in a concentration-dependent manner (Fig. 3b). In contrast, the amount of Erk-2 bound to GST-pepHePTP was unaffected by the addition of a control monobody, F0.1, that binds maltose binding protein, MBP (22). That F7.4 would also inhibit the binding of Erk-2 was unexpected, because the clone recognizes both wt and Erk-2(NHN) (Fig. S5), suggesting that the clone does not bind at the docking site. But it also raises an interesting possibility that the clone may bind near the CD domain and sterically hinder the binding of a D-peptide. By fine tuning the epitope, novel inhibitors may be engineered for other challenging targets, e.g. proteases, that specifically utilize this mechanism.
Figure 3. Functional assays of engineered Fn3.
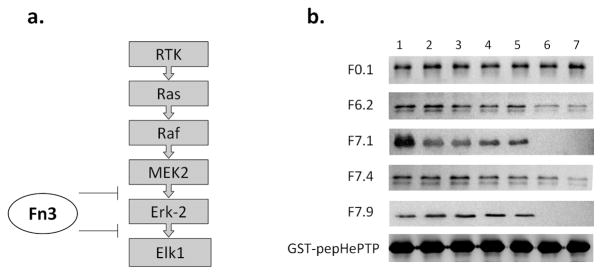

a. The components of mammalian ERK pathway. The binding of the MEK2 D-peptide mediates Erk-2 phosphorylation, and the binding of the Elk1 D-peptide mediates Elk1 phosphorylation. The engineered monobodies should disrupt both interactions.
b. GST-pepHePTP was used to pull down Erk-2 in the presence of Fn3 competitors. The precipitated Erk-2 was visualized by Western blot using M2. F0.1 is a control monobody that binds MBP (22). Competitor concentrations in Lanes 1 – 7: 0, 0.016, 0.08, 0.4, 2, 10, and 50 μM.
c. In vitro phosphorylation of Elk1(307-428) by Erk-2 in the presence of Fn3 competitors was analyzed by Western blot. Competitor concentrations: (For F0.1 and F6.2) 0, 0.0064, 0.032, 0.16, 0.8, 4, and 20 μM. (For F7.1, F7.4, and F7.9) 0, 0.0013, 0.0064, 0.032, 0.16, 0.8, 4, and 20 μM. The phospho-Elk1 band was quantified using ImageJ and normalized to Lane 1. Only the intensities corresponding to three highest Fn3 concentrations and Lane 1 are plotted for clarity.
d. In vitro phosphorylation of Erk-2 by MEK2 in the presence of Fn3 competitors. Competitor concentrations: 0, 0.0064, 0.032, 0.16, 0.8, 4 and 20 μM. The intensity of the phospho-Erk-2 bands was quantified and normalized to Lane 1. Only select intensities are plotted for clarity.
To test for inhibition of the kinase activity, we used activated Erk-2 to phosphorylate Elk1(307-428) in the presence of a monobody (23). Elk1 phosphorylation decreased in the presence of F6.2, F7.1, F7.4 or F7.9 but not in the presence of F0.1 (Fig. 3c). Similarly, the monobodies inhibited MEK2-dependent phosphorylation of Erk-2 (Fig. 3d), although higher monobody concentrations were needed to achieve similar inhibition. This may be because full length MEK2 contains two potential D-peptide sequences and also contacts Erk-2 within the N-terminal lobe (24).
Suppression of Erk-2 signaling in mammalian cells
To test the activity of designed monobodies in cells, we transiently expressed them in HEK293 using a dual expression vector (pCMVFn3-IEGFP) containing monobody and EGFP genes. Following addition of epidermal growth factor (EGF) to stimulate the Erk-2 pathway, phosphorylation of Erk-2 was quantified by Western blot. The monobodies did not significantly reduce the level of Erk-2 phosphorylation in stimulated HEK293 cells (Fig. 4a). There are several possible explanations for this, including the low level of Fn3 expression and the involvement of a scaffold protein that makes it difficult to disrupt the MEK2-Erk-2 interaction inside the cell (25). Also, only a subset of the cells are transfected and express monobodies, whereas the Western blot examines the entire cell population. Together, transient transfection may not have the sensitivity to detect Fn3-mediated inhibition of Erk-2 phosphorylation inside the cell.
Figure 4. Activity of engineered Fn3 in HEK293 cells.
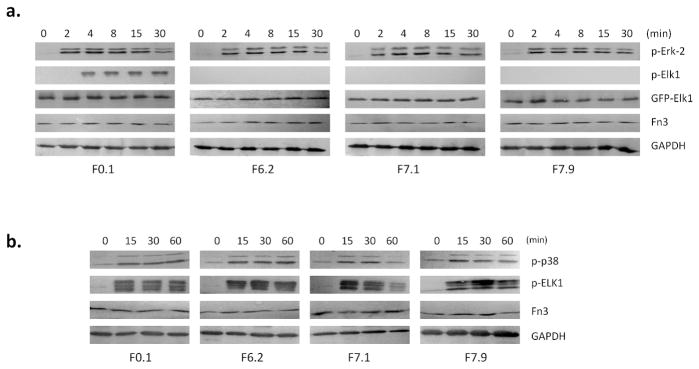
a. HEK293 cells were transiently transfected with pCMVFn3-IEGFP-Elk1(307-428) and stimulated with EGF to activate the ERK-2 pathway. in the resulting Elk1 phosphorylation was inhibited by F6.2, F7.1, and F7.9. The stable expression of EGFP-Elk1 was checked by fluorescence microscopy and by Western blot with anti-GFP antibody (Fig. S7). Fn3 expression was checked using 9E10 against the cMyc tag. The number of cells used in the analysis was normalized using GAPDH.
b. HEK293 cells expressing various Fn3 clones were stimulated with 100 mM NaCl to activate the p38 pathway. The monobodies had little or no effect on the p38-dependent phosphorylation of Elk1.
We examined if Elk1 phosphorylation is affected by expressed monobodies in stimulated HEK293 cells. Because endogenous Elk1 cannot be detected due to its low protein abundance (26), we fused Elk1(307-428) to the C-terminus of EGFP, thus placing both Fn3 and Elk1 on the same expression vector, pCMVFn3-IEGFP-Elk1(307-428). The co-expression of Fn3 and EGFP-Elk1(307-408) in the same cell facilitates the detection of Fn3 activity by ensuring that all cells that express Elk1(307-408) also express a monobody. There was a marked increase in phospho-Elk1 following EGF stimulation in control cells, but the phospho-Elk1 band was missing in the cells that co-express F6.2, F7.1 or F7.9 (Fig. 4a). The lack of phospho-Elk1 is not due to protein degradation, because the EGFP-Elk1(307-428) band of correct molecular weight can be seen with anti-GFP antibody (Fig. S7).
Alternatively, we treated the transfected cells with 100 mM NaCl to stimulate the p38 pathway and induce p38-dependent Elk1 phosphorylation (27). Elk1 phosphorylation was unaffected by the monobodies under this stimulation condition, showing that they do not interfere with the p38 pathway (Fig. 4b). Similarly, stimulating the cells with antibiotic anisomycin leads to p38-dependent phosphorylation of ATF2 (28), which remains unchanged by the expressed monobodies (Fig. S8). Therefore, the monobodies can selectively inhibit the Erk-2 pathway while leaving the p38 pathway unaffected.
Monobodies are active in yeast
Three MAPKs in S. cerevisiae, Fus3, Kss1, and Hog1, control the pheromone, filamentous growth, and hyperosmolarity response, respectively (29). Fus3 and Kss1 are Erk-2 orthologs, whereas Hog1 is orthologous to mammalian p38 (30). Biochemical and structural studies have shown that yeast MAPKs also use the binding of D-peptides to control the specificity and efficiency of phosphorylation (31, 32). These enzymes all have 45 – 60% sequence similarity to Erk-2 but show several key differences in the docking domain (Fig. S9). For example, 122-DH-123 of Erk-2 are conserved in Fus3 and Kss1 but mutated to QF in Hog1. Since the monobodies were screened against Erk-2(NHN) binding, they should similarly exhibit selectivity against Hog1. We transformed yeast with monobody expression vectors and quantified the activity of each MAPK pathway using three β-galactosidase reporter constructs containing pathway-specific promoters (33).
First, we stimulated the cells with the mating factor α to induce the pheromone response and measured the activity of the Fus3 pathway in the presence of different monobodies. The Fus3 pathway was strongly inhibited in the cells that express F6.2, F7.1, or F7.9, but not in the cells that express F7.4 (Fig. 5a). F7.8 weakly inhibited Fus3. Next, we deprived the cells of glucose and grew them in a galactose-containing medium to induce the filamentous growth response. The activity of the Kss1 pathway was lower in the cells expressing F6.2, F7.8 or F7.9 (Fig. 5b). The inhibition by F7.1 and F7.4 was more subdued, although the measured effect was statistically significant (e.g. p = 0.02 for F7.1 compared to F0.1 and no Fn3). Finally, we subjected the cells to a buffer containing 1M KCl to induce the Hog1 pathway that regulates the hyperosmolarity response. The activity of the Hog1 pathway was largely unaffected by the monobodies, although F7.8 reduced the activity of the pathway by roughly 30% (p < 0.01) (Fig. 5c). Incidentally, F7.8 is the only clone that showed any significant binding to p38α, which may explain its activity against Hog1. Taken together, the monobodies can be functionally expressed in yeast to inhibit various yeast MAPK pathways with high selectivity.
Figure 5. Specificity of monobody activity in yeast.

Yeast were transformed with reporter vectors to measure the activity of three yeast MAPK pathways that control a. mating, b. filamentous growth, and c. hyperosmolarity responses. The yeast cells were further transformed with an Fn3 expression vector (F0.1, F6.2, F7.1, F7.4, F7.8, and F7.9) or not (“No Fn3”). The cells were stimulated to activate each of the three pathways, and the resulting reporter activity (in arbitrary unit) was measured.
Monobodies are active in C. elegans
The Erk-2 pathway plays a prominent role in various forms of cancer, and upstream kinases, Raf and MEK, are both known targets of cancer therapy (34). In contrast, the possibility of modulating Erk-2 activity to alter cellular differentiation and developmental processes is less well established. In C. elegans, the Ras-MAPK pathway controls various physiological processes ranging from vulval differentiation to olfaction (35). The role of C. elegans Erk-1/2 ortholog MPK-1 (Ce-MPK-1) in vulval induction has been demonstrated (36, 37). During vulval induction, a gonadal cell releases a diffusible, EGF-like signal that stimulates Ce-MPK-1 signaling in six nearby vulval precursor cells (VPCs). The VPCs most proximal to the EGF-releasing cell are induced to develop into the vulva, while the distal cells remain uninduced. However, constitutive mutations in the Ras ortholog LET-60 bypass the EGF-signal requirement and several VPCs, including the distal VPCs, develop into pseudovulvae, or vulva-like projections (Fig. 6a) (38). Inactivating mutations downstream of LET-60, including Ce-MPK-1, suppress the constitutive let-60 gain of function (gof) multivulva (Muv) phenotype.
Figure 6. Monobodies inhibit MAP kinase function in C. elegans.

a. Nomarski images of wild-type (N2) and Ce-Ras let-60(gof) C. elegans. Whereas wt animals consistently display a single, properly positioned and functional vulva (black arrow), let-60(gof) animals are multivulval and display a range of additional ectopic pseudovulvae (white arrows).
b. Distribution of Muv phenotype among different strains and transgenic worms. The percentage of each population displaying a given number of vulvae is shown in pie charts. let-60(gof);mpk-1(lof) are significantly non-Muv compared to let-60(gof) worms (p = 2.5 × 10−72 based on χ2 statistics), indicating the let-60(gof) Muv phenotype is mediated primarily through downstream Ce-MPK-1 activity. Similarly, expression of F6.2 and F7.9 in let-60(gof) animals using the VPC-specific lin-31 promoter significantly reduced the prevalence of ectopic pseudovulvae compared to either let-60(gof) animals or the transgenic worms expressing the control monobody, F0.1 (p = 1.6 × 10−47 − 8.5 × 10−22). The combined data of two independent lines is shown. The total number of animals analyzed is n > 150 for control strains and n > 750 for each set of transgenic let-60(gof) worms.
Homology modeling shows that the docking site residues of Erk-2 are conserved in Ce-MPK-1. Therefore, the designed Fn3 inhibitors should similarly bind Ce-MPK-1 and inhibit its function in the animal. Because Ce-MPK-1 null mutation results in sterility and early larval lethality, adult animals with knockout Ce-MPK-1 cannot be stably constructed (39). A pleiotropy of mutant phenotypes associated with loss of function (lof) is common for critical enzymes within a multicellular organism. As such, development of engineered protein inhibitors that can be utilized in a cell- or tissue-specific manner to suppress the activity of an essential enzyme would provide an alternative approach to dissect the role of a given protein while avoiding the pleiotropy associated with lof genetic alleles. Using the let-60(gof) Muv phenotype as the readout, we tested whether Fn3 expression and corresponding Ce-MPK-1 inhibition could suppress let-60(gof) multivulva development. We cloned F0.1, F6.2, and F7.9 downstream of a VPC-specific promoter (lin-31p) and generated transgenic worms expressing engineered monobodies in the VPCs of let-60(gof) animals. The proportion of worms displaying ectopic vulvae was significantly reduced across populations of let-60(gof) worms expressing either F6.2 or F7.9 compared to let-60(gof) animals or let-60(gof) animals expressing F0.1 (Fig. 6b, S10).
That the inhibitors do not completely suppress the Muv phenotype is likely caused by multiple factors, including developmental timing and transgene expression. For example, the lin-31 promoter may not initiate Fn3 expression early enough to produce levels sufficient to completely block Ce-MPK-1 activity. Possible mosaic expression of the Fn3 clones among the VPCs can also result in partial inhibition of Ce-MPK-1 function. The incomplete suppression of the Muv phenotype suggests the challenges inherent to transgene expression and molecular manipulation in multicellular organisms using exogenously expressed protein binders. Nonetheless, our study clearly demonstrates that a specific MAPK inhibitor can be used in a whole organism to induce predictable phenotypic changes. Engineered Fn3-based protein inhibitors may be similarly useful for characterizing other physiologically relevant enzymes that were previously inaccessible through whole animal mutant studies. Finally, this study adds to a burgeoning role for C. elegans in the in vivo validation of biochemically described protein interactions as well as the application of engineered molecular tools (40, 41). Combinatorial use of such targeted approaches will therefore provide valuable insight into the mechanisms governing cellular behavior.
Use of protein inhibitors in basic research
The roles of other MAPKs in basic cellular processes continue to be important topics of investigation (42). The use of engineered monobodies for studying cell signaling in vivo is supported by their ability to inhibit the activity of Erk-2 orthologs in model organisms. We have shown that binders against a predetermined epitope can be engineered to induce selective inhibition of an intracellular kinase and provided evidence that targeting protein-protein interaction is a viable strategy for inhibiting MAPK signaling in vivo. Since the design strategy for epitope-guided engineering is general, a similar approach should be useful for isolating functional binders against other biologically relevant enzymes in the future.
Experimental procedures
Fn3 binder selection
The yeast-displayed Fn3 library was a gift from D. Wittrup (MIT). The construction and characterization of the library (G4) was previously described (15). The yeast library was grown in synthetic drop-out dextrose medium lacking tryptophan (SD/−Trp) and induced at 30 °C for 18 hrs by switching to a galactose-based medium. The induced cells (2 × 109) were labeled with biotinylated Erk-2 (b-Erk-2) and bound to streptavidin-coated magnetic dynabeads (Invitrogen). Bound cells were separated from unbound cells using a LS column filled with ferromagnetic particles (Miltenyi Biotec). The collected cells were grown in citrate based SD/−Trp medium (pH 4.5). For FACS sorting, the cells were labeled with b-Erk-2 (or Erk-2) and 9E10, and then with streptavidin-phycoerythrin (or M2 and anti-mouse-FITC). The double labeled cells were sorted at Roswell Park Cancer Institute (RPCI). The concentration of biotinylated Erk-2 was gradually decreased from 500 nM to 10 nM. A negative sort was performed using 100 nM Erk-2(NHN). 1 μM of biotinylated Erk-2, GST-p38α, or JNK1α1 was used to test for target specificity. The affinity was computed based on the mean fluorescence intensity, MFI (43).
In vitro assays
The D-peptide from hematopoietic protein tyrosine phosphatase (pepHePTP, RLQERRGSNVALMLDV, Ref. (14)) was fused to GST and purified from BL21(DE3) pLysS. The resin bound GST-pepHePTP was mixed with 5 μg Erk-2 and 16 nM – 50 μM Fn3 monobodies. The resin was washed and analyzed by SDS-PAGE and Western blot with M2. For the kinase inhibition assay, 0.1 unit of active GST-MEK2 (New England Biolabs, NEB) was mixed with 1 μM of Erk-2 in 20 μl of kinase assay buffer (50 mM Tris, pH 7.5, 10 mM MgCl2, 1 mM EGTA, and 2 mM DTT, 20 μM ATP) in the presence of Fn3 monobodies. After 20 min of incubation at 30°C, the reaction was stopped and phospho-Erk-2 was analyzed by Western blot. In vitro Elk1 phosphorylation was conducted with 10 units of active Erk-2 (NEB) and 820 ng of GST-Elk1(307-428) (Cell Signaling Technology).
Activity in HEK293
The Fn3 genes were cloned into pIRES2-EGFP to construct pCMVFn3-IEGFP. Elk1(307-428) was cloned from pCMV5FELK (Andrew Sharrocks) and fused to the C-terminus of EGFP. The resulting pCMVFn3-IEGFP-Elk1(307-408) was transfected into human embryonic kidney (HEK) 293 cells at 50 – 70% confluence. The transfection efficiency based on EGFP fluorescence 36 hr after transfection reached 70 – 80%. The cells were starved in the culture media without serum for 9 hr and stimulated with 10 nM of epidermal growth factor (EGF) for 0 – 30 min to stimulate the Erk-2 pathway. Alternatively, the cells were treated with 100 mM NaCl or 10 μg/ml of anisomycin to stimulate the p38 pathway. The cells were lysed in 200 μl of lysis buffer and 10 μl of clarified lysate was analyzed by Western blot.
Activity of Fn3 in yeast
The monobody activity was measured in yeast by transforming BJ5465 with a reporter plasmid—pFUS-LacZ (for Fus3), pFRE-LacZ (for Kss1), or pMSB2-LacZ (for Hog1) (P. Cullen)—and an Fn3 expression plasmid. The mating response was induced with 10 μM α-factor for 2 hr. Filamentous growth was induced by growing the cells in 2% galactose media for 6 hr. Hyperosmolarity response was induced with 1 M KCl in YPD for 4 hr. The β-galactosidase reporter activity was measured using 2-nitrophenyl β-D-galactopyranoside.
Activity of Fn3 in C. elegans
Fn3 function in worms was measured with the genetic strain MT2124 that carries the let-60(n1046) gof mutation in LET-60. SD366 carries the lof mpk-1(n2521) allele in addition to let-60(n1046). All Fn3 constructs were cloned downstream of the vulval cell-specific promoter lin-31p (4.3 kb of lin-31 5′ regulatory sequence). Transgenic worms were generated by germline transformation (44). All Fn3 constructs were injected into MT2124 at 500 ng/μl. Each construct was co-injected with 10 ng/μl pPD48.33 (myo-2p::gfp). The number of vulvae was quantified on two separate days at 8x magnification on a Zeiss dissection microscope with two independent transgenic lines per construct. Representative images of wt and let-60(n1046) worms were obtained using a Zeiss Axio Imager Z1 microscope (using a 40x Plan-APO oil objective and DIC optics), high resolution AxioCam MRm digital camera and Zeiss AxioVision software.
Supplementary Material
Acknowledgments
We would like to thank D. Wittrup, E. Goldsmith, J. Ashwell, A. Sharrocks, S. Andreadis, and P. Cullen for sharing materials with us. The work was supported by NSF CAREER (1053608) to SP, a NSF grant (MCB-0917896) to DMF, and by a contract (C024355) from the New York State Stem Cell Science Trust (NYSTEM) and an NIH grant (R01HL103709) to EST. The use of the core facilities at the Department of Pharmaceutical Sciences, UB is gratefully acknowledged.
Footnotes
Additional experimental details are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rockberg J, Lofblom J, Hjelm B, Uhlen M, Stahl S. Epitope mapping of antibodies using bacterial surface display. Nat Methods. 2008;5:1039–1045. doi: 10.1038/nmeth.1272. [DOI] [PubMed] [Google Scholar]
- 2.Boersma YL, Chao G, Steiner D, Wittrup KD, Pluckthun A. Bispecific Designed Ankyrin Repeat Proteins (DARPins) Targeting Epidermal Growth Factor Receptor Inhibit A431 Cell Proliferation and Receptor Recycling. J Biol Chem. 2011;286:41273–41285. doi: 10.1074/jbc.M111.293266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Georgiou G, Stathopoulos C, Daugherty PS, Nayak AR, Iverson BL, Curtiss R. Display of heterologous proteins on the surface of microorganisms: From the screening of combinatorial libraries to live recombinant vaccines. Nat Biotechnol. 1997;15:29–34. doi: 10.1038/nbt0197-29. [DOI] [PubMed] [Google Scholar]
- 4.Pepper LR, Cho YK, Boder ET, Shusta EV. A decade of yeast surface display technology: where are we now? Comb Chem High Throughput Screen. 2008;11:127–134. doi: 10.2174/138620708783744516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel. 2011;24:3–9. doi: 10.1093/protein/gzq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanoue T, Adachi M, Moriguchi T, Nishida E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol. 2000;2:110–116. doi: 10.1038/35000065. [DOI] [PubMed] [Google Scholar]
- 7.Zhou B, Wu L, Shen K, Zhang J, Lawrence DS, Zhang ZY. Multiple regions of MAP kinase phosphatase 3 are involved in its recognition and activation by ERK2. J Biol Chem. 2001;276:6506–6515. doi: 10.1074/jbc.M009753200. [DOI] [PubMed] [Google Scholar]
- 8.Bardwell AJ, Abdollahi M, Bardwell L. Docking sites on mitogen-activated protein kinase (MAPK) kinases, MAPK phosphatases and the Elk-1 transcription factor compete for MAPK binding and are crucial for enzymic activity. Biochem J. 2003;370:1077–1085. doi: 10.1042/BJ20021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell. 2004;14:43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- 10.Bardwell AJ, Frankson E, Bardwell L. Selectivity of docking sites in MAPK kinases. J Biol Chem. 2009;284:13165–13173. doi: 10.1074/jbc.M900080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen F, Mackerell AD, Jr, Luo Y, Shapiro P. Using Caenorhabditis elegans as a model organism for evaluating extracellular signal-regulated kinase docking domain inhibitors. J Cell Commun Signal. 2008;2:81–92. doi: 10.1007/s12079-008-0034-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hancock CN, Macias A, Lee EK, Yu SY, Mackerell AD, Jr, Shapiro P. Identification of novel extracellular signal-regulated kinase docking domain inhibitors. J Med Chem. 2005;48:4586–4595. doi: 10.1021/jm0501174. [DOI] [PubMed] [Google Scholar]
- 13.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 14.Zhou T, Sun L, Humphreys J, Goldsmith EJ. Docking interactions induce exposure of activation loop in the MAP kinase ERK2. Structure. 2006;14:1011–1019. doi: 10.1016/j.str.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 15.Hackel BJ, Ackerman ME, Howland SW, Wittrup KD. Stability and CDR composition biases enrich binder functionality landscapes. J Mol Biol. 2010;401:84–96. doi: 10.1016/j.jmb.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kummer L, Parizek P, Rube P, Millgramm B, Prinz A, Mittl PR, Kaufholz M, Zimmermann B, Herberg FW, Pluckthun A. Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries. Proc Natl Acad Sci U S A. 2012;109:E2248–2257. doi: 10.1073/pnas.1205399109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wojcik J, Hantschel O, Grebien F, Kaupe I, Bennett KL, Barkinge J, Jones RB, Koide A, Superti-Furga G, Koide S. A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat Struct Mol Biol. 2010;17:519–527. doi: 10.1038/nsmb.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbreth RN, Truong K, Madu I, Koide A, Wojcik JB, Li NS, Piccirilli JA, Chen Y, Koide S. Isoform-specific monobody inhibitors of small ubiquitin-related modifiers engineered using structure-guided library design. Proc Natl Acad Sci U S A. 2011;108:7751–7756. doi: 10.1073/pnas.1102294108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 1999;13:163–175. [PMC free article] [PubMed] [Google Scholar]
- 20.Colby DW, Kellogg BA, Graff CP, Yeung YA, Swers JS, Wittrup KD. Engineering antibody affinity by yeast surface display. Methods Enzymol. 2004;388:348–358. doi: 10.1016/S0076-6879(04)88027-3. [DOI] [PubMed] [Google Scholar]
- 21.Shifman JM, Mayo SL. Exploring the origins of binding specificity through the computational redesign of calmodulin. Proc Natl Acad Sci U S A. 2003;100:13274–13279. doi: 10.1073/pnas.2234277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilbreth RN, Esaki K, Koide A, Sidhu SS, Koide S. A dominant conformational role for amino acid diversity in minimalist protein-protein interfaces. J Mol Biol. 2008;381:407–418. doi: 10.1016/j.jmb.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cruzalegui FH, Cano E, Treisman R. ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene. 1999;18:7948–7957. doi: 10.1038/sj.onc.1203362. [DOI] [PubMed] [Google Scholar]
- 24.Robinson FL, Whitehurst AW, Raman M, Cobb MH. Identification of novel point mutations in ERK2 that selectively disrupt binding to MEK1. J Biol Chem. 2002;277:14844–14852. doi: 10.1074/jbc.M107776200. [DOI] [PubMed] [Google Scholar]
- 25.Sacks DB. The role of scaffold proteins in MEK/ERK signalling. Biochem Soc Trans. 2006;34:833–836. doi: 10.1042/BST0340833. [DOI] [PubMed] [Google Scholar]
- 26.Yang SH, Whitmarsh AJ, Davis RJ, Sharrocks AD. Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. Embo J. 1998;17:1740–1749. doi: 10.1093/emboj/17.6.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferreiro I, Joaquin M, Islam A, Gomez-Lopez G, Barragan M, Lombardia L, Dominguez O, Pisano DG, Lopez-Bigas N, Nebreda AR, Posas F. Whole genome analysis of p38 SAPK-mediated gene expression upon stress. BMC Genomics. 2010;11:144. doi: 10.1186/1471-2164-11-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breitwieser W, Lyons S, Flenniken AM, Ashton G, Bruder G, Willington M, Lacaud G, Kouskoff V, Jones N. Feedback regulation of p38 activity via ATF2 is essential for survival of embryonic liver cells. Genes Dev. 2007;21:2069–2082. doi: 10.1101/gad.430207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gustin MC, Albertyn J, Alexander M, Davenport K. MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1998;62:1264–1300. doi: 10.1128/mmbr.62.4.1264-1300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caffrey DR, O’Neill LA, Shields DC. The evolution of the MAP kinase pathways: coduplication of interacting proteins leads to new signaling cascades. J Mol Evol. 1999;49:567–582. doi: 10.1007/pl00006578. [DOI] [PubMed] [Google Scholar]
- 31.Bardwell L, Cook JG, Chang EC, Cairns BR, Thorner J. Signaling in the yeast pheromone response pathway: specific and high-affinity interaction of the mitogen-activated protein (MAP) kinases Kss1 and Fus3 with the upstream MAP kinase kinase Ste7. Mol Cell Biol. 1996;16:3637–3650. doi: 10.1128/mcb.16.7.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Remenyi A, Good MC, Bhattacharyya RP, Lim WA. The role of docking interactions in mediating signaling input, output, and discrimination in the yeast MAPK network. Mol Cell. 2005;20:951–962. doi: 10.1016/j.molcel.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 33.Pitoniak A, Birkaya B, Dionne HM, Vadaie N, Cullen PJ. The signaling mucins Msb2 and Hkr1 differentially regulate the filamentation mitogen-activated protein kinase pathway and contribute to a multimodal response. Mol Biol Cell. 2009;20:3101–3114. doi: 10.1091/mbc.E08-07-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meier F, Schittek B, Busch S, Garbe C, Smalley K, Satyamoorthy K, Li G, Herlyn M. The RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the effective treatment of advanced melanoma. Front Biosci. 2005;10:2986–3001. doi: 10.2741/1755. [DOI] [PubMed] [Google Scholar]
- 35.Sternberg PW, editor. Vulval development. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han M, Golden A, Han Y, Sternberg PW. C. elegans lin-45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature. 1993;363:133–140. doi: 10.1038/363133a0. [DOI] [PubMed] [Google Scholar]
- 37.Lackner MR, Kornfeld K, Miller LM, Horvitz HR, Kim SK. A MAP kinase homolog, mpk-1, is involved in ras-mediated induction of vulval cell fates in Caenorhabditis elegans. Genes Dev. 1994;8:160–173. doi: 10.1101/gad.8.2.160. [DOI] [PubMed] [Google Scholar]
- 38.Ferguson EL, Horvitz HR. Identification and characterization of 22 genes that affect the vulval cell lineages of the nematode Caenorhabditis elegans. Genetics. 1985;110:17–72. doi: 10.1093/genetics/110.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lackner MR, Kim SK. Genetic analysis of the Caenorhabditis elegans MAP kinase gene mpk-1. Genetics. 1998;150:103–117. doi: 10.1093/genetics/150.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leifer AM, Fang-Yen C, Gershow M, Alkema MJ, Samuel AD. Optogenetic manipulation of neural activity in freely moving Caenorhabditis elegans. Nat Methods. 2011;8:147–152. doi: 10.1038/nmeth.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wood JF, Wang J, Benovic JL, Ferkey DM. Structural domains required for Caenorhabditis elegans G protein-coupled receptor kinase 2 (GRK-2) function in vivo. J Biol Chem. 2012;287:12634–12644. doi: 10.1074/jbc.M111.336818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drew BA, Burow ME, Beckman BS. MEK5/ERK5 pathway: the first fifteen years. Biochim Biophys Acta. 2012;1825:37–48. doi: 10.1016/j.bbcan.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papo N, Silverman AP, Lahti JL, Cochran JR. Antagonistic VEGF variants engineered to simultaneously bind to and inhibit VEGFR2 and alphavbeta3 integrin. Proc Natl Acad Sci U S A. 2011;108:14067–14072. doi: 10.1073/pnas.1016635108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.