

Abstract
Methane-producing Archaea are of interest due to their contribution to atmospheric change and for their roles in technological applications including waste treatment and biofuel production. Although restricted to anaerobic environments, methanogens are found in a wide variety of habitats, where they commonly live in syntrophic relationships with bacterial partners. Owing to tight thermodynamic constraints of methanogenesis alone or in syntrophic metabolism, methanogens must carefully regulate their catabolic pathways including the regulation of RNA transcripts. The transcriptome is a dynamic and important control point in microbial systems. This paper assesses the impact of mRNA (transcriptome) studies on the understanding of methanogenesis with special consideration given to how methanogenesis is regulated to cope with nutrient limitation, environmental variability, and interactions with syntrophic partners. In comparison with traditional microarray-based transcriptome analyses, next-generation high-throughput RNA sequencing is greatly advantageous in assessing transcription start sites, the extent of 5′ untranslated regions, operonic structure, and the presence of small RNAs. We are still in the early stages of understanding RNA regulation but it is already clear that determinants beyond transcript abundance are highly relevant to the lifestyles of methanogens, requiring further study.
1. Introduction
Methane- (CH4-) producing Archaea occupy an important position in the global carbon cycle and in atmospheric change by performing the final steps of biomass degradation in anaerobic systems, and releasing significant amounts of CH4 to the atmosphere every year [1]. Also, methanogenic Archaea are of interest due to their role in anaerobic degradation including waste treatment, biogenic gas production from coal, and other substrates that have potential for CH4 to be harvested for use as a fuel. Therefore, considerable environmental and economic benefits may come from understanding biological CH4 production.
In terms of physiology, three major, partially overlapping, methanogenesis pathways are recognized: (i) methanogenesis from carbon dioxide (CO2) reduction with hydrogen (H2) (hydrogenotrophic pathway), (ii) methanogenesis from methylated compounds such as methanol and methylated amines (methylotrophic pathway), and (iii) methanogenesis from acetate cleavage (aceticlastic pathway). The biochemistry of methanogenesis was reviewed elsewhere [2–4] and is summarized in Figure 1. The only known biological producers of CH4 are a diverse range of anaerobic Archaea within the Euryarchaeota phylum including the following orders: Methanobacteriales, Methanococcales, Methanocellales, Methanosarcinales, Methanomicrobiales, and Methanopyrales [3, 5] and the recently proposed Methanoplasmatales [6]. Members of the Methanosarcinales order have the widest substrate range where all three major methanogenic pathways are represented, while the other orders generally perform methanogenesis only via CO2 reduction [3, 4].
Figure 1.
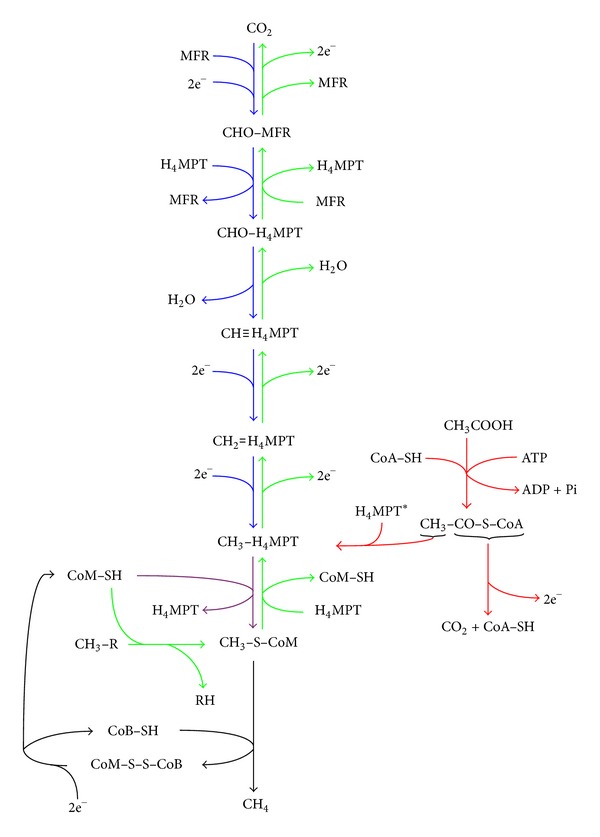
Overview of the three major known methanogenic pathways in Archaea. Color coding indicates the steps common to all three types (black), unique to the methylotrophic pathway (green), unique to the hydrogenotrophic (or CO2 reducing) pathway (blue), unique to the aceticlastic pathway (red), and shared between hydrogenotrophic and aceticlastic methanogenesis (purple). 2e− represents reducing equivalents, produced or consumed during each reaction. MFR: methanofuran; H4MPT: tetrahydromethanopterin; CoM-SH: coenzyme M; CoB-SH: coenzyme B; CoA-SH: coenzyme A; CoM-S-S-CoB: heterodisulfide of coenzyme M and coenzyme B; ATP: adenosine triphosphate; R: ligand bound to methylated compound that serves as substrate for methylotrophic methanogenesis. *Tetrahydrosarcinapterin is a functional analogue of H4MPT found in the Methanosarcinales order of methanogens.
Because of a lower bioenergetic yield compared to other reactions used by microbial groups, methanogens generally thrive in environments or conditions lacking terminal electron acceptors other than CO2 [7]. Methanogens are found in a diverse range of habitats including wetlands, rice paddies, fresh and marine water sediments, digestive tracts of ruminants and termites, anaerobic waste digesters, and geothermal vents. These habitats feature a broad range of environmental conditions with strong variation of temperature, salinity, nutrient availability, and pH. Methanogens generally grow syntrophically with fermentative bacteria which produce methanogenic substrates. Careful regulation of metabolism on the part of both syntrophic partners is required owing to the tight thermodynamic constraints of conversion of biomass to CH4.
In order to understand the activity of methanogenic Archaea and their contribution in different ecosystems, it is important to address how methanogenesis is regulated at the genetic and cellular systems levels and how these regulations impact upon the adaptation of methanogens to their environments and their interactions with their syntrophic partners. Gene expression and regulation in methanogenic Archaea has not been completely characterized and is thus an important missing link in the understanding of these organisms.
RNA plays a central role in gene expression as an intermediate between genes and proteins, as an adapter during translation, and as a component in ribosomes. Transcriptomic approaches aim to characterize the RNA content of a sample of interest, facilitating estimations of gene expression levels and identification of differentially expressed genes between different treatments. This paper aims to discuss the ways in which transcriptome analyses have broadened the understanding of the regulation of methanogenesis and the ecology of methanogens. To this end, the effects of reducing power supply, the significance of isofunctional enzymes, and the use of alternate energy conserving pathways during methanogenesis will be major points of discussion. Transcriptome-based systems-level assessments of how methanogens adapt to stresses and limitations in their environments and niches are also reviewed. Furthermore, this paper will summarize the current body of knowledge about RNA regulation in methanogenesis and how emerging technologies are advancing research in this area.
2. Methanogenesis Is Fine Tuned to Meet Niche Requirements
2.1. Response of Methanogenic Pathways to Substrate Supply
Methanogenesis-related mRNAs are amongst the most highly abundant in methanogens. For example, it has been shown that 33 of the 100 most highly abundant mRNAs detected in Methanosarcina barkeri DSM 804 grown on methanol are related to methanogenesis [8]. However, for reasons of efficiency, methanogenesis-related mRNAs are not constitutively present at high levels, but rather their levels are carefully regulated in order to facilitate optimization of metabolism relative to growth conditions. A common strategy of gene regulation is transcriptional upregulation only in the presence of the substrate of the gene product. This is especially important in the metabolically diverse Methanosarcinales order. When methanol-grown Methanosarcina mazei or Methanosarcina acetivorans (both members of the Methanosarcinales) were compared with their acetate-grown counterparts, transcriptome analyses showed that the genes specific to the methylotrophic and aceticlastic methanogenic pathways were regulated in substrate-dependent manners [9, 10]. Transcript levels of genes encoding different isozymes of methanol-specific methanol transferase and corrinoid proteins are subject to transcriptional control by promoter activity and posttranscriptional control by means of differential transcript stabilities, as indicated by transcriptional fusions, translational fusions, and qRT-PCR [11]. Transcriptome sequencing revealed that a regulator, known as MreA, was responsible for regulating 280 genes, either directly or indirectly, when M. acetivorans was grown with acetate [12]. DNA-binding experiments reported with this transcriptome data set indicated that MreA can directly upregulate transcription of acetate catabolism genes and directly downregulate methanol catabolism genes [12].
The pathways of methanogenesis from methanol and from methylamines are identical apart from the use of substrate-specific methyltransferases and corrinoid proteins involved in the initial transfer of the methyl group from the substrate to coenzyme M (CoM-SH) (Figure 1). The utilization of trimethylamine (TMA) as a methanogenic substrate is complicated further since it is degraded to two other methanogenic substrates, dimethylamine (DMA) and monomethylamine (MMA), during its conversion into CH4, CO2, and ammonia. Each of TMA, DMA, and MMA also requires substrate-specific methyltransferases and corrinoid proteins, each of which is encoded by two to three homologous genes in M. mazei. Microarray-based transcriptome analysis indicated that the mRNA levels of 72 genes were different in TMA-grown versus methanol-grown M. mazei [13]. None of the differences occurred in the core methanogenesis pathway or in energy conservation. The major substrate dependent differences occurred in the mRNA levels of the specific methyltransferases, with different homologues regulated to different extents. Monitoring of transcript levels of the methyltransferase and corrinoid genes via qRT-PCR in conjunction with chemical analysis of the culture medium indicated that M. mazei features a gene expression program to firstly utilize TMA followed by DMA and finally MMA, as would be expected. Although it is expected that genes involved in TMA degradation would be upregulated in the presence of TMA, it is vital, nonetheless, to understand the dynamics of primary metabolism and these results exemplified the validity of this approach. One of the strengths of global transcriptome analyses (and other global analyses) is the identification of non-intuitive cellular responses. For example, TMA grown M. mazei also evidenced higher transcript levels of genes involved in aromatic amino acid biosynthesis and ether lipid synthesis [13], the latter possibly being a response to combat the ability of TMA to depolarize the cell membrane.
Thermodynamically, hydrogenotrophic and methylotrophic methanogenesis are more favorable than aceticlastic methanogenesis. Consequently, acetate-grown Methanosarcina spp. grow slower than their methanol- or TMA-grown counterparts and utilize methylated compounds in preference to acetate [14]. The precise mechanisms of energy conservation operating in the various methanogenesis pathways are still a topic of active investigation. However key differences in energy conservation exist between the hydrogenotrophic, methylotrophic, and aceticlastic methanogenesis pathways. In the Methanosarcinales order, the use of a cytochrome-containing, membrane-bound HdrED type heterodisulfide reductase conserves some of the free energy of coenzyme M-coenzyme B heterodisulfide (CoM-S-S-CoB) reduction through the generation of a transmembrane proton gradient [2, 4]. On the other hand, hydrogenotrophic methanogens outside of the Methanosarcinales order utilize a soluble HdrABC type heterodisulfide reductase that couples the exergonic heterodisulfide reduction with the endergonic reduction of CO2 during the formyl-methanofuran (formyl-MFR) formation step [15].
The trafficking of electrons to the different Hdr complexes gives rise to key differences in energy conservation between the different methanogenic pathways [4]. Microarray-based transcriptome analyses of methanogens grown with either acetate or a methylotrophic substrate are shedding light on some of these differences [9, 10, 16]. The higher expression of Ech hydrogenase, an A1A0-type ATP synthase, and genes coding for flavoproteins and ferredoxins in acetate-grown cells, as opposed to methanol-grown cells, indicate that these components are part of a different pathway of electron trafficking and energy conservation during aceticlastic methanogenesis in M. mazei [9]. In comparison with the wild type strain, an ech mutant of M. mazei had a reduced growth rate and yield whilst utilizing TMA and was completely unable to grow using acetate as a sole methanogenic substrate [17]. The role of Ech hydrogenase is thought to be in accepting electrons from reduced ferredoxin, contributing to the transmembrane proton gradient and producing H2, the reducing power of which will eventually be used to reduce CoM-S-S-CoB [17, 18]. Higher transcript levels of the M. mazei HdrABC complex were reported in nitrogen-fixing conditions versus nitrogen- (ammonium) sufficient conditions in the simultaneous presence of methanol and acetate [19]. The biological significance of this phenomenon is unclear but it may be an adaptation to supply the energy and reducing equivalents necessary for nitrogen fixation.
In contrast to M. mazei, transcriptome analyses indicated that M. acetivorans possesses a different pathway that does not utilize an Ech hydrogenase, for electron flow and energy conservation associated with the reduction of CoM-S-S-CoB during aceticlastic methanogenesis [10, 16]. In this case, it was proposed that reduced ferredoxin donates its electrons to HdrED via the membrane-bound Rnf complex, with methanophenazine being a common electron trafficking intermediate. In this alternate pathway, energy is conserved through the generation of transmembrane ion gradients without using H2 as an intermediate. A freshwater Methanosarcina spp. also utilized H2 as an intermediate during aceticlastic methanogenesis, much like M. mazei and in contrast to the marine isolate M. acetivorans [18]. Avoiding the use of H2 as an intermediate during aceticlastic methanogenesis represents a competitive adaption to marine environments [18], where sulfate concentrations are relatively high (>20 mM), and competition for H2 by sulfate reducers is consequently higher than that in freshwater environments [20].
Although HdrED plays roles in both methylotrophic and aceticlastic methanogenesis, these two pathways employ very different energy conservation strategies. Transcript and mutant analyses indicate that the oxidation of reduced F420 by the membrane-bound F420 dehydrogenase (Fpo) plays a major role in electron trafficking in methylotrophic methanogenesis [16, 21]. Furthermore, the HdrABC complex plays a role in methylotrophic methanogenesis but not in aceticlastic methanogenesis [22]. Transcriptome analysis suggested that, in the absence of HdrABC, methanol-grown M. acetivorans is partially deficient in the ability to reduce CoM-S-S-CoB [22]. It has been suggested that electrons from formyl-MFR are donated to the HdrABC type heterodisulfide reductase, although this possibility remains untested [16].
In the CO2-reducing methanogenic pathway, some methanogenic steps are catalyzed by isofunctional enzymes where the same reduction step is coupled to the oxidation of a different electron donor. For example, the reduction of methenyl-tetrahydromethanopterin (methenyl-H4MPT) to methylene-H4MPT is catalyzed by both the F420-dependent methylene-H4MPT dehydrogenase (Mtd) and the H2-dependent methylene-H4MPT dehydrogenase (Hmd). These steps are often regulated in response to the supply of H2 or formate (Figure 2). For instance, in Methanothermobacter thermautotrophicus, microarray-based transcriptome analysis revealed that mRNA levels of F420-related targets (F420 reducing hydrogenase (frh), mtd, F420-dependent methylene-H4MPT reductase (mer), and methyl-CoM reductase (mcr)) were higher in H2-limited versus H2-sufficient conditions [23]. The upregulation of F420-related targets indicates that M. thermautotrophicus attempts to scavenge and use H2 more efficiently under conditions of H2 limitation where ferredoxin reduction is less thermodynamically favorable. Similarly, it was shown that genes related to F420 redox reactions in the hydrogenotrophic pathway of Methanococcus maripaludis were upregulated in response to H2 limitation [24]. Also, another study found that formate dehydrogenase (fdh) was upregulated during H2 limitation [25]. Interestingly, hmd showed higher levels of mRNA abundance under more rapid growth rate with H2 limitation, while growth rate did not regulate mRNA levels of mtd [25]. Hmd utilizes H2 directly with low affinity while Mtd uses reduced F420 as the electron donor (Figure 2). The preferential use of Mtd over Hmd at low H2 availability was also observed in a proteomics study, supporting the trends seen in the transcriptome analyses [26]. It has been suggested that Hmd and Mtd can act cyclically and produce H2 when, for example, reduced F420 is produced during formate oxidation [27]. This cyclical nature of Hmd and Mtd suggests that their regulation may be tied in with balancing the pools of oxidized and reduced F420 and to maintain a suitable H2 pool for ferredoxin reduction in order to optimize energy producing metabolism. However, in making such assertions from transcriptomics or proteomics data, it must be borne in mind that posttranscriptional levels of regulation of primary metabolism (e.g., allosterism) are very common, and biochemical studies are ultimately required to assess such hypotheses as mentioned previously.
Figure 2.
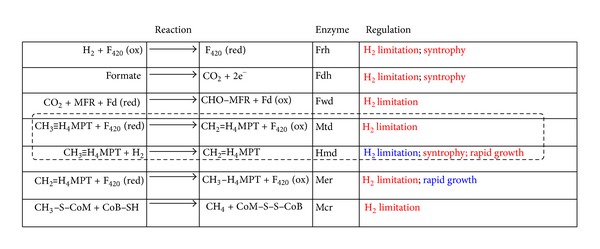
Observed regulatory patterns in hydrogenotrophic methanogenesis. Regulation steps for hydrogenotrophic methanogenesis are summarized from published sources discussed in this paper. Conditions, (syntrophic interaction or H2 limitation) causing upregulation and downregulation of enzyme transcript levels are indicated in red and blue, respectively. Abbreviations are as per Figure 1 with the addition of the following: F420: coenzyme F420; Fd: ferredoxin; Frh: F420-reducing hydrogenase; Fdh: formate dehydrogenase; Fwd: formyl-MFR dehydrogenase; Mtd: F420-dependent methylene-H4MPT dehydrogenase; Hmd: H2-dependent methylene-H4MPT dehydrogenase; Mer: F420-dependent methylene-H4MPT reductase; Mcr: methylcoenzyme M reductase. (The dotted lined box highlights two isofunctional enzymes oppositely regulated by H2 limitation).
Other methanogen optimizations in response to increased growth rate were the moderately increased mRNA levels of the genes for formyl-MFR:H4MPT methyltransferase and methenyl-H4MPT cyclohydrolase and moderately decreased mRNA levels of genes for heterodisulfide reductase [25]. It was shown, via a transcriptome sequencing approach in Methanobrevibacter smithii, that some components of the hydrogenotrophic pathway have strain-specific differences in mRNA levels [28]. These occurred for the mtd and mer genes and also for genes encoding components for an ABC-type cobalt transporter. Cobalt is an important part of corrinoid cofactors in methanogenesis-related enzymes, such as methyl-H4MPT-CoM methyltransferase (Mtr). These specific differences in methanogenesis pathways of M. smithii strains perhaps represent adaptations to various microhabitats within the heterogeneous human gastrointestinal tract, where M. smithii strains are the dominant Archaea [28].
Evidence for tight regulation of hydrogenotrophic methanogenesis was also demonstrated during syntrophic growth. Higher transcript levels of genes encoding H2-dependent methanogenesis-related targets were evident in M. maripaludis when grown in coculture with Desulfovibrio vulgaris than when the methanogen was grown alone under H2 limitation [29]. This indicates that M. maripaludis regulates its methanogenic pathway in order to facilitate syntrophy with D. vulgaris where the thermodynamics of both fermentative and methanogenic catabolisms must be carefully balanced. M. thermautotrophicus has two isofunctional Mcr enzymes, MRI and MRII. Northern blot analysis and proteomics show that both are expressed when M. thermautotrophicus is grown in pure culture in H2-sufficient conditions while only MRI was expressed during coculture with Syntrophothermus lipocalidus where H2 partial pressures were between 20 and 80 Pa [30].
2.2. Response of Methanogens to Environmental Perturbations
Transcriptome analyses of methanogens are useful not only for studying the process of methanogenesis but also for understanding the physiology and ecological adaptations of methanogens to their environments. Methanogens exhibit general adaptations to different growth rates that are brought about by various stresses such as nutrient limitation [9, 10, 19], heat stress, and oxidative stress [31] that generally involve downregulation of the translation apparatus in response to reduced cellular demands at low growth rates [24]. However, methanogens do not feature a universal stress response [23].
Nutrient limitation is very relevant when characterizing the ability of an organism to survive and adapt to changes in its environment. Transcriptomics indicated that M. mazei increased transcript levels of the core genes of nitrogen metabolism (nitrogenase, ammonium transporters, and glutamine synthetase) and several other genes including a cobalt transporter and genes with potential regulatory functions [19]. Similarly, a proteomics investigation revealed that M. maripaludis increased protein levels of nitrogenase, glutamine synthetase, ammonia transporters, and a nitrogen sensor/regulator in response to nitrogen limitation [26]. As mentioned previously, M. mazei showed higher levels of transcripts for HdrABC in nitrogen-fixing conditions, possibly related to increased demand for reducing equivalents for N2 reduction. Interestingly, a homologue of Hmd (HmdII) also increased in abundance in M. maripaludis in response to nitrogen-limiting conditions. Whether or not the alteration in HmdII plays any role in altering the dynamics of reducing equivalent pools remains unknown. For M. mazei, it was possible to predict a DNA motif involved in nitrogen responsiveness, and in the case of M. maripaludis, it was possible expand to upon the range of nitrogen responsive genes for a previously known motif. This is a good demonstration of the potential for omics technologies to generate hypotheses for further investigations, exemplified by investigations into DNA binding by the nitrogen-related transcriptional repressor, known as NrpR [32]. Complementary transcriptomics and proteomics studies identified three phosphate responsive phosphate transporters and a phosphate responsive putative phosphate transport regulator in M. maripaludis [24, 26]. Carbon monoxide dehydrogenase/acetyl coenzyme A synthase also appears to be downregulated during phosphate limitation [26].
Adaptation to temperature stress often involves alteration of cell surface components and the synthesis of molecular chaperones. In M. barkeri, heat shock resulted in the alteration of the levels of 168 transcripts [31]. Most notable was the increase in transcript abundance of Hsp70 and Hsp60. In M. thermautotrophicus, Hsp70 (but not Hsp60) was also responsive to heat stress and other stresses, such as oxidative stress and high pH [23]. The synthesis of chaperones was an adaptation to heat and cold stress by Methanococcoides burtonii [33, 34]. M. burtonii also showed increased levels of transcripts for RNA-binding proteins in response to cold stress, and it was suggested that this was an adaptive mechanism whereby RNA was maintained in a state suitable for translation [33, 34]. Transcriptome and proteome analyses of M. burtonii both indicated that remodeling of the cell surface was an important adaptation to heat and cold stress [33, 34]. Interestingly, 7 of the 10 most differentially abundant transcripts in response to cold stress for M. burtonii encoded genes of unknown function [33]. This suggests that M. burtonii also utilizes novel cold adaptive mechanisms.
Sodium stress is an important factor to consider particularly for methanogens inhabiting a wide range of sites including freshwater, marine, or transitional environments. In M. mazei, the most highly differentially expressed gene during sodium chloride stress was a hypothetical protein with no known homologues outside of the Methanosarcina genus [35]. The second most differentially expressed gene encoded a putative hypothetical protein. This indicates that some novel salt adaptive mechanisms may be present in M. mazei. However, conventional salt adaptive mechanisms were also evident in M. mazei with the upregulation of transcript levels related to solute transport and biosynthesis and Na+ export in response to high sodium chloride concentrations [35]. Interestingly, among nonmarine methanogens, their sensitivity to sodium levels is rather variable from highly to less sensitive [36]. The role of such a variable response has not been systematically evaluated but can play a role in the abundance of methanogenic groups in oligotrophic and minerotrophic environments as seen among different types of wetlands (e.g., [37]).
Methanogens grow under strictly anaerobic conditions and are highly sensitive to oxidative stress. Superoxide dismutase and catalase activities are typical components of oxidative stress responses. The activities of catalase and superoxide dismutase and the levels of their corresponding mRNAs were shown to be altered in M. barkeri in response to the oxidative stress inducing agents paraquat (N,N ′-dimethyl-4,4′-bipyridinium dichloride) and hydrogen peroxide [38]. However, air exposure did not result in the upregulation of transcript levels of superoxide dismutase, catalase, or other nonspecific peroxidases in M. barkeri [31]. Rather, exposure of M. barkeri to air resulted in widespread changes in gene expression including upregulation of transposases and the downregulation of genes related to translation functions, amino acid transporters, energy metabolism, and signal transduction [31]. The oxidative stress response (induced by exposure to air) of M. barkeri, Methanospirillum hungatei, and two syntrophic bacteria, D. vulgaris and Syntrophobacter fumaroxidans, was studied in a defined coculture via a multispecies microarray transcriptome analysis approach [39]. In coculture, M. barkeri responded in a similar fashion as it did in pure culture with the downregulation of energy production and methanogenesis. M. hungatei responded to oxidative stress by upregulating thioredoxin and a heat shock protein (Hsp20) indicating that it deals with reactive oxygen species directly and attempts to protect its proteins from the effects of oxidative damage. In M. thermautotrophicus, an operon encoding a superoxide dismutase gene and other antioxidant enzymes did not alter in transcript level after exposure to hydrogen peroxide [23]. However, M. thermautotrophicus responded to hydrogen peroxide stress much like M. barkeri responded to air exposure, with the alteration of large numbers of transcript levels for processes related to translation, energy metabolism, amino acid transporters and metabolism, and coenzyme transport and metabolism [23]. From the above, it is clear that oxidative stress responses are varied, rather than conserved, between different methanogens. Given the extremely low oxygen tolerances typical of methanogens, it is not surprising that the most conserved aspect of oxidative stress responses is the downregulation of growth and metabolism, indicating that methanogens attempt to merely survive brief exposures to oxidative conditions.
2.3. Understanding Ecological Interaction through Transcripts
The regulations of the methanogenic pathways of M. maripaludis [29] and M. thermautotrophicus [30] in pure culture versus coculture were discussed before (see “Section 2.1”). Here we discuss other facets of these syntrophic interactions. M. maripaludis responded to syntrophic growth with D. vulgaris by downregulating transcripts associated with biosynthetic functions such as CO2 fixation [29]. This may seem counterintuitive since acetate was provided as a carbon source, in place of lactate, during monoculture, and suggests that syntrophically grown M. maripaludis may have received an assimilable carbon source from D. vulgaris. Evidence of transfer of alanine from D. vulgaris to M. maripaludis and an increase in transcript abundance of M. maripaludis alanine dehydrogenase suggests that alanine may be used by M. maripaludis as a carbon and nitrogen source during coculture [29]. The fact that alanine dehydrogenase activity produces reducing equivalents suggests the occurrence of a novel interspecies electron transfer mechanism. Generally, electron transfer between methanogens and their syntrophic partners involves formate and H2 [40, 41]. Interestingly, two seemingly isofunctional enzymes, the energy conserving hydrogenases Eha and Ehb, seemed to have opposite regulation, with Eha being upregulated and Ehb being down-regulated during coculture of M. maripaludis [29]. However, it was previously shown that Ehb functions in anabolism, while Eha plays a role in energy-generating metabolism [42]. This suggests that methanogens must carefully regulate both their growth and their metabolism in order to optimize the thermodynamics associated with the metabolism of the interdependent partners in the syntrophic relationship. Proteomic analysis also indicated that biosynthetic functions of M. thermautotrophicus are down-regulated during coculture with S. lipocalidus where levels of proteins involved in carbon fixation, amino acid biosynthesis, and RNA/DNA metabolism were decreased [43]. The possibility of interspecies carbon transfer was not discussed, and it is suggested that growth of M. thermautotrophicus is restrained in coculture. Interestingly, the α-subunit of the proteasome of M. thermautotrophicus was N-acylated during coculture which suggests that modification of global protein turnover dynamics is an adaptation to coculture [43].
In another case of methanogen-bacteria syntrophy between Pelotomaculum thermopropionicum and M. thermautotrophicus, it was shown that physical contact was key for the syntrophic interaction and that this interaction was mediated by a flagellar cap protein, FliD, where the FliD of P. thermopropionicum bound to M. thermautotrophicus [44]. Transcriptome analysis indicated that in the presence of FliD, M. thermautotrophicus had higher transcript levels of genes associated with methanogenesis, ATP synthesis, and hydrogenases. This is evidence of pili-mediated signaling involved in the onset of syntrophic interactions. Cell surface components are important in survival and establishment of relationships with bacterial partners within the gastrointestinal environment. Different M. smithii strains exhibited strain-specific differences in the transcript levels of their repertoire of adhesin-like proteins [28]. A qRT-PCR approach also indicated that cell surface components such as glycans and adhesin-like proteins were important in host colonization and in the establishment of syntrophic relationships in M. smithii [45]. The several examples presented in this section show that the consequences of syntrophic interactions amongst methanogens and bacterial partners can be significant by directly or indirectly regulating the activity of methanogens, hence deserving further attention for future studies.
Other studies have aimed to investigate the ecological dynamics of methanogens by analyzing the transcriptome of environmental samples. It was found that the mcrA transcript/gene ratio correlated weakly (regression coefficient = 0.76) with the CH4 flux from a peat soil [46]. In another study, it was shown that mcrA transcript levels had a positive relationship with CH4 flux in a CH4-emitting peat soil site while the transcript levels of particulate CH4 monooxygenase (a key gene in CH4 oxidation) had a negative relationship with CH4 flux at a CH4-oxidizing site [47]. In the CH4-oxidizing site, mcrA transcript levels had no correlation with CH4 flux [47]. Monitoring the transcript levels of key genes involved in CH4 flux, although informative to a small extent, is not sufficient to adequately predict CH4 flux dynamics. Methanogens are complex biological systems that interact with a wide variety of other microorganisms within a complex food web. Community level high-throughput sequence analysis has the potential to characterize the genetic potential, transcriptional activity (depending on whether it is the DNA or RNA that is sequenced), and diversity and abundance of microorganisms in biogas-producing microbial consortia [48]. The challenge, however, remains in interpreting such a large quantity of data in order to predict nutrient fluxes and responses to perturbations made to the system. To meet this challenge, greater knowledge is required in systems biology, at the level of microorganisms' cells and at the level of microbial communities. A significant milestone towards this goal will be the elucidation of the factors that regulate translation of mRNA, since transcript abundance alone is not sufficient to predict activity.
3. From Transcriptome to Phenotype
The synthesis of mRNA is merely the first step in gene expression. After transcription, the mRNA must be translated to form a protein product, and, following this, various post-translational regulatory events may alter the activity of proteins. It is a common biological phenomenon that the absolute levels of transcript abundances are poor indicators of protein levels [49, 50]. Rather, other attributes, including codon bias, gene ontology, and coding sequence length are better predictors of protein abundance [51, 52].
However, it is when comparing global responses of an organism to two or more test conditions that transcriptome studies identify profiles of differentially expressed targets in good approximation to that obtained using other “omics” technologies. For instance, reasonably good correlations are observed between differential transcript abundance and differential protein abundance for wild type M. maripaludis versus a mutant deficient in Ehb hydrogenase activity [50], for acetate versus methanol grown M. acetivorans [10] and for M. burtonii grown at 4°C versus 23°C [33]. Thus, methanogens use the regulation of transcript abundance as a major point of gene regulation in response to the environment. In cases where relative changes in transcript levels disagree with proteomics data in such comparisons, there is the possibility that a post-transcriptional mechanism of gene regulation is being employed. For instance, differences between transcriptomics and proteomics data of M. burtonii indicated that 16 genes encoding ribosomal proteins and 10 genes involved in methanogenesis were posttranscriptionally regulated in response to changes in incubation temperature [33]. Also, comparison of M. maripaludis transcriptome data with measurements of cellular pools of amino acids indicated that post-transcriptional regulation plays a major role in branched-chain amino acid biosynthesis [24]. Such multi-omics approaches can yield hypotheses regarding the role of posttranscriptional regulation in adapting to the conditions tested and stimulate further studies focused on particular regulatory mechanisms. Care must be taken, however, to account for the technical limitations of both transcriptomic and proteomic technologies and to account for the fact that mRNA generally has a short half-life relative to proteins.
Traditionally, transcriptome analyses, particularly those utilizing microarray technology, focus on evaluating the transcript abundance of coding sequences, although other applications exist. However, transcriptome sequencing studies are now becoming more prevalent. In comparison with microarray technology, transcriptome sequencing has greater dynamic range and is more suitable for mapping transcription start sites (TSSs) and detecting other unknown transcripts that may either be unannotated coding sequences or small RNAs (sRNAs) [53, 54]. Deep sequencing transcriptome analyses allow the identification of RNA degradation hotspots, facilitating the prediction of sequence motifs associated with RNA destabilization [55]. Information on TSSs and sRNAs will be instrumental in furthering the understanding of the role of RNA regulation in biological systems since RNA plays other crucial, though less documented roles in regulating gene expression. sRNAs may interact with mRNAs causing up- or downregulation of translation or altering the rate of mRNA turnover. sRNAs may also interact with proteins and modify their activities [56]. The ability to document a very large portion of TSSs for an organism will greatly aid investigations into how 5′- and 3′-untranslated regions (UTRs) influence RNA structure, stability, and translation.
Transcriptome sequencing of M. mazei mapped 876 TSSs, 208 sRNAs, and 40 small open reading frames of less than 31 amino acids [57]. This also led to the observation that M. mazei features long 5′ UTRs. Most of the discovered sRNAs in the transcriptome sequence analysis of M. mazei were located in intergenic regions, although some sRNAs were antisense to mRNAs [57]. The presence of 135 of the sRNAs depended upon nitrogen availability indicating that they play a regulatory role in nitrogen metabolism of M. mazei. One of the sRNAs, designated sRNA154, was subsequently shown to be important for optimal growth rate in nitrogen fixation conditions [58]. This indicates that sRNAs play an important role in posttranscriptional regulation of nitrogen metabolism in M. mazei.
Anther of the sRNAs discovered in M. mazei, designated sRNA162, was studied in greater detail [59]. Transcriptome analysis of wild type M. mazei versus an sRNA162 overexpressing derivative revealed that transcript levels of 185 open reading frames (ORFs) were differentially regulated including 48 ORFs involved in metabolism [59]. It was shown that sRNA162 binds to the 5′ UTR of the MM2241 transcript. By doing so, it masked the ribosome-binding site and caused translational level regulation of MM2241 which is postulated to encode a product involved in transcriptional repression of genes involved in MMA utilization. The sRNA162 overexpressing M. mazei strain adapted from growth on methanol to growth on TMA faster than the wild type strain did, further exemplifying its role as a regulator of methanogenesis.
The aforementioned discovery of long 5′ UTRs in M. mazei [57] is highly significant since these are involved in the regulation of translation rate and transcript stability through various different mechanisms [60]. A 5′ UTR was important in the methanogenic substrate-dependent regulation of CODH/ACS activity in Methanosarcina spp. by an uncharacterized mechanism that was likely either early transcriptional termination or endoribonuclease activity against the 5′ UTR [61]. In M. acetivorans, 5′ UTRs are also known to be involved in regulating the expression of different isozymes of methanol specific methyltransferases [62].
Bioinformatics work previously inferred that methanogens generally carry 5′ UTRs while other groups of Archaea often feature leaderless mRNAs [63]. Since then it was shown that haloarchaeal transcripts are mostly leaderless [64], and a recent transcriptome sequencing study showed that most Sulfolobus solfataricus mRNAs completely lack 5′ UTRs [55]. In S. solfataricus, leadered mRNAs required Shine-Dalgarno (SD) motifs to direct the 30S ribosome subunit to the translation initiation region, while correct positioning of the 30S subunit on leaderless mRNAs required a prebound initiator tRNA [65]. It is, however, worth noting that 70S ribosomes bind with higher affinity to leaderless mRNA than do 30S ribosomal subunits [66]. It was also suggested that SD-dependent initiation would operate during the translation of distal cistrons of polycistronic mRNAs [65]. However, the 5′ UTRs of most leadered haloarchaeal transcripts lacked an SD motif [64]. It is significant that methanogens may generally feature 5′ UTRs while most transcripts within the Haloarchaea and Crenarchaea lack 5′ UTRs and that the Haloarchaea and Crenarchaea seem to differ in their requirement for SD motifs in their 5′ UTR-containing mRNAs. Thus, it appears that there are differences in the mechanisms of translation initiation between these three groups of Archaea.
Further evidence of key differences in posttranscriptional control within the Archaea concerns 3′ UTRs. Polyadenylation at the 3′ end of mRNAs influenced RNA degradation in hyperthermophilic Archaea and in some methanogens but not in other methanogens nor the Haloarchaea [67]. Most of what is known about the role of 3′ UTRs in Archaea comes from the study of nonmethanogenic Archaea. For instance, the simultaneous presence of both the 5′ and the 3′ UTRs was shown to be required for translational regulation for two genes of Haloferax volcanii [68]. It was the 3′ UTR that dictated the direction of translational regulation [68], though this regulatory mechanism remains uncharacterized.
Differences in posttranscriptional control between different archaeal groups are significant when it is considered that methanogens are generally found to be difficult to manipulate in the laboratory for biochemical studies due to their high oxygen sensitivity. Consequently, other archaeal systems are often used as models to investigate a variety of fundamental processes, including translation related processes. However, owing to differences in posttranscriptional control and mRNA features between different archaeal groups, the use of methanogen models must be considered. The roles of 5′ UTRs and SD motifs in regulating the translation of methanogen mRNAs need to be clarified in future studies. The roles of the 3′ UTRs in methanogenic Archaea are also unclear. Current sequence annotation methods are insufficient to reliably predict TSSs, small open reading frames, and sRNAs. However, sequence annotation methods will likely improve due to transcriptome-sequencing work providing a wealth of training sets for the development of new bioinformatic tool sets. These advances will likely be very influential in ongoing research into methanogens and biological systems in general. Ultimately, the paucity of experimental data related to RNA regulation of translation in the Archaea must be addressed.
Acknowledgments
The authors would like to thank Stephen Zinder and Michal Ziv-El for critical reading and useful discussion during the preparation of this paper.
References
- 1.Conrad R. Soil microorganisms as controllers of atmospheric trace gases (H2, CO, CH4, OCS, N2O, and NO) Microbiological Reviews. 1996;60(4):609–640. doi: 10.1128/mr.60.4.609-640.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferry JG. Fundamentals of methanogenic pathways that are key to the biomethanation of complex biomass. Current Opinion in Biotechnology. 2011;22(3):351–357. doi: 10.1016/j.copbio.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, Whitman WB. Metabolic, phylogenetic, and ecological diversity of the Methanogenic archaea . Annals of the New York Academy of Sciences. 2008;1125:171–189. doi: 10.1196/annals.1419.019. [DOI] [PubMed] [Google Scholar]
- 4.Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nature Reviews Microbiology. 2008;6(8):579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- 5.Sakai S, Imachi H, Hanada S, Ohashi A, Harada H, Kamagata Y. Methanocella paludicola gen. nov., sp. nov., a methane-producing archaeon, the first isolate of the lineage “Rice Cluster I”, and proposal of the new archaeal order Methanocellales ord. nov. International Journal of Systematic and Evolutionary Microbiology. 2008;58(4):929–936. doi: 10.1099/ijs.0.65571-0. [DOI] [PubMed] [Google Scholar]
- 6.Paul K, Nonoh JO, Mikulski L, Brune A. ‘Methanoplasmatales,’ thermoplasmatales-related archaea in termite guts and other environments, are the seventh order of methanogens. Applied and Environmental Microbiology. 2012;78(23):8245–8253. doi: 10.1128/AEM.02193-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sowers KR. Methanogenesisin topics. In: Schmidt TM, Schaechter M, editors. Ecological and Environmental Microbiology. Academic Press; 2011. pp. 149–173. [Google Scholar]
- 8.Culley DE, Kovacik WP, Brockman FJ, Zhang W. Optimization of RNA isolation from the archaebacterium Methanosarcina barkeri and validation for oligonucleotide microarray analysis. Journal of Microbiological Methods. 2006;67(1):36–43. doi: 10.1016/j.mimet.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 9.Hovey R, Lentes S, Ehrenreich A, et al. DNA microarray analysis of Methanosarcina mazei Gö1 reveals adaptation to different methanogenic substrates. Molecular Genetics and Genomics. 2005;273(3):225–239. doi: 10.1007/s00438-005-1126-9. [DOI] [PubMed] [Google Scholar]
- 10.Li L, Li Q, Rohlin L, et al. Quantitative proteomic and microarray analysis of the archaeon Methanosarcina acetivorans grown with acetate versus methanol. Journal of Proteome Research. 2007;6(2):759–771. doi: 10.1021/pr060383l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Opulencia RB, Bose A, Metcalf WW. Physiology and posttranscriptional regulation of methanol:coenzyme M methyltransferase isozymes in Methanosarcina acetivorans C2A. Journal of Bacteriology. 2009;191(22):6928–6935. doi: 10.1128/JB.00947-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reichlen MJ, Vepachedu VR, Murakami KS, Ferry JG. MreA functions in the global regulation of methanogenic pathways in Methanosarcina acetivorans . mBio. 2012;3(4):e00189–e00112. doi: 10.1128/mBio.00189-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krätzer C, Carini P, Hovey R, Deppenmeier U. Transcriptional profiling of methyltransferase genes during growth of Methanosarcina mazei on trimethylamine. Journal of Bacteriology. 2009;191(16):5108–5115. doi: 10.1128/JB.00420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith MR, Mah RA. Growth and methanogenesis by Methanosarcina strain 227 on acetate and methanol. Applied and Environmental Microbiology. 1978;36(6):870–879. doi: 10.1128/aem.36.6.870-879.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costa KC, Wong PM, Wang T, et al. Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(24):11050–11055. doi: 10.1073/pnas.1003653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rohlin L, Gunsalus RP. Carbon-dependent control of electron transfer and central carbon pathway genes for methane biosynthesis in the Archaean, Methanosarcina acetivorans strain C2A. BMC Microbiology. 2010;10, article 62 doi: 10.1186/1471-2180-10-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welte C, Kallnik V, Grapp M, Bender G, Ragsdale S, Deppenmeier U. Function of Ech hydrogenase in ferredoxin-dependent, membrane-bound electron transport in Methanosarcina mazei . Journal of Bacteriology. 2010;192(3):674–678. doi: 10.1128/JB.01307-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferry JG, Lessner DJ. Methanogenesis in marine sediments. Annals of the New York Academy of Sciences. 2008;1125:147–157. doi: 10.1196/annals.1419.007. [DOI] [PubMed] [Google Scholar]
- 19.Veit K, Ehlers C, Ehrenreich A, et al. Global transcriptional analysis of Methanosarcina mazei strain Gö1 under different nitrogen availabilities. Molecular Genetics and Genomics. 2006;276(1):41–55. doi: 10.1007/s00438-006-0117-9. [DOI] [PubMed] [Google Scholar]
- 20.Habicht KS, Gade M, Thamdrup B, Berg P, Canfield DE. Calibration of sulfate levels in the Archean ocean. Science. 2002;298(5602):2372–2374. doi: 10.1126/science.1078265. [DOI] [PubMed] [Google Scholar]
- 21.Welte C, Deppenmeier U. Re-evaluation of the function of the F420 dehydrogenase in electron transport of Methanosarcina mazei . FEBS Journal. 2011;278(8):1277–1287. doi: 10.1111/j.1742-4658.2011.08048.x. [DOI] [PubMed] [Google Scholar]
- 22.Buan NR, Metcalf WW. Methanogenesis by Methanosarcina acetivorans involves two structurally and functionally distinct classes of heterodisulfide reductase. Molecular Microbiology. 2010;75(4):843–853. doi: 10.1111/j.1365-2958.2009.06990.x. [DOI] [PubMed] [Google Scholar]
- 23.Kato S, Kosaka T, Watanabe K. Comparative transcriptome analysis of responses of Methanothermobacter thermautotrophicus to different environmental stimuli. Environmental Microbiology. 2008;10(4):893–905. doi: 10.1111/j.1462-2920.2007.01508.x. [DOI] [PubMed] [Google Scholar]
- 24.Hendrickson EL, Liu Y, Rosas-Sandoval G, et al. Global responses of Methanococcus maripaludis to specific nutrient limitations and growth rate. Journal of Bacteriology. 2008;190(6):2198–2205. doi: 10.1128/JB.01805-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hendrickson EL, Haydock AK, Moore BC, Whitman WB, Leigh JA. Functionally distinct genes regulated by hydrogen limitation and growth rate in Methanogenic archaea . Proceedings of the National Academy of Sciences of the United States of America. 2007;104(21):8930–8934. doi: 10.1073/pnas.0701157104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia Q, Wang T, Hendrickson EL, Lie TJ, Hackett M, Leigh JA. Quantitative proteomics of nutrient limitation in the hydrogenotrophic methanogen Methanococcus maripaludis . BMC Microbiology. 2009;9, article 149 doi: 10.1186/1471-2180-9-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hendrickson EL, Leigh JA. Roles of coenzyme F420-reducing hydrogenases and hydrogen- and F420-dependent methylenetetrahydromethanopterin dehydrogenases in reduction of F420 and production of hydrogen during methanogenesis. Journal of Bacteriology. 2008;190(14):4818–4821. doi: 10.1128/JB.00255-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen EE, Lozupone CA, Rey FE, et al. Pan-genome of the dominant human gut-associated archaeon, Methanobrevibacter smithii, studied in twins. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(1):4599–4606. doi: 10.1073/pnas.1000071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker CB, Redding-Johanson AM, Baidoo EE, et al. Functional responses of Methanogenic archaea to syntrophic growth. The ISME Journal. 2012;6(11):2045–2055. doi: 10.1038/ismej.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo HW, Zhang H, Suzuki T, Hattori S, Kamagata Y. Differential expression of methanogenesis genes of Methanothermobacter thermoautotrophicus (formerly Methanobacterium thermoautotrophicum) in pure culture and in cocultures with fatty acid-oxidizing syntrophs. Applied and Environmental Microbiology. 2002;68(3):1173–1179. doi: 10.1128/AEM.68.3.1173-1179.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Culley DE, Nie L, Brockman FJ. DNA microarray analysis of anaerobic Methanosarcina barkeri reveals responses to heat shock and air exposure. Journal of Industrial Microbiology and Biotechnology. 2006;33(9):784–790. doi: 10.1007/s10295-006-0114-3. [DOI] [PubMed] [Google Scholar]
- 32.Lie TJ, Hendrickson EL, Niess UM, Moore BC, Haydock AK, Leigh JA. Overlapping repressor binding sites regulate expression of the Methanococcus maripaludisglnK1 operon. Molecular Microbiology. 2010;75(3):755–762. doi: 10.1111/j.1365-2958.2009.07016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campanaro S, Williams TJ, Burg DW, et al. Temperature-dependent global gene expression in the Antarctic archaeon Methanococcoides burtonii . Environmental Microbiology. 2011;13(8):2018–2038. doi: 10.1111/j.1462-2920.2010.02367.x. [DOI] [PubMed] [Google Scholar]
- 34.Williams TJ, Lauro FM, Ertan H, et al. Defining the response of a microorganism to temperatures that span its complete growth temperature range (-2°C to 28°C) using multiplex quantitative proteomics. Environmental Microbiology. 2011;13(8):2186–2203. doi: 10.1111/j.1462-2920.2011.02467.x. [DOI] [PubMed] [Google Scholar]
- 35.Pflüger K, Ehrenreich A, Salmon K, et al. Identification of genes involved in salt adaptation in the archaeon Methanosarcina mazei Gö1 using genome-wide gene expression profiling. FEMS Microbiology Letters. 2007;277(1):79–89. doi: 10.1111/j.1574-6968.2007.00941.x. [DOI] [PubMed] [Google Scholar]
- 36.Bräuer SL, Cadillo-Quiroz H, Ward RJ, Yavitt JB, Zinder SH. Methanoregula boonei gen. nov., sp. nov., an acidiphilic methanogen isolated from an acidic peat bog. International Journal of Systematic and Evolutionary Microbiology. 2011;61(1):45–52. doi: 10.1099/ijs.0.021782-0. [DOI] [PubMed] [Google Scholar]
- 37.Cadillo-Quiroz H, Yashiro E, Yavitt JB, Zinder SH. Characterization of the archaeal community in a minerotrophic fen and terminal restriction fragment length polymorphism-directed isolation of a novel hydrogenotrophic methanogen. Applied and Environmental Microbiology. 2008;74(7):2059–2068. doi: 10.1128/AEM.02222-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brioukhanov AL, Netrusov AI, Eggen RIL. The catalase and superoxide dismutase genes are transcriptionally up-regulated upon oxidative stress in the strictly anaerobic archaeon Methanosarcina barkeri . Microbiology. 2006;152(6):1671–1677. doi: 10.1099/mic.0.28542-0. [DOI] [PubMed] [Google Scholar]
- 39.Scholten JCM, Culley DE, Nie L, et al. Development and assessment of whole-genome oligonucleotide microarrays to analyze an anaerobic microbial community and its responses to oxidative stress. Biochemical and Biophysical Research Communications. 2007;358(2):571–577. doi: 10.1016/j.bbrc.2007.04.160. [DOI] [PubMed] [Google Scholar]
- 40.De Bok FAM, Luijten MLGC, Stams AJM. Biochemical evidence for formate transfer in syntrophic propionate-oxidizing cocultures of Syntrophobacter fumaroxidans and Methanospirillum hungatei . Applied and Environmental Microbiology. 2002;68(9):4247–4252. doi: 10.1128/AEM.68.9.4247-4252.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Worm P, Stams AJM, Cheng X, Plugge CM. Growth- and substrate-dependent transcription of formate dehydrogenase and hydrogenase coding genes in Syntrophobacter fumaroxidans and Methanospirillum hungatei . Microbiology. 2011;157(1):280–289. doi: 10.1099/mic.0.043927-0. [DOI] [PubMed] [Google Scholar]
- 42.Porat I, Kim W, Hendrickson EL, et al. Disruption of the operon encoding Ehb hydrogenase limits anabolic CO2 assimilation in the archaeon Methanococcus maripaludis . Journal of Bacteriology. 2006;188(4):1373–1380. doi: 10.1128/JB.188.4.1373-1380.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Enoki M, Shinzato N, Sato H, Nakamura K, Kamagata Y. Comparative proteomic analysis of Methanothermobacter themautotrophicus ΔH in pure culture and in co-culture with a butyrate-oxidizing bacterium. PloS ONE. 2011;6(8) doi: 10.1371/journal.pone.0024309.e24309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimoyama T, Kato S, Ishii S, Watanabe K. Flagellum mediates symbiosis. Science. 2009;323(5921):p. 1574. doi: 10.1126/science.1170086. [DOI] [PubMed] [Google Scholar]
- 45.Samuel BS, Hansen EE, Manchester JK, et al. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(25):10643–10648. doi: 10.1073/pnas.0704189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freitag TE, Prosser JI. Correlation of methane production and functional gene transcriptional activity in a peat soil. Applied and Environmental Microbiology. 2009;75(21):6679–6687. doi: 10.1128/AEM.01021-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freitag TE, Toet S, Ineson P, Prosser JI. Links between methane flux and transcriptional activities of methanogens and methane oxidizers in a blanket peat bog. FEMS Microbiology Ecology. 2010;73(1):157–165. doi: 10.1111/j.1574-6941.2010.00871.x. [DOI] [PubMed] [Google Scholar]
- 48.Wirth R, Kovács E, Maráti G, Bagi Z, Rákhely G, Kovács KL. Characterization of a biogas-producing microbial community by short-read next generation DNA sequencing. Biotechnology for Biofuels. 2012;5, article 41 doi: 10.1186/1754-6834-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lange C, Zaigler A, Hammelmann M, et al. Genome-wide analysis of growth phase-dependent translational and transcriptional regulation in halophilic archaea. BMC Genomics. 2007;8, article 415 doi: 10.1186/1471-2164-8-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xia Q, Hendrickson EL, Zhang Y, et al. Quantitative proteomics of the archaeon Methanococcus maripaludis validated by microarray analysis and real time PCR. Molecular and Cellular Proteomics. 2006;5(5):868–881. doi: 10.1074/mcp.M500369-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang T, Wan S, Xu Z, et al. Analysis and prediction of translation rate based on sequence and functional features of the mRNA. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0016036.e16036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dressaire C, Laurent B, Loubière P, Besse P, Cocaign-Bousquet M. Linear covariance models to examine the determinants of protein levels in Lactococcus lactis . Molecular BioSystems. 2010;6(7):1255–1264. doi: 10.1039/c001702g. [DOI] [PubMed] [Google Scholar]
- 53.Güell M, Yus E, Lluch-Senar M, Serrano L. Bacterial transcriptomics: what is beyond the RNA horiz-ome? Nature Reviews Microbiology. 2011;9(9):658–669. doi: 10.1038/nrmicro2620. [DOI] [PubMed] [Google Scholar]
- 54.Marguerat S, Wilhelm BT, Bähler J. Next-generation sequencing: applications beyond genomes. Biochemical Society Transactions. 2008;36(5):1091–1096. doi: 10.1042/BST0361091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wurtzel O, Sapra R, Chen F, Zhu Y, Simmons BA, Sorek R. A single-base resolution map of an archaeal transcriptome. Genome Research. 2010;20(1):133–141. doi: 10.1101/gr.100396.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sonnleitner E, Abdou L, Haas D. Small RNA as global regulator of carbon catabolite repression in Pseudomonas aeruginosa . Proceedings of the National Academy of Sciences of the United States of America. 2009;106(51):21866–21871. doi: 10.1073/pnas.pnas.0910308106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jäger D, Sharma CM, Thomsen J, Ehlers C, Vogel J, Schmitz RA. Deep sequencing analysis of the Methanosarcina mazei Gö 1 transcriptome in response to nitrogen availability. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(51):21878–21882. doi: 10.1073/pnas.0909051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ehlers C, Jäger D, Schmitz RA. Establishing a markerless genetic exchange system for Methanosarcina mazei strain Gö1 for constructing chromosomal mutants of small RNA genes. Archaea. 2011;2011:7 pages. doi: 10.1155/2011/439608.439608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jäger D, Pernitzsch SR, Richter AS, Backofen R, Sharma CM, Schmitz RA. An archaeal sRNA targeting cis-and trans-encoded mRNAs via two distinct domains. Nucleic Acids Research. 2012;40(21):10964–10979. doi: 10.1093/nar/gks847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Londei P. Evolution of translational initiation: new insights from the archaea. FEMS Microbiology Reviews. 2005;29(2):185–200. doi: 10.1016/j.femsre.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 61.Anderson KL, Apolinario EE, MacAuley SR, Sowers KR. A 5′ leader sequence regulates expression of methanosarcinal CO dehydrogenase/acetyl coenzyme A synthase. Journal of Bacteriology. 2009;191(22):7123–7128. doi: 10.1128/JB.00731-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bose A, Metcalf WW. Distinct regulators control the expression of methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. Molecular Microbiology. 2008;67(3):649–661. doi: 10.1111/j.1365-2958.2007.06075.x. [DOI] [PubMed] [Google Scholar]
- 63.Torarinsson E, Klenk HP, Garrett RA. Divergent transcriptional and translational signals in Archaea. Environmental Microbiology. 2005;7(1):47–54. doi: 10.1111/j.1462-2920.2004.00674.x. [DOI] [PubMed] [Google Scholar]
- 64.Brenneis M, Hering O, Lange C, Soppa J. Experimental characterization of Cis-acting elements important for translation and transcription in halophilic archaea. PLoS Genetics. 2007;3(12) doi: 10.1371/journal.pgen.0030229.e229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benelli D, Maone E, Londei P. Two different mechanisms for ribosome/mRNA interaction in archaeal translation initiation. Molecular Microbiology. 2003;50(2):635–643. doi: 10.1046/j.1365-2958.2003.03721.x. [DOI] [PubMed] [Google Scholar]
- 66.O’Donnell SM, Janssen GR. Leaderless mRNAs bind 70S ribosomes more strongly than 30S ribosomal subunits in Escherichia coli . Journal of Bacteriology. 2002;184(23):6730–6733. doi: 10.1128/JB.184.23.6730-6733.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Portnoy V, Schuster G. RNA polyadenylation and degradation in different Archaea; roles of the exosome and RNase R. Nucleic Acids Research. 2006;34(20):5923–5931. doi: 10.1093/nar/gkl763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brenneis M, Soppa J. Regulation of translation in haloarchaea: 5′-and 3′-UTRs are essential and have to functionally interact in vivo . PLoS ONE. 2009;4(2) doi: 10.1371/journal.pone.0004484.e4484 [DOI] [PMC free article] [PubMed] [Google Scholar]