

Abstract
Adhesion to the extracellular matrix (ECM) is critical for epithelial tissue homeostasis and function. ECM detachment induces metabolic stress and programmed cell death via anoikis. ECM-detached mammary epithelial cells are able to rapidly activate autophagy allowing for survival and an opportunity for re-attachment. However, the mechanisms controlling detachment-induced autophagy remain unclear. Here we uncover that the kinase PERK rapidly promotes autophagy in ECM-detached cells by activating AMP-activated protein kinase (AMPK), resulting in downstream inhibition of mTORC1-p70S6K signaling. LKB1 and TSC2, but not TSC1, are required for PERK-mediated inhibition of mammalian target of rapamycinin MCF10A cells and mouse embryo fibroblast cells. Importantly, this pathway shows fast kinetics, is transcription-independent and is exclusively activated during ECM detachment, but not by canonical endoplasmic reticulum stressors. Moreover, enforced PERK or AMPK activation upregulates autophagy and causes luminal filling during acinar morphogenesis by perpetuating a population of surviving autophagic luminal cells that resist anoikis. Hence, we identify a novel pathway in which suspension-activated PERK promotes the activation of LKB1, AMPK and TSC2, leading to the rapid induction of detachment-induced autophagy. We propose that increased autophagy, secondary to persistent PERK and LKB1-AMPK signaling, can robustly protect cells from anoikis and promote luminal filling during early carcinoma progression.
Keywords: anoikis, breast cancer, unfolded protein response
INTRODUCTION
Adhesion signaling has a key role in developmental processes by coordinating cell survival, polarity and growth factor signaling among other events.1 In epithelial cells, the loss of adhesion to the extracellular matrix (ECM) induces metabolic stress that results in a decrease in the ATP:ADP ratio.2 This leads to AMP-activated protein kinase (AMPK) activation, an intracellular energy-sensing pathway that controls cell metabolism.3 AMPK can also induce autophagy via inhibition of mTOR (mammalian target of rapamycin) signaling. mTOR is a protein kinase that coordinately orchestrates multiple cellular functions, including protein synthesis, nutrient transport to promote cell growth and proliferation; moreover, mTOR is an archetypal negative regulator of autophagy.4 Two distinct mTOR complexes, mTORC1 and mTORC2, exists in a cell. mTORC1, which is sensitive to allosteric inhibition by rapamycin, is assembled by binding of Raptor and mLST8 to mTOR, whereas mTORC2 is rapamycin-resistant and assembled by binding of Rictor, Sin1 and mLST8 to mTOR. Under high nutrient availability, mTOR is stimulated and promotes eIF4E-binding protein (4E-BP1) and p70S6K phosphorylation to activate translation initiation. Protein synthesis is the most energy-expensive cellular process in the cell. For that reason, in response to low nutrient availability or diverse environmental insults, a cell commonly diminishes global translation as an energy-conserving mechanism; two important signaling pathways implicated in the attenuation of protein synthesis are as follows: (1) the LKB1-AMPK-mediated inhibition of the mTOR pathway and (2) the integrated stress response, in which multiple stress-induced kinases phosphorylate eukaryotic initiation factor 2α (eIF2α) at Ser51, which acts as a potent suppressor of translational initiation.5
One important eIF2α kinase, the endoplasmic reticulum (ER) kinase PERK, is critical for mounting an effective unfolded protein response. When the protein folding capacity of the ER is compromised during the unfolded protein response, the canonical function of PERK is to reduce protein load in the ER by phosphorylating eIF2α at Ser51, and thereby attenuating translation initiation.6 Interestingly, during hypoxia, the mTOR pathway is deactivated at the same time as PERK is activated; both responses in these pathways are proposed to diminish translation initiation and save energy in hypoxic cells.7 Furthermore, experimental ER stress (for example, thapsigargin treatment) triggers the phosphorylation of the mTORC2 complex member Rictor in a GSK-3β-dependent manner to oppose mTORC2 activation of AKT.8 However, in both of these studies it remained unclear whether the unfolded protein response signaling machinery, and specifically PERK, directly regulate the mTORC1 complex. In fact, whether PERK can actively inhibit mTOR signaling and its impact on cell and tissue biology has never been rigorously scrutinized.
Here we show that PERK induces autophagy and enhances the survival of ECM-detached mammary epithelial cells (MECs) by promoting rapid and transcription-independent activation of AMPK and TSC2. We also demonstrate that PERK activation of AMPK requires LKB1 signaling, and that this mechanism is specifically triggered by the loss of adhesion rather than by canonical ER stress signals that activate PERK. These data reveal a previously unrecognized mechanism by which PERK can actively inhibit the mTOR pathway to rapidly optimize energy consumption via autophagy induction and maintain cellular homeostasis during loss of ECM adhesion. Our data using mammary gland organogenesis and mouse models suggest that these findings may have important implications for mammary tissue homeostasis and possibly early cancer progression.
RESULTS
PERK negatively regulates mTOR signaling in lactating mammary glands
We recently reported that tissue-specific deletion of PERK results in autophagy inhibition in ECM-detached cells and enhanced luminal cell (that is, cells occupying the luminal space) apoptosis.9 These results motivated us to scrutinize whether mTOR signaling was affected in this physiological setting; thus, we monitored AMPK and p70S6K phosphorylation, two key components of mTORC1 signaling, in lactating female mammary glands isolated from PERK conditional knockout (ko; PERKΔ/Δ) and wild-type (wt; PERKloxP/loxP) mice. p-AMPK expression was significantly reduced in PERK-deficient mammary tissue in comparison with controls (Figure 1a, upper row); this difference was evident in the vast majority of detached luminal cells, as well as in cells in contact with the underlying ECM (Figure 1a, lower row). Furthermore, the phosphorylation levels of the mTORC1 downstream substrate p70S6K were remarkably enhanced in PERKΔ/Δ mammary tissue; once again, both ECM-attached and -detached luminal cells showed increased signal (Figure 1b, upper row). This suggest that PERK inhibition can alter AMPK/mTORC1 signaling in both ECM-attached and lumen-occupying MECs. Overall, these results establish a genetic requirement for PERK to sustain both AMPK activation as well as suppress mTOR signaling in mouse mammary tissue; moreover, PERK is able to negatively regulate mTOR signaling in both ECM-attached and -detached MECs in vivo.
Figure 1.

PERK negatively regulates mTOR signaling in lactating mammary glands. (a, b) Immunohistochemical detection of p-AMPK and p-p70S6K on day 12 lactating mammary glands sections from control PERKloxP/loxP and mammary gland-specific PERK ko mice (PERKΔ/Δ). (a) Images showing enhanced p-AMPK staining in the control tissues along with strong staining in luminal cells and cell clusters (lower panels) that is reduced in PERKΔ/Δ tissues. (b) Images showing reduced p-p70S6K staining in PERKloxP/loxP control tissues in comparison with PERKΔ/Δ tissues.
Adhesion to the ECM prevents PERK-mediated inhibition of mTORC1 signaling via AMPK activation
As the above results broached a functional link between PERK and AMPK-mTOR signaling, we sought to mechanistically analyze how PERK inhibited mTORC1 signaling in ECM-detached cells. We had shown that PERK is activated after ECM detachment.10 We now show that simultaneously, ECM detachment resulted in the activation of AMPK that was accompanied by mTOR inhibition, evidenced by reduced p70S6K phosphorylation (Figure 2a, left panel and Supplementary Figure 1A). Increased AMPK activation was also accompanied by an increase in LC3-II lipidation (Figure 2a, left panel). Next, we determined whether these effects were indeed dependent on AMPK. We transfected adhered and suspended MFC10A cells with small interference RNA (siRNA) targeting AMPKα and scramble control (scRNA). We found that AMPKα depletion could revert, albeit not fully with the indicated knockdown level, mTOR inhibition and LC3-II lipidation in suspension (Figure 2a, right panel). Strikingly, in contrast to the AMPK activation observed upon loss of cell-matrix adhesion, canonical ER stress inducers (that is, thapsigargin) did not modify basal AMPK phosphorylation over a range of doses (Figure 2b, left panel). Nevertheless, thapsigargin did suppress mTOR activation, suggesting alternative mechanisms for mTOR inhibition upon disruption of the ER folding capacity versus ECM detachment (Figure 2b, left panel and Supplementary Figure 1B). To assess whether AMPK activation and mTOR inhibition resulted from the actual loss of integrin-mediated cell adhesion, we disrupted cell adhesion by treating MCF10A cells with the β1-integrin function-blocking antibody (AIIB2; Figure 2b, middle panel). We found that AIIB2 mimicked suspension-induced activation of AMPK and inhibition of mTOR signaling. Restoration of cell–ECM interactions by adding laminin-rich reconstituted basement membrane (Matrigel) to suspended cells was able to partially deactivate AMPK and restore p70S6K activity (Figure 2b, right panel). These data argue that upon loss of integrin–ECM attachment, there is rapid PERK activation and simultaneous AMPK-mediated inhibition of mTOR signaling.
Figure 2.

Adhesion to the ECM prevents PERK-mediated inhibition of mTORC1 signaling via AMPK activation. (a) MCF10A cells grown attached (A) or suspended for 12 h (S; left panel) and transfected with AMPK or control (scRNA) siRNAs (right panel) were lysed and immunoblotted (IB) with indicated antibodies (Abs). (b) Lysates from MCF10A were cultured in attached (A) or suspended (S) conditions or treated with 10 μM thapsigargin (Tg), 10 μg/ml AIIB2 or 5% matrigel (MGel) and IB with the indicated Abs. (c) Lysates of PERK +/+ and PERK −/− MEFs were collected from attached (left panel) and attached (A) and suspended (S) conditions (right panel), treated with or without 10 μM compound C (C.C.) as indicated and IB against the indicated antigens. (d) MCF10A lysates from attached (A) and in suspended (S) cells that were transfected with PERK or control (scRNA) siRNAs were treated with or without 10 μM CC as indicated and IB against the indicated antigens. (e) Adherent Fv2E-ΔNPERK MCF10A treated with or without 100 pM AP were collected at the indicated time points (minutes) and analyzed by IB with the indicated Abs.
The tight correlation between PERK and AMPK activation and mTORC1 inhibition, both in vivo, and during ECM detachment prompted us to test whether these two signals were functionally related. Similar to the mammary gland, tissue PERK-deficient mouse embryo fibroblast cells (MEFs) PERK−/− exhibited reduced AMPK activity and higher levels of p70S6K when compared with wt control MEFs (Figure 2c, left panel). This supports that at a basal level PERK activation is required to maintain a homeostatic level of AMPK and in mTOR activation. Importantly, PERK deletion was sufficient to prevent suspension-induced AMPK phosphorylation, as well as sustain mTORC1 activity in detached cells (Figure 2c, right panel), and this was not solely related to the lower basal level of activation of the pathway in the PERK−/− MEFs (Supplementary Figure 1A). Next we tested whether AMPK activity might be required for PERK-dependent downregulation of mTOR. For that purpose we treated PERK−/− and wt MEFs with compound C (CC), an ATP competitive inhibitor of AMPK.11 We found that AMPK inhibition could rescue p70S6K phosphorylation in wt suspended MEFs; this effect was even more pronounced in PERK-deficient MEFs (Figure 2c, right panel). To further validate these results, we transfected MCF10A cells with control (scRNA) or two different siRNAs targeting PERK (Figure 2d and Supplementary Figure 1C), placed them in adherent or suspended conditions, and treated them with CC. As predicted, suspension-induced activation of AMPK, measured by p-AMPK and phosphoacetyl CoA carboxylase levels, was significantly reversed by PERK depletion; moreover, reduced AMPK activation correlated with increased p70S6K phosphorylation in detached cells (Figure 2d). Furthermore, CC inhibited AMPK phosphorylation and activity, and rescued p70S6K phosphorylation in control MCF10A cells and the slight p70S6K observed in PERK knockdown cells. These results suggest that specific suspension-induced inhibition of mTORC1 requires AMPK phosphorylation at Thr172 activity and PERK signaling. We further substantiated the requirement for PERK kinase activity using MCF10A cells stably expressing the dominant-negative mutant PERKΔC; compared with controls (β-gal), these cells also exhibited increased mTOR activation during suspension (Supplementary Figure 1D). Finally, we used a gain-of-function approach to conclusively determine whether PERK activation is sufficient to inhibit mTOR signaling via AMPK activation and independently of other cell detachment-induced signals. To this end we used adhered Fv2E-PERK MCF10A cells engineered to express the Fv2E–PERK fusion protein that is activated with the dimerizing molecule AP20187 (AP).12 We found that 100 pM of AP was able to induce Fv2E-PERK activation as well as AMPK phosphorylation at different times points. This was accompanied by decreased p70S6K phosphorylation, yet no effect was observed on ERK1/2 or AKT phosphorylation, or total levels (Figure 2e), arguing against a role for either PI3K or mTORC2 signaling as downstream events of Fv2E-PERK activation. Thus, PERK activation is necessary and sufficient to both activate AMPK and inhibit the mTORC1 pathway; this signaling cascade is observed upon inducible PERK activation in adhered cells using the Fv2E-PERK system or in response to suspension-induced stress.
LKB1 and TSC2, but not TSC1, are required for suspension-induced inhibition of mTORC1 signaling
We next wished to determine the specific components of the AMPK pathway that served as entry points for PERK signaling to inhibit mTOR. AMPK is activated its upstream kinase LKB1, a tumor suppressor, and TSC1/2 transduces the inhibitory signal of AMPK on mTOR by promoting Rheb GAP activity.13 LKB1 phosphorylates a conserved threonine in the regulatory T loop of AMPK that is required for full activation of this kinase.14,15 We validated our observations in MCF10As cells and confirmed that PERK could have effectively been activated after ECM detachment in MEFs (Figure 3a). Next, using wt or LBK1−/− MEFs, we found that p70S6K deactivation in suspension was lost in the LKB1 ko versus wt MEFs (Figure 3a, left panel). This argues that suspension-induced inhibition of mTORC1, which requires AMPK and PERK, also requires LKB1. When comparing TSC1 and TSC2 ko MEFs with wt counterparts, we found that TSC2, but not TSC1, was required for suspension-induced mTORC1 inhibition, as MEFs deficient for the former were not able to diminish mTORC1 activation in suspension (Figure 3a, middle and right panel). The enhanced PERK phosphorylation in LKB1−/−, TSC1−/− and TSC2−/− MEFs in basal-adhered conditions is consistent with an increased unfolded protein response due to enhanced protein translation and higher ER client protein load.16 These data suggests that the LKB1-AMPK-TSC2 axis has an essential role in inhibiting mTOR signaling upon loss of integrin–ECM engagement and PERK activation.
Figure 3.

LKB1 and TSC2, but not TSC1, are required for suspension-induced inhibition of mTORC1 signaling. (a) Lysates of LKB1 +/+, LKB1 −/− and TSC2 +/+, LKB1 −/− MEFs were collected from attached (A) and suspended (S) conditions, and immunoblotted (IB) against the indicated antigens. (b) MFC10A Fv2E-PERK cells were transfected with TSC2 (upper panel), LKB1 (lower panel) siRNAs or scramble control (scRNA), and treated with or without 100 pM AP at the indicated time points (min) and IB against the indicated antibodies (Abs). Right graphs show TSC2 and LKB1 knocking down controls by mRNA transcript level analysis normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (c) Adhered (A) or suspended (S) LKB1+/+, LKB1−/− and TSC2+/+, LKB1−/− MEFs were treated with 0.1 Leupeptin and 20 mM NH4 as indicated and analyzed by IB for the indicated antigens. Densitometric analysis for LC3 flux was determined using Image J software (N = 3). (d, e) Images showing adhered Fv2E-PERK/GFP-LC3 (d) or GFP-LC3 (e) MCF10A cells transfected with empty vector (EV) or p70S6K mutant Δ2-46 ΔCT104 (p70CA) for 48 h and treated with or without AP (d) or AICAR (e) for 4 h. After that, cells were fixed and stained with anti-HA antibodies for p70CA expression. Lower graphs, percentage of LC3-positive (five or more puncta/cell) cells were scored (n = 50) and plotted in the p70CA- and EV- (see Supplementary Figure 1F) transfected cells. Scale bars indicate 10 μm. P-values were determined by the Student’s t-test.
To determine whether the PERK-induced inhibition of mTORC1 requires LKB1 and/or TSC2 in MECs, we transfected Fv2E-PERK MCF10A cells with siRNAs targeting TSC2 or LKB1, and treated them with AP for 10 or 20 min. We found that AP-mediated activation of Fv2E-PERK inhibited p70S6K phosphorylation in control (scRNA) cells. However, in TSC2 or LKB1 siRNA-transfected MECs, PERK activation was unable to inhibit p70S6K phosphorylation (Figure 3b). We conclude that suspension-induced activation of PERK and AMPK results in mTORC1 inhibition. We further conclude that enforced PERK activation can lead to mTOR inhibition via TSC2 and LKB1 in MEFs and MECs. Finally, inhibition of p70S6K phosphorylation in MECs detached from the ECM also requires LKB1 and TSC2.
We had shown that PERK can activate autophagy in ECM-detached cells and it is well established that inhibition of mTOR by AMPK induces autophagy.17 Thus, we sought to test the role of the upstream mediators LBK1 and TSC2 in ECM-detachment-induced autophagy. We found that both negative regulators of mTOR are required for suspension-induced autophagy (Figure 3c). Next we tested whether LKB1 and TSC2 were able to modulate LC3-II turnover. For that purpose we blocked lysosomal degradation with ammonium chloride and leupeptine.18 When looking to the LC3-II/LC3-I ratio, we found that both genes affected autophagic flux in suspension to a similar extent (Figure 3c). Our data suggest that the LKB1–AMPK complex is most likely the entry point for PERK-induced inhibition of mTORC1, and that TSC2 is a critical mediator of PERK-induced mTOR inhibition.
Although p70S6K can function as a negative as well as positive regulator of autophagy,19 its potential functions in regulating autophagy during ECM detachment has not been previously tested. To address the role during of p70S6K in PERK-induced mTORC1 inhibition and autophagy, we transfected Fv2E-PERK/GFP-LC3 MCF10A cells with the constitutively active p70S6K mutant Δ2-46 ΔCT104 (referred to as p70CA hereafter) or empty vector as control. Upon AP-mediated activation of PERK for 30 min, increased GFP-LC3 puncta were observed in cells, consistent with increased autophagosome formation (Figure 3d and Supplementary Figures 1E and F). Remarkably, this punctate GFP-LC3 staining was completely lost in cells co-expressing p70CA (Figure 3d, graph). Similarly, we found that transient expression of p70CA resulted in inhibition of AMPK-mediated induction of autophagy stimulated by AICAR (5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide), an AMP analog that stimulates AMPK activity (Figure 3e, graph and Supplementary Figure 1F). We conclude that inhibition of p70S6K is required for PERK-induced autophagy in an LKB1-, AMPK- and TSC2-dependent manner.
Impact of modulating AMPK activity on lumen formation in three-dimensional epithelial culture
The LKB1-AMPK pathway is tumor suppressive.20 PERK has a dual role in cancer and it can aid tumorigenesis by inducing cytoprotection and oxidative stress relief. This opened the possibility that PERK signals may subvert LKB1-AMPK signaling to serve as a survival signal in immortalized MECs. During three-dimensional (3D) acinar morphogenesis, ECM-detached luminal cells in which PERK forced to remain constitutively activated undergo autophagy, survive and fill the luminal space; moreover, PERK is heavily phosphorylated in human ductal carcinoma in situ (DCIS) lesions that exhibit luminal filling.9 Remarkably, treatment of MCF10A cells with the AMPK-selective activator AICAR elicited acini with filled lumen, which mimicked, albeit with a lower penetrance, the phenotype due to inducible PERK activation (Figure 4a). We observed that AICAR treatment could effectively induce AMPK activity preferentially in luminal cells (Figure 4b, left set). AICAR could also induce autophagy during a 3D morphogenesis as measured by LC3 staining, and this also occurred in luminal cells preferentially (Figure 4b, right set). This suggests that autophagy has an important role in AICAR-induced survival in luminal cells. Interestingly, PERK and AICAR have been shown to promote cell cycle withdrawal,9,21 and MECs in G1 are less sensitive to anoikis signals.22 Thus, we cannot rule out that other signals linked to cell cycle inhibition might cooperate in promoting ECM-independent survival. Nevertheless, our results strongly support that AMPK activation by PERK could have a deleterious effect on acinar structure and function by positively regulating the survival of luminal cells at least via autophagy.
Figure 4.

Impact of modulating AMPK activity on luminal filling in 3D culture. (a) Representative confocal images of day 12 MCF10A acini, treated from day 6 to day 12 with 10 μM AICAR showing intraluminal cell accumulation. Scale bars indicate 10 μm. (b) Confocal equatorial images from parental (left set) or GFP-LC3 (right set) MCF10A acini treated from day 6 to day 12 with 10 μM AICAR and stained with the indicated antibodies (Abs). Magnifications show intraluminal p-AMPK (red) or GFP-LC3 (green) staining in AICAR-treated acini. The percent of positive events per acinus was scored and plotted (right graphs, n = 20). Scale bars indicate 25 μm. (c) Confocal equatorial images from Fv2E-PERK MCF10A acini treated with or without 100 pM AP and/or 10 μM compound C (CC), and/or 50 nM rapamycin (Rap) fixed at day 12 of morphogenesis showing intraluminal filling. Right graphs show the distribution and mean of intraluminal cells for each equatorial section of a single acinus (n = 20). Scale bars indicate 15 μm. P-values were determined by the Student’s t-test.
We previously reported that AP-mediated activation of the Fv2E–PERK fusion protein promotes survival of luminal cells (Avivar-Valderas et al.9 and Figure 4c, upper middle picture). Thus, we interrogated whether PERK-mediated luminal filling was dependent on AMPK signaling. Treatment with CC reverted AP-induced luminal filling in response to Fv2E-PERK activation (Figure 4c, upper right panel). As expected, treatment with the mTORC1 inhibitor rapamycin resulted in enhanced survival and an increase in luminal cell accumulation (Figure 4c, lower right panel). Together, these results suggest that PERK negatively regulates mTOR signaling in luminal cells via the AMPK activation. If deregulated, this signal can in fact induce prolonged survival of cells in the 3D lumen.
DISCUSSION
Here we delineate the first clear negative crosstalk between PERK signaling and mTOR pathway. Specifically, we demonstrate that the loss of ECM adhesion is a source of stress that immediately triggers PERK activation. PERK is required for the activation of the metabolic stress master regulator AMPK, which mediates PERK antagonism on mTOR and autophagy induction in ECM-detached cells. Moreover, we also determine that PERK inhibition of mTOR via the LKB1-AMPK activation is specific for loss of cell adhesion-induced stress and not canonical ER stress signaling through PERK.
Protein synthesis demands high levels of energy. Thus, cells have inbuilt tightly regulated mechanisms to limit protein synthesis that is energy demanding. mTOR has an essential role by phosphorylating the translational inhibitor 4E-BP1,23 thus favoring CAP-dependent translation. On the other hand, PERK antagonizes this process by phosphorylating eIF2α at Ser51; by inhibiting eIF2B activity, it blocks the binding of the initiator tRNA (tRNAi Met) to the 40S ribosome, thereby attenuating translation initiation. Some reports suggest crosstalk between these two pathways. For example, mTOR activation or loss of TSC1/2 activity16 results in constitutive PERK-eIF2α signaling. This is presumably due to increased protein synthesis that augments the demand on the ER folding capacity. Others have shown that ER stress (that is, thapsigargin, dithiothreitol) negatively regulates mTORC2 and AKT to enhance autophagy by inhibiting the AKT upstream activator IRS1.24 However, these studies did not identify how the ER stress signaling components (that is, PERK, IRE1α) communicates with the mTOR pathway. In our hands, PERK inhibits mTORC1 signaling without decreasing AKT phosphorylation. This suggests that AMPK activation is the main transducer of the PERK signal that inhibits mTORC1 upon loss of integrin signaling. Our work reveals that the ER-kinase PERK is required for downstream activation of the LKB1–AMPK complex, which leads to TSC1/2-dependent inhibition of mTOR. Thus, we provide the first evidence for a negative fast kinetics signaling crosstalk from PERK to mTOR signaling. We propose that this rapid inhibition of mTOR by PERK might be required to promote autophagy in ECM-deprived cells.
Interestingly, we observed that perturbations on PERK signaling might alter AMPK-mTORC1 signaling in adhered cells as well. This signaling may be required for ECM-attached cells for efficiently promote autophagy and/or anti-oxidative response to maintain cellular fitness. We propose that transient changes in adhesion might engage this cytoprotective pathway or that other functions of PERK, related to its canonical role, are also functioning via AMPK activation and mTOR inhibition.
Our work raises important questions as to how does PERK signal to the LKB1–AMPK complex to affect cell fate? Autophagy can induce survival during ECM detachment, presumably by mitigating metabolic stress caused by a drastic drop in ATP production in suspended cells.2,25 In our model, PERK activates AMPK to promote pro-survival autophagy, to rapidly provide an ATP source.9 Recently, a pro-survival role of AMPK has also been shown in ECM-detached cells.3 In agreement with our findings, Ng et al.3 showed that AMPK activation mitigates ATP reduction by inhibiting mTORC1 and subsequent global protein synthesis. We also uncover that PERK and AMPK-dependent activation of autophagy is dependent of p70S6K inhibition, as constitutive activation of this kinase rescued from PERK- or AICAR-induced autophagy. p70S6K has dual roles, as it is essential for catabolic (autophagy)26 and also anabolic (ribosomal biosynthesis) processes.27 Our results indicate that in the context of suspension-induced PERK signaling, p70S6K must be inhibited to favor autophagy. Furthermore, we show that PERK activation can readily activate AMPK phosphorylation at a site that is a substrate for LKB1. Although how this happens is not precisely clear, it is possible that PERK regulates a putative AMPK phosphatase that allows for more prolonged phosphorylation by LKB1. Potentially, CaMMK-dependent activation of AMPK in response to ER Ca2+ release could mediate this signal.28,29 Further studies are required to elucidate how specifically PERK activates AMPK; in particular, it will be important to investigate whether PERK activates CaMMK activation during ECM-detachment.
PERK-mediated activation of AMPK may have important and multifaceted implications for cancer development. Our previous work indicates that forced activation of PERK can lead to growth arrest by regulating cell cycle inhibitors, as well as robust survival and luminal filling in 3D culture. During the 3D acinar morphogenesis, AICAR-mediated activation of AMPK causes luminal filling similar to enforced PERK activation. Thus, it is possible that autophagy and PERK-induced AMPK activation during early cancer progression not only maintains cells in a growth-arrested or slow-cycling9 state, but also serve as a robust survival pathway that protects cancer cells from diverse micro-environmental stresses, including ECM detachment. It is possible that the protective role of growth arrest from anoikis shown by others22 may also be linked to PERK-dependent coordination of growth arrest and survival via autophagy9 and this study. Our findings may have specific implications for pre-invasive breast lesions, such as DCIS, which commonly do not exhibit high rates proliferation and are refractory to chemotherapy.30 Remarkably, autophagy is frequent in DCIS9,31 and we have found PERK phosphorylation to be frequently upregulated in these lesions.9 Thus, as proposed in our earlier studies in DCIS lesions, PERK signaling via induction of growth arrest10,32 and autophagy9 may contribute to dormancy of these tumors. Thus, although LKB1 functions as a tumor suppressor, PERK activation may tap into the LKB1-AMPK signaling to protect early progressed cells via autophagy, and this may provide a window of opportunity to acquire additional mutations and propel tumor growth. LKB1 has been shown to maintain Myc-expressing MECs in a quiescent state by preventing loss of polarity.33,34 It is possible that in early breast cancer lesions, LKB1 serves as an antagonist of oncogene signaling while still serving as a mediator of PERK-induced survival. This may happen in cells that could translocate to the lumen due to loss of cell junctions or gain of MT1-MMP for example.35 Thus, only after loss of LKB1 or other components of the pathway, luminal cells become permissive to oncogene-induction of fully proliferation.
Our study identifies a previously unrecognized context-specific negative signal from PERK to mTOR via activation of LKB1 and AMPK signaling. We demonstrate that this mechanism is specifically triggered by the loss of adhesion, rather than conventional ER stress activation of PERK. This triggers an effective adaptation pathway orchestrated by PERK to induce autophagy. Adaptation pathways that favor anchorage-independent growth and survival are critical during cancer initiation and progression.36 We propose that during early cancer progression, tumor cells can co-opt stress-adaptive pathways to support survival. Our study suggests that PERK-dependent phosphorylation of AMPK may represent an important mechanism of addiction to these cytoprotective pathways and, hence, a potential weakness to target slow-cycling or dormant tumor cells in lesions such as DCIS that resist treatment.
MATERIALS AND METHODS
Cell lines and treatments
Low-passage MCF10A cells were cultured as described.37 For Fv2E-PERK stable cell lines, cells were treated with 100 pM AP (added daily) or an equal volume of ethanol (vehicle) as control. Anoikis assays were performed as described.9 Stable GFP-LC3 MCF10A cell line was a kind gift from Jayanta Debnath (University of California, San Francisco). The Δ2-46 ΔCT104 p70S6K mutant was purchased from Addgene (Cambridge, MA, USA) (plasmid 1898). The wt and TSC1, TSC2, LKB1 and ko MEFs were kindly provided by Dr Nabeel Bardeesy (Harvard Medical School, Boston, MA, USA). Function-blocking AIIB2 was obtained form Developmental Study Hybridoma Bank (University of Iowa, Iowa City, IA, USA). For 3D cultures, cells were plated in commercially available growth factor reduced Matrigel (BD Biosciences, San Diego, CA, USA) and grown as described.37 For cell viability quantification, detached cells were washed with phosphate-buffered saline, disaggregated and collected as single cell suspensions using cell-strained cap (BD Falcon, San Jose, CA, USA) to be incubated in 1:2 Trypan blue stain (BioWhittaker, Bridgeport, NJ, USA). Total and non-viable cells were manually determined using counting chamber.
Immunoblots
MCF10A and MEF cells were lysed, and protein was analyzed by immunoblotting as described.38 Membranes were blotted using antibodies against: p-PERK (Thr981), PERK and p-ERK1/2 (Tyr204) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); glyceraldehyde 3-phosphate dehydrogenase (Calbiochem, Billerica, MA, USA); LC3 (Axxora, LLC, Farmingdale, NY, USA); ERK1 (BD Biosciences); HA (Roche, Nutley, NJ, USA); p-AKT (Ser473), AKT, p-p70S6K (Thr389), p70S6K, p-S6 Ribosomal Protein (Ser240/244), phospho-acetyl CoA carboxylase (Ser79), p-AMPKα (Thr172) and AMPK (Cell Signaling Technology, Danvers, MA, USA). Bound antibodies were detected with HRP secondary antibodies and chemiluminiscence assays as described.10
RNAi and cDNA transfections
Cells were transiently transfected using Lipofectamine RNAiMax (Invitrogen, Grand Island, NY, USA) and FuGENE HD (Roche). The siRNA for AMPKα was purchased from Dharmacon (Lafayette, CO, USA), TSC2 and LKB1 from Cell Signaling and PERK (ID number 18102), PERK2 (ID number 103593) and Silencer negative control from Ambion (Grand Island, NY, USA). Transfections of plasmids and siRNA oligonucleotides were done using 3 μg DNA mixed with 6 μl of Fugene HD or 100 nM AMPKα, PERK, LKB1 or TSC2 siRNA oligonucleotides with 8 μl Lipofectamine RNAiMax, respectively, and incubated for 24 and 48 h.
Immunofluorescence microscopy
Rhodamine-conjugated secondary antibodies (Molecular Probes, Grand Island, NY, USA) were used for two-dimensional and 3D culture immunofluorescence assays. For detection of autophagosomes in adhered or suspended conditions, stably expressing GFP-LC3 or GFP-LC3 Fv2E-PERK MCF10A cells were grown on glass coverslips and fixed with 3% paraformaldehyde following standard protocols.38 The 3D Matrigel MCF10A acinar structures were fixed at day 12 and processed for immunofluorescence microscopy analysis as established previously.10 Confocal analyses were performed using the Leica SP5 Multi-photon confocal microscope equipped with UV diode (405 nm), argon (458, 476, 488 and 514 nm), HeNe (543 nm) and a HeNe (633 nm) lasers. All images were taken using × 63 magnification.
Animal tissue and immunohistochemistry
Mammary gland epithelium tissues were obtained from control PERKloxP/loxP and mammary gland-specific PERK ko mice (PERKΔ/Δ) generated as described.39 Immunohistochemistry from embedded paraffin sections was performed as described.10 The sections were processed using VectaStain ABC Elite Kit (Vector Laboratories, Bulingame, CA, USA), and the signal was detected using DAB Substrate Kit for peroxidase (Vector Laboratories). p-p70S6K (Thr389) and p-AMPKα (Thr172) were purchased from (Cell Signaling Technology).
Real-time PCR primers
RNA was isolated from cells with TRIzol reagent following the manufacturer’s indications (Invitrogen). Reverse transcription was performed using M-MuLV Reverse Transcriptase (New England Biolabs, Ipswich, MA, USA). Real-time was performed on a DNA Engine Opticon System using Power SYBR green PCR Master Mix (Bio-Rad, Hercules, CA, USA). The human forward and reverse primer sequences used were as follows: TSC2, 5′-CGGGACACAGCCCTGTACAAGT-3′ and 5′-GGTACAGGGAGGAGAAAGAG GCC-3′; LKB1, 5′-GTTGCGAAGGATCCCCAACGG-3′ and 5′-CACCAGCTGGAT GACATTTTTGTGC-3′; glyceraldehyde 3-phosphate dehydrogenase 5′-CCCC TGGCCAAGGTCATCCA-3′ and 5′-ACAGCCTTGGCAGCGCCAGT-3′.
Statistics
Statistical analysis was performed using GraphPad Prism 5.0 software (GraphPad software, Inc., La Jolla, CA, USA) and P-values were calculated using one-way analysis of variance followed by the Bonferroni multiple comparison post test or the unpaired Student’s t-test with P < 0.05 considered statistically significant; N.S., not significant.
Supplementary Material
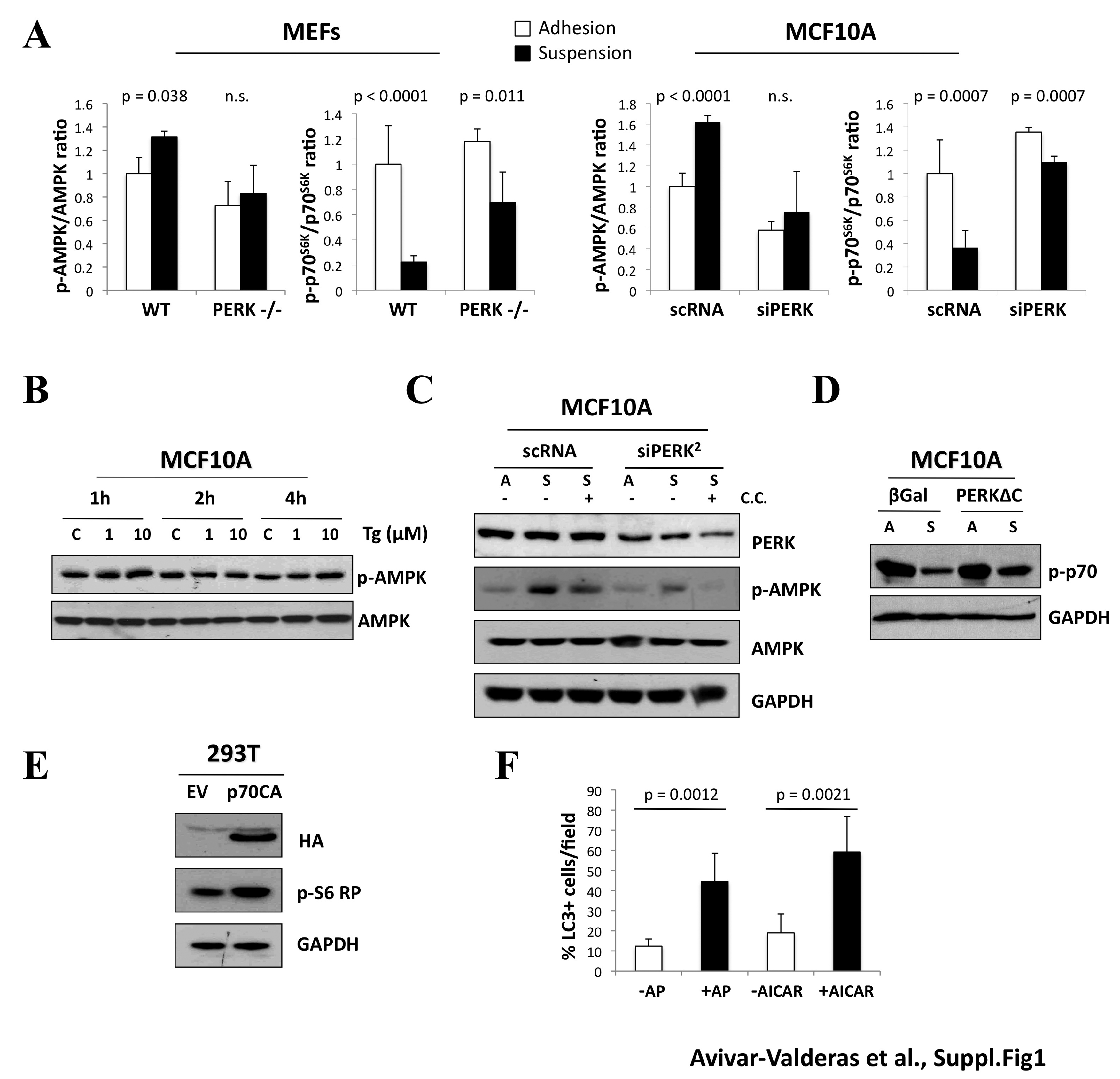
A, Graphs showing densitometric analysis of the ratio p-AMPK/AMPK and p-p70S6K/p70S6K of PERK+/+ (WT) and PERK−/− (PERK−/−) mouse embryonic fibroblast (MEFs) and MCF10A cells grown in adhered or suspended conditions. The analysis was determined using Image J software (MEFs n=3 and MCF10As n=5). B, MCF10A cells were grown in adhered conditions and treated with DMSO (C), 1 μM or 10 μM thapsigargin at the indicated time points (hours). Lysates were collected and IB with the indicated Abs. C, MCF10A cultures were collected from attached (A) or suspended (S) conditions after transfection with PERK2 or control (scRNA) siRNAs and treated with 10μM compound C. Cells were lysed and IB against the indicated antigens. D, Lysates from attached (A) or suspended 12h (S) βGal and PERKΔC MCF10A cells were IB with indicated Abs. E, HEK293T cells were transfected with control empty vector (EV) or p70S6K mutant Δ2-46 ΔCT104 (p70CA), lysates were collected after 48h and IB with the indicated Abs. F, Graphs showing percentage of LC3 positive cell on adhered Fv2E-PERK/GFP-LC3 or GFP-LC3 MCF10A cells transfected with empty vector for 48h and treated with or without AP or AICAR respectively for 4h. p values were determined by the t-test.
{kind=link}
Acknowledgments
Confocal laser scanning microscopy was performed at the MSSM-Microscopy Shared Resources Facility, supported with funding from NIH-NCI shared resources grant (5R24CA095823-04), NSF Major Research Instrumentation grant (DBI-9724504) and NIH shared instrumentation grant (1 S10RR0 9145-01). This work is supported by grants from the Samuel Waxman Cancer Research Foundation Tumor Dormancy Program, NIH/National Cancer Institute (CA109182, CA163131), NIEHS (ES017146) and NYSTEM to J.A.A-G, NIH RO1 CA126792, CA126792-S1 (ARRA) to JD, and P01 CA104838 and a Leukemia and Lymphoma Scholar award to JAD.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng TL, Leprivier G, Robertson MD, Chow C, Martin MJ, Laderoute KR, et al. The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 2012;19:501–510. doi: 10.1038/cdd.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(Part 20):3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raven JF, Koromilas AE. PERK and PKR: old kinases learn new tricks. Cell Cycle. 2008;7:1146–1150. doi: 10.4161/cc.7.9.5811. [DOI] [PubMed] [Google Scholar]
- 6.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen CH, Shaikenov T, Peterson TR, Aimbetov R, Bissenbaev AK, Lee SW, et al. ER stress inhibits mTORC2 and Akt signaling through GSK-3beta-mediated phosphorylation of rictor. Sci Signal. 2011;4:ra10. doi: 10.1126/scisignal.2001731. [DOI] [PubMed] [Google Scholar]
- 9.Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, et al. PERK integrates autophagy and oxidative stress responses to promote survival during ECM detachment. Mol Cell Biol. 2011;31:3616–3629. doi: 10.1128/MCB.05164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sequeira SJ, Ranganathan AC, Adam AP, Iglesias BV, Farias EF, Aguirre-Ghiso JA. Inhibition of proliferation by PERK regulates mammary acinar morphogenesis and tumor formation. PLoS ONE. 2007;2:e615. doi: 10.1371/journal.pone.0000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, et al. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004;23:169–179. doi: 10.1038/sj.emboj.7600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 16.Ozcan U, Ozcan L, Yilmaz E, Duvel K, Sahin M, Manning BD, et al. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. 2008;29:541–551. doi: 10.1016/j.molcel.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuertes G, Martin De Llano JJ, Villarroya A, Rivett AJ, Knecht E. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem J. 2003;375(Part 1):75–86. doi: 10.1042/BJ20030282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7:167–178. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 20.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 21.Peyton KJ, Liu XM, Yu Y, Yates B, Durante W. Activation of AMP-activated protein kinase inhibits the proliferation of human endothelial cells. J Pharmacol Exp Ther. 2012;342:827–834. doi: 10.1124/jpet.112.194712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins NL, Reginato MJ, Paulus JK, Sgroi DC, Labaer J, Brugge JS. G1/S cell cycle arrest provides anoikis resistance through Erk-mediated Bim suppression. Mol Cell Biol. 2005;25:5282–5291. doi: 10.1128/MCB.25.12.5282-5291.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, et al. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 1999;13:1422–1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6:239–247. doi: 10.4161/auto.6.2.11062. [DOI] [PubMed] [Google Scholar]
- 25.Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008;19:797–806. doi: 10.1091/mbc.E07-10-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klionsky DJ. Cell biology: regulated self-cannibalism. Nature. 2004;431:31–32. doi: 10.1038/431031a. [DOI] [PubMed] [Google Scholar]
- 27.Dennis PB, Fumagalli S, Thomas G. Target of rapamycin (TOR): balancing the opposing forces of protein synthesis and degradation. Curr Opin Genet Dev. 1999;9:49–54. doi: 10.1016/s0959-437x(99)80007-0. [DOI] [PubMed] [Google Scholar]
- 28.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, et al. Ca2 +/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Kerlikowske K, Molinaro AM, Gauthier ML, Berman HK, Waldman F, Bennington J, et al. Biomarker expression and risk of subsequent tumors after initial ductal carcinoma in situ diagnosis. J Natl Cancer Inst. 2010;102:627–637. doi: 10.1093/jnci/djq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Espina V, Mariani BD, Gallagher RI, Tran K, Banks S, Wiedemann J, et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PLoS ONE. 2010;5:e10240. doi: 10.1371/journal.pone.0010240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ranganathan AC, Ojha S, Kourtidis A, Conklin DS, Aguirre-Ghiso JA. Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res. 2008;68:3260–3268. doi: 10.1158/0008-5472.CAN-07-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Partanen JI, Nieminen AI, Makela TP, Klefstrom J. Suppression of oncogenic properties of c-Myc by LKB1-controlled epithelial organization. Proc Natl Acad Sci USA. 2007;104:14694–14699. doi: 10.1073/pnas.0704677104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Partanen JI, Tervonen TA, Myllynen M, Lind E, Imai M, Katajisto P, et al. Tumor suppressor function of Liver kinase B1 (Lkb1) is linked to regulation of epithelial integrity. Proc Natl Acad Sci USA. 2012;109:E388–E397. doi: 10.1073/pnas.1120421109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung CT, Brugge JS. Outgrowth of single oncogene-expressing cells from suppressive epithelial environments. Nature. 2012;482:410–413. doi: 10.1038/nature10826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165–178. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 38.Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66:1702–1711. doi: 10.1158/0008-5472.CAN-05-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bobrovnikova-Marjon E, Hatzivassiliou G, Grigoriadou C, Romero M, Cavener DR, Thompson CB, et al. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc Natl Acad Sci USA. 2008;105:16314–16319. doi: 10.1073/pnas.0808517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, Graphs showing densitometric analysis of the ratio p-AMPK/AMPK and p-p70S6K/p70S6K of PERK+/+ (WT) and PERK−/− (PERK−/−) mouse embryonic fibroblast (MEFs) and MCF10A cells grown in adhered or suspended conditions. The analysis was determined using Image J software (MEFs n=3 and MCF10As n=5). B, MCF10A cells were grown in adhered conditions and treated with DMSO (C), 1 μM or 10 μM thapsigargin at the indicated time points (hours). Lysates were collected and IB with the indicated Abs. C, MCF10A cultures were collected from attached (A) or suspended (S) conditions after transfection with PERK2 or control (scRNA) siRNAs and treated with 10μM compound C. Cells were lysed and IB against the indicated antigens. D, Lysates from attached (A) or suspended 12h (S) βGal and PERKΔC MCF10A cells were IB with indicated Abs. E, HEK293T cells were transfected with control empty vector (EV) or p70S6K mutant Δ2-46 ΔCT104 (p70CA), lysates were collected after 48h and IB with the indicated Abs. F, Graphs showing percentage of LC3 positive cell on adhered Fv2E-PERK/GFP-LC3 or GFP-LC3 MCF10A cells transfected with empty vector for 48h and treated with or without AP or AICAR respectively for 4h. p values were determined by the t-test.