

Abstract
Activation-induced deoxycytidine deaminase (AID) and Apobec 3G (Apo3G) cause mutational diversity by initiating mutations on regions of single-stranded (ss) DNA. Expressed in B cells, AID deaminates C → U in actively transcribed immunoglobulin (Ig) variable and switch regions to initiate the somatic hypermutation (SHM) and class switch recombination (CSR) that are essential for antibody diversity. Apo3G expressed in T cells catalyzes C deaminations on reverse transcribed cDNA causing HIV-1 retroviral inactivation. When operating properly, AID- and Apo3G-initiated mutations boost human fitness. Yet, both enzymes are potentially powerful somatic cell “mutators”. Loss of regulated expression and proper genome targeting can cause human cancer. Here, we review well-established biological roles of AID and Apo3G. We provide a synopsis of AID partnering proteins during SHM and CSR, and describe how an Apo2 crystal structure provides “surrogate” insight for AID and Apo3G biochemical behavior. However, large gaps remain in our understanding of how dC deaminases search ssDNA to identify trinucleotide motifs to deaminate. We discuss two recent methods to analyze ssDNA scanning and deamination. Apo3G scanning and deamination is visualized in real-time using single-molecule FRET, and AID deamination efficiencies are determined with a random walk analysis. AID and Apo3G encounter many candidate deamination sites while scanning ssDNA. Generating mutational diversity is a principal aim of AID and an important ancillary property of Apo3G. Success seems likely to involve hit and miss deamination motif targeting, biased strongly toward miss.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-012-1212-1) contains supplementary material, which is available to authorized users.
Keywords: Activation-induced deoxycytidine deaminase (AID), Apo3G, Somatic hypermutation, ssDNA scanning, Ig diversity, HIV-1 inactivation
Introduction
A seemingly endless array of antigens ready to attack susceptible hosts engendered the development of powerful immune systems in mammals. To counteract foreign invasion, the immune system is able to detect and neutralize antigens using a repertoire of about a trillion immunoglobulin (Ig) antibody variants produced in B cells during a humoral immune response [1]. These vast numbers of Ig variants ensure that at least one antibody is available to recognize virtually any invading antigen. Antibody diversification is achieved by three processes: V(D)J recombination, somatic hypermutation (SHM), and class switch recombination (CSR). During V(D)J recombination variable (V), diverse (D) and joining (J) segments of the Ig gene randomly recombine to generate low affinity antibodies [2]. When an antigen is recognized, B cells undergo affinity maturation via SHM. SHM is characterized by an exceptionally high mutation rate of ~10−3 to 10−4/base pair/cell division within the V(D)J rearranged Ig genes (Fig. 1) [3, 4]. CSR is a unique recombination event in which the Ig heavy chain Cμ region (coding for an IgM antibody) is replaced with a downstream C region, Cγ, Cα, or Cε (Fig. 1) to produce IgG, IgA or IgE antibody isotypes [5].
Fig. 1.

AID-targeted deamination at V(D)J and S regions of Ig genes initiates SHM and CSR. Low affinity IgM-producing IgVH genes undergo SHM and CSR which require C → U deamination by AID at V(D)J and switch (S) regions, respectively. Transcription from the V promoter (P) is needed for AID access to V(D)J regions, whereas germline transcription from the I promoters is required for AID targeting to the donor switch (Sμ) and a downstream acceptor S (shown in this case Sε). Error-prone processing of G:U mispair by MMR, BER and replication introduces a huge number of mutations (~10−4 to 10−3 per base per generation) in the antigen-binding V(D)J region as well as the rejoining Sμ–Sε regions. CSR combines the V(D)J exon with one of the appropriate downstream constant C regions γ3, (γ1, γ2b, and γ2a not shown), ε or α, converting IgM or IgD to the other isotypes IgG3 (IgG1, IgG2b, and IgG2a not shown), IgE, or IgA. The graph on top shows a typical SHM profile. Mutations start ~150 bp downstream from the promoter (P) at the leader sequence (L) reaching a maximum frequency over the V(D)J coding exon decaying exponentially towards the 3′ end. Eμ is an intronic enhancer. E3′s are enhancers at 3′ regulatory regions (3′RR)
Activation-induced deoxycytidine deaminase (AID) is a B cell-specific protein required for both SHM and CSR [6, 7]. Upon antigen encounter, AID is upregulated and recruited to actively transcribed variable (V) and switch (S) regions of the Ig loci initiating SHM and CSR in activated germinal center (GC) B cells [4, 5]. In V regions, AID deaminates multiple cytosine (C) bases on single-stranded regions of DNA creating uracils that are then processed by error-prone replication, base excision repair (BER), and mismatch repair (MMR) to create the diversity seen in SHM (reviewed in [4, 8–10]). In CSR, deamination by AID initiates double-strand breaks in S regions forming substrates for non-homologous end-joining that transform IgM-producing cells into B cells that make the IgG, IgA, or IgE isotypes (reviewed in [5, 11]) (Fig. 1).
AID belongs to an APOBEC family of polynucleotide deaminases that can act on ssDNA or RNA substrates. For example, Apo1 is essential for regulation of apolipoprotein B expression through C → U deamination editing of its mRNA. APOBEC proteins (Apo3A-H) provide innate resistance against retroviruses and endogenous retroelements [12, 13], including Apo3G that restricts HIV-1 replication in the absence of the viral infectivity factor (vif) [14]. In T cells, Apo3G deaminates C residues on HIV-1 reverse transcribed cDNA (Fig. 2) [15].
Fig. 2.

Apo3G-dependent restriction of HIV-1. Apo3G is encapsidated into Vif-deficient (∆vif) HIV-1 virions. Upon infection, Apo3G enters the cytoplasm of the targeted T cell and deaminates multiple C on the HIV-1 reverse transcribed minus (−) cDNA strand creating a massive number of U residues. Synthesis of the (+) strand, using U as templates, introduces hypermutation in essential genes, effectively inactivating HIV-1 infectivity. Alternatively, abasic sites resulted from the removal of U by the cellular enzyme, uracil DNA glycosylase, could inhibit the synthesis of the HIV-1 (+) strand cDNA or could serve as substrates for apurinic-apyrimidinic endonucleases leading to degradation of the cDNA reverse transcribed intermediate
Although AID and Apo3G have different functions and targets in B and T immune cells, both enzymes exhibit processive behavior on ssDNA substrates. Processive behavior occurs when a single AID or Apo3G enzyme molecule (present as a monomer, dimer, or perhaps multimer) catalyzes multiple C → U deaminations on an ssDNA substrate prior to acting on a different substrate [16–20]. Since AID and Apo3G are essential proteins for adaptive and innate immunity, deciphering the biochemical properties that regulate their action during SHM, CSR, and restriction of HIV-1virus is essential.
There have been recent reviews in the literature placing human, mouse, chicken, and cell lines data on AID-induced mutations in broad biological and immunological perspective, see, e.g., [4, 9–11, 21, 22], and we will briefly summarize these. We will mainly concentrate on the action of AID and Apo3G on ssDNA, which involves scanning to locate deamination motifs and ensuing catalysis. There is a large amount of biochemical mechanistic information available on scanning and catalysis by enzymes that act on dsDNA, e.g., DNA glycosylases [23–29], and restriction endonucleases [30–33]. Far less is known about scanning and catalysis by enzymes that scan ssDNA such as the APOBEC family enzymes, AID and Apo3G.
In this review, we will discuss recent advances made in the areas of global AID targeting, which include the targeting of Ig V- and S-regions, and the inadvertent and potentially deleterious targeting of other regions of the B cell genome. We will also discuss the application of structural data from Apo2 to the functional properties of AID and Apo3G. However, our principal aim is to highlight the newly emerging field of ssDNA targeting and scanning enzymes, including the application of single-molecule microscopy to visualize the motion of APOBEC enzymes on ssDNA in real-time, and random walk modeling to analyze clonal deamination patterns and deamination efficiencies. Enzymes that scan dsDNA have evolved as high efficiency catalysts, designed to ensure the absence of mutations. We will address the possibility that enzymes that scan ssDNA have evolved as low efficiency catalysts, designed to ensure mutational diversity.
Targeting of AID to V and S regions of Ig
AID site-specific targeting in B cells
Under normal conditions, high levels of AID expression are tightly regulated and primarily localized to activated B cells in GCs [6]. Mice and humans that are deficient in AID fail to class switch and cannot produce high affinity antibodies [7, 34]. Although SHM and CSR are mechanistically distinct processes, both are initiated by AID-catalyzed C → U deamination at Ig V and S regions undergoing active transcription [16, 35–40]. Error-prone DNA repair (BER and MMR) or replication of G:U mispairs in V regions produce SHM [10] (Fig. 1). In contrast, repair in S regions leads to DNA double-stranded breaks required for CSR [5, 10, 41]. Direct AID action in GC B cells have been observed as high frequency G:C → A:T transition mutations in the V and S regions in mice that are deficient in both BER and MMR (ung −/− msh2 −/− or ung −/− msh6 −/−) [42–46]. Analysis of mutations in V regions revealed that AID targets WRC hot motifs (W = A/T, R = A/G) on both DNA strands, and that SHM mutations start at about 100–200 bp downstream from a transcription initiation site, gradually decaying to zero 1.5–2 kb downstream prior to reaching intronic enhancer and C regions [42, 43]. AID-initiated mutations in S regions begin ~150 bp after the S region germline promoter initiation site, continuing throughout the entire 2–12 kb S region sequence, deaminating both strands of DNA [44].
Although AID acts principally in SHM and CSR, it has also been reported to initiate “inadvertent” mutations at numerous non-Ig loci. Using ung −/− msh2 −/− mice, genomic sequencing analysis of 83 actively transcribed genes of GC B cells showed that approximately half the expressed genes were deaminated by AID [47]. For example, mutations of BCL6, CD79 [48], CD 95 [49], FAS, c-MYC, PIM1, and PAX5 [50–52] were detected. A genome-wide ChIP assay coupled with a deep sequencing (ChIP-Seq) assay revealed that AID associates with as many as 5,910 genes in activated B cells [53]. However, a recent study [54] has raised objections regarding the ChIP-Seq statistical analysis that disputes not only the association of AID with “non-targeted” genes but also disagrees with an earlier ChIP-Seq finding that AID is preferentially bound at stalled promoter sites [55]. In response to these objections, Yamane et al. [56] have provided a variety of counter arguments supporting AID “off-targeting”. These currently contentious issues regarding inadvertent action of AID, and more generally other Apobec deaminases, are both timely and obviously vital, since they impact directly on the potential for misdirected Apobecs to cause disease. And, although we do not have a “dog in this fight”, as intellectually engaged spectators we are rooting for a rapid and unambiguous resolution.
In GC B cells, AID targeting occurs in the 3′ regulatory regions of the Ig locus [57] that contains 11 switch-like sequences 0.5–1 kb long. AID can mutate this region with high frequency leading to “suicide recombination” between the Sμ switch and 3′ regulatory regions resulting in deletion of the entire C gene and elimination of B cell receptor expression [57]. Also, AID targeting to non-Ig sites has been implicated as a source of genomic instability in cancer cells where deamination leads to mutations that initiate double-stranded breaks in non-Ig genes that serve as substrates for translocation or gene deletion in mature B cell lymphomas [58]. AID has also been reported to act at 5′ regulatory regions of proto-oncogenes to alter their transcription activities potentially contributing to B cell malignant transformation [59]. In BL2 cells that overexpress AID, there are genes with short clustered repeat sequences containing WRC motifs, e.g., SNHG3, MALAT1, BCL7A, and CUX1, that have features common to V and S regions and are heavily mutated with frequencies comparable to SHM at IgV regions (2.2 × 10−4 to 6.1 × 10−4) [60]. Therefore, AID can act on non-Ig loci, yet, even allowing for the controversy mentioned in the previous paragraph, it is nonetheless clear that endogenous AID targeting during SHM and CSR is typically 10- to 100-fold more efficient at V and S regions compared to at non-Ig genes [47–49].
Transcription is required for AID targeting
AID is not active on dsDNA but deaminates actively transcribed DNA in vivo and in vitro [16, 36–40]. Transcription generates regions of ssDNA on the non-transcribed strand that can be targeted by AID. The relationship between active transcription and SHM was first observed in studies connecting the distribution of mutations in the V region with the Ig gene promoter [61–63]. Mutations in the IgV region always began ~150 bp downstream of the promoter [62, 63], ending about 1.5–2 kb further downstream, and stopping prior to reaching the C region regardless of which Jκ gene segment was rearranged [62, 64, 65]. Another critical finding came by inserting a promoter upstream of the C region. In this transgenic Igk construct, SHM occurs in the C region at similar levels as previously observed in V regions [66, 67]. When a VH promoter was moved 750 bp upstream of its normal location, the pattern of SHM shifted to non-Ig DNA that was inserted into the VH leader intron used to make the transgene [68]. Transcription levels of the Ig transgenes showed a strong correlation with levels of SHM [69–71]. Finally, a study examining the structure of IgVH regions in human B cell lines undergoing SHM identified multiple ssDNA patches averaging ~11 nt long on both DNA strands [72]. Active transcription in conjunction with DNA-associated proteins is required for the detection of ssDNA patches thus expanding the link between SHM and transcription [72]. Mutations are found on both strands of DNA in V regions with about equal frequency, so AID must gain access, possibly through bidirectional transcription [73], recruitment of an exosome complex to the transcription bubble [74], or through negative supercoiling that unwinds DNA during stalled transcription [75].
AID-initiated mutations of S regions for CSR also require active transcription. When primary B cells are stimulated with cytokines, germline transcripts are produced that originate from the Sμ promoter (I) and a corresponding acceptor S region promoter for each specific IgE, IgA, and IgG antibody isotype (Fig. 1) [5]. For example, the germline transcripts IgE, IgG2b, and IgG3 are correlated with induced switching of IgE, IgG2b, and IgG3, respectively [76–78]. When germline promoters are deleted, CSR is aborted [79, 80]. One unique feature of transcribed S-regions is their ability to create R-loops that form when RNA is transcribed from G-rich S regions to create stable RNA–DNA hybrids with the C-rich template strand leaving an exposed G-rich ssDNA [81, 82]. Although CSR is affected by the presence and location of R-loops in the Sμ region [81], the targeting of AID to S regions is not nearly as pronounced, as shown in ung −/− msh2 −/− mice that exhibit no bias for R-loop-forming regions [44], and CSR is still observed in mouse B cells when the Sγ1 locus is replaced by a Xenopus A:T-rich Sμ region [83].
cis- and trans-factors involved in the regulation of AID targeting
The large majority of transcribed genes in B cells have at most sporadic association with AID [53], with typically negative consequences, so factors other than transcription must be important for targeting AID to Ig loci. Since AID is targeted to particular regions of DNA during SHM and CSR (Fig. 1), experiments were designed to determine if a cis-DNA element within the V region is required. In studies using transgenic mice, substitution of the endogenous V sequence with unrelated DNA, e.g., neomycin-resistance or β-globin sequences, did not affect SHM in B cells [61, 84]. Consistent with the absence of a cis-targeting element within a V region, purified AID appears to target V- and non-V-DNA (e.g., lacZ) with similar efficiency in vitro [85]. There is no evidence that the presence of specific cis-acting elements are responsible for the recruitment of AID to S-regions [86, 87]. Nevertheless, the unique feature of multiple G-rich repetitive motifs could recruit a 5′-AGCT-3′ repeat-binding protein, known as the 14-3-3 adaptor protein, to attract AID through protein–protein interaction [88], and S region sequences could enhance AID access through the formation of DNA secondary structure, such as R-loops or G-quartets that contain transient ssDNA regions [89].
An examination of DNA regions surrounding Ig loci have shown that cis-acting elements, such as the intronic enhancer (iE) and the 3′ intronic enhancers (3′ E), are involved in both positive and negative regulation of SHM and CSR [61, 90–95]. The potential role of enhancers in AID targeting is difficult to dissect because transcription is required for AID-induced mutations so that any changes in cis-acting sequences that decrease transcription will also decrease AID targeting. Nevertheless, the presence of two or three E-box motifs (CAGGTG) within the Ig enhancers is sufficient to target AID to a nearby transcribed transgene in B cells without altering the rate of transcription [96, 97]. The E47 spliced isoform of the transcription factor E2A binds the E-box motif in the nuclear extract of hypermutating B cells [97]. Although conditional inactivation of E2A in transgenic mice did not have an effect on SHM and CSR, its overexpression enhanced AID targeting in chicken DT40 cells [98, 99]. The mechanism of E-box-mediated targeting to V and S regions is not clear, but the importance of E-box motifs in AID targeting is reinforced by their presence in “hyper”-mutated non-Ig genes in activated B cells in normal mice [47] and in T cell lymphomas in AID transgenic mice [52].
AID can target non-Ig genes, including itself [17, 39, 75, 85, 100, 101], and has similar ssDNA binding constants regardless of the presence of C [102, 103], suggesting that AID is not recruited to a specific Ig enhancer DNA sequence. Ig enhancers are likely used to recruit proteins that can interact with and target AID to V and S regions. Possible AID–interacting targeting partners include RNA polymerase II [104], transcription pausing factor Spt5, transcription repressor KAP1/Trim28 [105], UNG, and mismatch repair MSH2/MSH6 complex [106], RNA exosome subunits [74], a number of proteins involved in RNA processing CTNNBL1 [107], PTBP2 [108], SRSF1 [109], GANP [110], RPA [111], DNA-PKcs [112], and Protein Kinase A [113]. This impressive list in terms of length and breadth offers challenging opportunities for future biochemical studies.
In activated B cells, AID genome-wide occupancy is correlated with RNA pol II distribution [53]. AID interacts and colocalizes with the transcription pausing factor Spt5 [55] and RNA exosome complex [74]. These interactions suggest that AID could be recruited to the stalled transcription bubbles to deaminate both transcribed and non-transcribed strands [74]. The molecular mechanisms responsible for targeting AID to specific regions of the genome in B cells are not clear. Proteins, such as 14-3-3 [88], KAP1 [105], UNG, and the MSH2/MSH6 complex [106] were found to interact with AID specifically at S regions, and SRSF1 [109], and GANP [110] appear to target AID to V-regions. Chromatin structure at V and S regions, regulated through epigenetic modification of histones, can play an important role in the targeting of AID [114], since both positioning and stability of nucleosomes at IgV regions were shown to affect AID-initiated SHM patterns [115].
Apo3G targets HIV-1 in T cells
Apo3G is a deoxycytidine deaminase that restricts HIV-1 infection in T cells in the absence of the viral infectivity factor (vif), a protein that targets Apo3G for polyubiquitination and proteasomal degradation [14, 116]. Through interactions with the nucleopcapsid domain of the viral Gag protein and RNA from the HIV-1 virus and cell cytoplasm, Apo3G can be encapsulated into HIV-1Δvif virus particles and transported to a naive T cell [117–130]. Upon infection, Apo3G is released into the cytoplasm inhibiting HIV-1 replication by deaminating C → U throughout the viral minus (−) reverse transcribed cDNA (Fig. 2) [131–133]. The U-rich (−) cDNA is used as a template for (+) strand DNA synthesis, where A is inserted opposite U to generate potentially detrimental C → T mutations, especially in regions needed for HIV-1 replication [132, 134–136]. Although the U-rich viral cDNA could be degraded by the combined action of cellular uracil DNA glycosylases (UDG) and apurinic/apyrimidinic endonuclease [137], recent data argue against a role for UDG in viral restriction [138–140]. In cells that overexpress Apo3G, deamination-independent mechanisms for HIV-1 restriction have been observed, which could involve a blockage of reverse transcription, an inhibition of (+) strand DNA synthesis, or perhaps the elimination of proviral formation [141, 142]. However, non-catalytic viral restriction has only been observed to date when Apo3G is present at elevated levels in T cells. Deamination activity appears to be required for viral inactivation when Apo3G is expressed normally in T cells [143–145].
The deamination activity signatures of Apo3G and other APOBEC proteins have also been identified in locations outside their designated targets [146, 147]. Apo3G appears to be involved in hepatic metastasis of colorectal cancer [146], and the sequence analysis of the complete genomes of 21 breast cancers showed the presence of hypermutated regions where base substitutions were found almost exclusively at TpC dinucleotides, reflecting APOBEC deamination specificity [147].
Biochemical properties and structural features of AID and APOBEC proteins
Structural studies of deoxycytidine deaminases and HIGM-2 mutants
The APOBEC family consists of 11 members, all characterized by the presence of either one or two deaminase domains (Fig. 3a). Each domain contains a unique Zn2+ coordinating motif (X-Glu-X23-28-Pro-Cys-X2-4-Cys) [15, 148, 149]. There are four high-resolution crystal structures for APOBEC proteins, one of Apo2 (amino acids 41–224), whose structure was determined to be a rod-shaped tetramer (Fig. 3b), two for the catalytic C-terminal domain of Apo3G (Apo3G-CD2) (amino acids 197–380 or 191–384) (Fig. 3c), and one for Apo3C [150–153]. The structure of Apo3G-CD2 has also been solved using NMR [154–156]. A core structure, characteristic of APOBEC proteins, is preserved throughout the family, although there are differences between the Apo3G-CD2 structures (reviewed in [157, 158]). For example, the crystal structure of Apo3G-CD2 mutant (PDB number 3IR2) has a discontinuous β2 strand where residues L235 to R239 form a bulge that is not present in the native Apo3G-CD2 structure (PDB number 3E1U). Among the Apo3G-CD2 structures, there are also differences in the positions of active center loops 1 and 3 and the position of helix 1 [150, 152, 154–156]. These structural differences may arise from different mutations introduced into the crystallized protein that may influence the final folding of the CD2 domain or perhaps from different protein packing within the crystal. What is needed is a full-length crystal structure of native Apo3G.
Fig. 3.

Apobec protein domains, structures and sequence alignments. a AID and six other Apobec’s contain a single Zn2+ coordinating deoxycytidine/cytidine deaminase domain, whereas four other members are double-domain deaminases. The N-terminal domain (CD1) of Apo3G, Apo3F, and Apo3DE is inactive while the C-terminal domain (CD2) is catalytically active. b The crystal structure of Apo2 tetramer (Protein Data Bank 2NYT). Head-to-head interaction of two Apo2 dimers is mediated by tetramer interface amino acid residues of α-helix 6, active center loop1, and flexible loop 7. The Apo2 dimer is formed by combining two β2 strands to make one wide β-sheet structure (indicated by arrow). Each monomer is highlighted in a different color. The monomer active site residues C128, C131, and H98 (red) coordinate Zn2+ molecule (red). c The crystal structure of the Apo3G-CD2 (Protein Data Bank 3E1U). The α-helixes are marked in blue and β-strands in pink. The C288, C291, and H257 (red) coordinate Zn2+ molecule (red), whereas E259 (red) acts as a proton shuffler during the hydrolytic deamination reaction. d The AID model was generated using Apo3G-CD2 as a template. Active site residues H56, E58, C87, and C90 (red) are important for catalysis. H56, C87, and C90 are involved in coordinating the Zn ion (red sphere) while E58 is involved in proton shuffling. Predicted residues at the dimeric and tetrameric interfaces are indicated by arrows. e Alignment of AID, Apo3G-CD2, and Apo2. HIGM-2 base substitution mutations and C-terminal deletion mutations are indicated on the top. The amino acids from the protein active site necessary for catalysis are marked in red. The specificity loop is marked in red both in the Apo3G-CD2 structure and amino acid alignment
AID is notoriously difficult to obtain in high yields when expressed and purified from B cells and baculovirus-infected insect cells. Human AID can be isolated from Escherichia coli in high yield, but its specific activity is reduced by ~50-fold compared to baculovirus-expressed AID [159]. Once isolated from any source, AID has proven difficult to purify—it figuratively sticks to “everything”, possibly owing to the presence of a highly localized positive charge (+11) in its N-terminal region. Thus, not surprisingly, there is as yet no crystal structure for AID. Nevertheless, the high amino acid sequence homology within the APOBEC family (Fig. 3e) for the Apo2 and Apo3G-CD2 structures can be used to model (Fig. 3d), predict, and evaluate deamination activities for wild-type and mutant AID [103, 151].
In the crystal structures of Apo2 and Apo3G-CD2, a common β-sheet core, consisting of five β-strands (β1-5), surrounded by six α-helices (α1-6) was observed (Fig. 3b, c) [150, 151]. The structures overlap significantly. Differences in the two structures, such as the position and lengths of the active center loops 1 and 3, most likely reflect the difference in biochemical properties of the enzymes. The active site of Apo2 and Apo3G-CD2 contains a histidine and two cysteine residues involved in the coordination of a Zn molecule (Cys128, Cys131, His98 for Apo2 and Cys288, Cys291, His257 for Apo3G) (Fig. 3b, c). Catalysis for Apo3G depends on a water molecule serving as a hydrogen donor to a conserved glutamate residue (E259) that acts as a proton shuffler during the hydrolytic deamination reaction. No activity has been detected for Apo2, yet a water molecule is present in the structure, while a fourth Zn coordination bond provided by E60 replaces the water molecule in the inner monomers of the tetramer [151].
Mutations made to any of the conserved residues in the active site of AID (E58, H56, C87, or C90), produced a catalytically inactive enzyme [21, 103, 160, 161]. The motif specificity of APOBEC proteins is determined by 9–11 amino acids in loop 7, designated as the “specificity loop” (Fig. 3c, e). It is thought that the amino acids in the specificity loop interact with ssDNA through electrostatic, hydrophobic, or base stacking interactions [157]. The favored specificities for each APOBEC (e.g., WRC for AID and CCC for Apo3G) are specified by differences in specificity loops [150, 154, 162]. A swap of specificity loops for AID, Apo3G, and Apo3F resulted in a concomitant swap of deamination specificities [163, 164] that switched the specificity of the chimeric protein to that of the donor enzyme [163, 164]. The AID chimeric proteins in DT40 and mouse B cell lines were observed to depress CSR [164, 165], while concomitantly altering the V-gene mutation spectra and reducing the mutation frequency in complementarity-determining regions [164, 165]. Therefore, while not absolutely necessary, WRC motif targeting is nonetheless functionally important. The biological effects were less pronounced for chimeric Apo3G and Apo3F where the change in C deamination specificities did not prevent the restriction of HIV-1 infectivity [164].
Multiple forms of AID and Apo3G in aqueous solution
APOBEC proteins have been identified in solution as monomers, dimers, and higher order oligomers. The role of oligomerization on biological function has not been determined, although it has been suggested that dimerization of Apo3G is important for RNA-dependent incorporation of the enzyme into the HIV-1 budding virons [118, 120, 123, 126, 166]. Size exclusion chromatography has shown that human Apo2 protein in solution is present as a mix of dimers and tetramers [151]. The crystal structure of Apo2 was determined to be a rod-shaped tetramer with a unique butterfly-like shape formed by pairing two dimers in a head-to-head interaction via residues from h6, AC-loop1, and a flexible loop (Loop 7) connected to h4 on two inner monomers (Fig. 3b). The Apo2 dimer is formed by combining two β2 strands to make one wide β-sheet structure (Fig. 3b). Experimental studies using various analytical approaches show that AID is present in solution as a monomer [167], tetramer [161], or multimer (“pull-down” assay) [168]. Atomic force microscopy (AFM) showed that AID in the presence of ssDNA appears to be a monomer [167]. Nevertheless, the effect that oligomerization has on the activity of AID is not clear.
An informative result was obtained when amino acids corresponding to the Apo2 dimer (F46, Y48) and tetramer interfaces (R112, L113, N168) were mutated in AID (Fig. 3d), which resulted in a reduction or elimination of activity [103, 151]. Apo3G is also a mix of monomers, dimers, and higher order oligomers as shown by AFM; however, the composition of Apo3G dramatically changes in the presence of ssDNA and salt [19, 20, 169, 170]. The monomeric Apo3G F/W mutant (F126A/W127A) that interferes with head-to-head dimerization retains many of the salient biochemical properties observed in the native protein, such as enzyme-specific activity, high processivity, lengthy residence time on ssDNA, which can be approximately several minutes, and strong 3′ → 5′ deamination polarity [18–20, 169]. But there are many additional unknowns for Apo3G, such as scanning motion on ssDNA that might involve sliding, hopping, jumping, or intersegmental transfer [18, 29, 171–174], all of which may be amenable to analysis by single-molecule laser and atomic force microscopy techniques (see, e.g., [170, 174–176]). Given that AID and Apo3G as well as other APOBEC family members can likely exist in solution in a variety of oligomeric states, it will be important to characterize each oligomer form using physical biochemical techniques and to identify their biological roles.
Physiological and structural aspects of Hyper-IgM-2 (HIGM-2) syndrome
Hyper-IgM syndrome (HIGM) is a heterogeneous genetic immunodeficiency disorder characterized by normal or high serum levels of IgM and the absence of or decreased levels of IgG, IgA, and IgE antibodies [34, 177]. HIGM syndromes are identified by defects in factors important for proper antibody diversification [177], such as HIGM-1, the first HIGM syndrome discovered, which is an X-linked disorder caused by mutations in the gene encoding CD40 ligand (CD40L/CD154), a protein transiently expressed on the surface of activated T cells [178–182]. In HIGM-2 syndrome, all defects are caused by mutations in AID. The clinical features are well characterized with at least 39 different mutations discovered in the AID gene so far [5, 22, 34, 183–185]. Most mutations are autosomal recessive, where patients develop deficiencies in CSR and SHM and exhibit high susceptibility to bacterial infections mostly in the upper respiratory and digestive tracts [34, 186, 187].
A total of 23 different missense mutations have been identified located at 20 distinct loci (Fig. 3e) [103]. A few rare autosomal dominant HIGM-2 patients appear to have normal SHM but are defective in CSR [168, 188]. Two autosomal dominant HIGM-2 mutations result in C-terminal deletions lacking 9 and 17 amino acids, respectively (Fig. 3e) [168, 188]. The inability of the C-terminal deletion mutants to initiate CSR indicates that the C-terminal region is necessary for binding factors that target AID to the Ig switch regions [88, 106, 112]. AID mutants made in vitro with 18 or fewer C-terminal amino acids deleted are catalytically active and act with high processivity, with ΔC10 and ΔC15 having 2- to 3-fold higher specific activities than native AID [103, 189]. Higher activity of the C-terminal deletion mutants have been found to correlate with an increase in mutagenic activity at V regions in B cells [168, 189]. An in vitro biochemical assay showed that a deletion of 19 C-terminal amino acids of AID completely inactivates its deamination activity [103]. The structural model (Fig. 3d) shows that C-terminal amino-acids 165–180 form α-helix 6, suggesting that the entire helix 6 is required for the structural integrity of the core deaminase domain of AID [103]. AID “up-mutants” created in vitro by PCR mutagenesis [190] have mutations distributed over the entire gene resulting in increased specific activity. Mutations were found in residues that are implicated in AID/substrate interaction (e.g., T82I, K120R) and several that bring the sequence of AID closer to that of APOBEC3s (e.g., K34E and F115Y). Using a chicken DT40 B cell line, it was shown that AID up-mutants increased antibody diversification, but they also increased the frequency of chromosomal translocations, suggesting that AID specific activity may need to be tightly regulated in B cells.
Recently, the high-resolution crystal structures for Apo2 and Apo3G-CD2 have been used to predict how mutations in the conserved corresponding residues of AID impair enzyme function [103, 151]. The HIGM-2 mutations are spread over the entire AID gene (Fig. 3d, e) [34, 103, 168, 183]. Based on structural analysis, the HIGM-2 missense mutants were placed into three classes [103]. Mutants belonging to Class I, the catalysis class, have mutations in the active site of the enzyme (H56Y, E58K, C87R, C87S). As predicted, these mutants have no detectable activity, which is attributed to defects in Zn molecule coordination and proton shuffling necessary for C deamination [103, 151]. The Class II proteins contain eight mutants (R24W, S83P, S85N, A111E, R112C, R112H, L113P, R174S) that are suggested to alter interactions with ssDNA; these mutations are either on or near the predicted surface of AID. Two mutants from this class, R112C and R174S, are still catalytically active and bind ssDNA but with reduced affinity. Class III structural mutants (M6T, F11L, F15L, W80R, L98R, L106P, I136K, M139K, M139V, F151S, S43P) have mostly hydrophobic residues located inside the AID protein that might be necessary for proper folding of the enzyme. In summary, the structural analysis of AID mutants responsible for HIGM-2 indicates that not only is maintaining the structural integrity of AID important for proper substrate binding and deamination but it is also essential for immunological function.
Activity and Specificity of AID and Apo3G on ssDNA
AID deaminates C → U on ssDNA favoring 5′WRC (W = A/T, R = A/G) hot motifs [16, 191]. Preference for 5′WRC motifs, mimicking in vivo data, was first observed in an in vitro plaque assay where a lacZ reporter gene was inserted into circular M13mp2 DNA leaving a 365-nt ss gap [16]. After treatment with purified AID, deamination is detected as C → T mutations in the DNA of phage particles. Though 5′WRC hot motifs are favored, 5′SYC cold (S = A/G, Y = C/T) motifs are also deaminated by AID but with reduced (~5- to 10-fold) activity [16, 191]. The APOBEC protein, Apo3G also deaminates C → U on ssDNA, but favors 5′CCC target motifs with a preference for the 3′C, and its specific activity is strongly influenced by nucleotides surrounding the 5′CCC target motif [18, 133, 135, 192, 193]. The importance of surrounding nucleotides is illustrated by a 40-fold reduction in Apo3G specific activity when the extended 5′aaaCCCaaa motif is replaced by 5′tttCCCttt [20]. Neither AID nor Apo3G show activity on dsDNA or RNA substrates [18, 129, 194].
When acting on a model ssDNA substrate with two deamination motifs, AID deaminates both motifs with equal efficiency, whereas Apo3G deaminates the 5′ motif preferentially over the 3′ motif (Fig. 4a) [18–20, 159]. Mutational gradients consistent with the properties of Apo3G described above have been observed in the HIV-1 genome at two polypurine tracts that remain single-stranded on cDNA for an extended period of time [133, 192]. Directional deamination in vitro occurs in the absence of an external energy source (e.g., ATP or GTP) [18], and so deamination polarity cannot be caused by preferential motion of Apo3G in a 3′ → 5′ direction, but must instead result from a catalytic asymmetry determined by the structure of Apo3G. Apo3G contains two deaminase domains, a catalytically inactive N-region CD1 domain and a catalytically active C-region CD2 domain (Fig. 3a) [128].
Fig. 4.

Model for Apo3G deamination polarity. a A Fluorescein (F)-labeled 85-nt ssDNA containing 2 identical deamination motifs (AGC for AID, left and CCC for Apo3G, right) was incubated with AID or Apo3G. Following treatment with Uracil-DNA Glycosylase and hot alkali, single deaminations at the 5′ site (5′C) or the 3′ site (3′C) are detected as the appearance of the labeled 67-nt or 48-nt fragment, respectively. Processive deamination at both the 5′ and 3′ sites (5′C and 3′C) on the same substrate is detected as the presence of a 30-nt fragment. AID deaminates 5′ and 3′ target motifs with equal efficiency (left gel), whereas Apo3G deaminatates the 5′ target motif preferentially (right gel). b Proposed model for Apo3G deamination polarity. Apo3G can bind ssDNA, with equal probability, in active or inactive orientations. In the active orientation, the active domain CD2 (charge = −4.5) is positioned toward the 5′-end and CD1 (+11) is facing the 3′-end. In the inactive orientation, Apo3G residues responsible for substrate specificity are distant from the 5′-CCC target motif and away from zinc ion, thereby precluding catalysis. The inability of the CD2 domain to bind to 30 nt at the 3′end in the active orientation results in a “deamination dead zone”
A model proposed to explain favored 5′-deamination is that Apo3G can bind in two orientations, but that Apo3G is active mainly, perhaps even solely, when bound with CD2 facing the 5′-end of the ssDNA (Fig. 4b) [19, 176]. As a consequence, there should be a deamination “dead zone” when catalytically inactive CD1 binds at the 3′-end (Fig. 4b). A 3′ dead zone spanning ~30 nt is observed when Apo3G acts on ssDNA in vitro [19, 20, 176]. Binding with CD1 facing the 5′-end would allow unrestricted access of CD2 to the 3′-end, but in a catalytically inactive orientation (Fig. 4b). Consistent with this idea is that the 3′-C of the 5′-CCC-3′ motif is almost always deaminated, whereas the 5′C is never deaminated [20, 133]. Based on an X-ray structural analysis of CD2 [150], it was suggested that the 3′-C is properly positioned near the catalytic Zn hydroxylation site only when CD2 is facing the 3′-C of the 5′-CCC motif (Fig. 4b, see legend).
Processivity of AID and Apo3G
Biochemical studies of AID and Apo3G suggest that both are processive enzymes that can catalyze multiple C → U reactions on the same ssDNA substrate before dissociation [16–18, 195]. The first indication that AID acted processively came from results using the in vitro plaque assay discussed earlier, where conditions were set to limit M13mp2 DNA to a single encounter with AID [16]. When DNA from each mutant plaque was sequenced, many individual clones had multiple C → T mutations. In similar studies using Apo3G, processivity was also observed [176]. Recent studies, measuring AID activity on ssDNA containing multiple target motifs cloned within the gap region of M13 DNA, showed that the number of AID mutations increased linearly with time with a slow average rate of about 1 deamination every 46–63 s for hot spot target motifs [17]. In an assay using linear ssDNA containing two hot spot target motifs, reactions performed under enzyme single hit conditions to measure correlated deamination events, showed processive behavior for both AID and Apo3G (Fig. 4a) [18, 20, 159].
How might processivity be useful in vivo? Apo3G acting on HIV-1 cDNA, presumably while reverse transcription is taking place, can catalyze numerous C → U deaminations in variable locations. A wide diversity of mutations would reduce the ways in which HIV-1 could mutate itself to restore viability. And, as mentioned above, there are mutational gradients present in HIV-1 genome [131] that reflect the biochemical properties of Apo3G, high processivity, and catalytically asymmetry [18, 19].
In contrast to Apo3G, AID is seemingly tightly constrained to act within a transcription bubble. High processivity could ensure that AID remains physically bound to the bubble throughout the transcription process. We have shown that AID is able to track along with a moving transcription bubble in vitro, resulting in mutations concentrated on the non-transcribed ssDNA [17, 191]. During CSR, multiple mismatches could be occurring in switch regions to serve as foci for introducing dsDNA breaks. In contrast to CSR, multiple mismatches occurring during SHM may be likely to serve as targets for error-free BER or MMR [196], with one or perhaps a few unrepaired U·Gs responsible for generating IgV and S region mutations. There are in vivo data showing consecutively mutated IgV C and G sites attributed to AID processivity [196]. Ostensibly, one or perhaps a few mutations per cell division allow the selection of those cells making a higher affinity Ab, without accumulating inactivating mutations that would lead to a loss of surface Ig and ultimately to apoptosis [197]. Then, those B cells making higher affinity antibodies would undergo another round of mutation and selection.
Miss and hit targeting when AID and Apo3G act on single-stranded DNA
Studies of scanning and catalysis on ssDNA represent “virgin” territory. In contrast to dsDNA in solution, which has well-defined B, A, and Z forms and persistence lengths (~150 base pairs) in aqueous solution, ssDNA has no comparable structural properties. Nonetheless, despite the absence of spatially defined forms, it is important to begin to come to grips with the properties of ssDNA because these serve as APOBEC dC deaminase substrates. It could be argued that free ssDNA has little if any biological relevance. AID, for example, acts on transcribed DNA in the cell, and short regions of ssDNA are likely to be tightly constrained within a transcription bubble and by chromatin structure. On the other hand, performing rigorous biochemical studies on the simplest substrates to learn how APOBEC enzymes scan and deaminate unconstrained ssDNA is prerequisite to understanding their mechanisms at the most fundamental level, and will more than likely contribute to a deeper appreciation of how APOBEC-initiated mutational diversity occurs in vivo.
The biochemical mechanisms for dsDNA scanning are well understood. Enzymes that scan dsDNA typically search for rare targets on long stretches of DNA, perhaps one aberrant base in ~105–106 base pairs. Base excision repair enzymes, UDG and Ogg1, slide and/or hop along the DNA and, upon locating a U or oxidized G, cleave the glycosidic linkage with high efficiency (e.g., [24–29, 171]; e.g., ~70 % with UDG [26]. The situation is different for ssDNA scanning enzymes. Unlike UDG or Ogg1, where high resolution glycosylase-DNA co-crystal structures depict a target base flipped out of the double helix into a glycosidic bond cleavage site [198, 199], there are no Apobec–DNA cocrystal structures showing the orientation of a target C motif in relation to the Apobec catalytic Zn ion hydroxylation site to guide us [19, 150].
Diverse deaminations by AID are needed for immunoglobulin maturation or by Apo3G for HIV-1 retroviral inactivation (Apo3G). Both enzymes encounter numerous trinucleotide target motifs, including favored WRC motifs for AID and YCC motifs for Apo3G. The diversity observed would seem to imply that AID and Apo3G targeting is a “miss and hit” process, with far stronger emphasis on “miss”. But what are the deamination efficiencies and how might they be determined?
An analysis of AID scanning and deamination of ssDNA using a one-dimensional random walk model
Distributions of AID-catalyzed deaminations have been measured in reporter cassettes composed of arbitrary types of motifs, e.g., two different hot spot motifs arranged in alternating patterns as (AACAGC)n, imbedded in an M13mp2-lacZ reporter assay [17] (described above in the “Processivity of AID and Apo3G” subsection). Hundreds of M13 mutant phage clones were sequenced to determine the distributions of C → T mutations (originating from C → U deaminations)—a subset of mutant clones is used to illustrate the haphazard behavior of AID (Fig. 5a). The mutational patterns contain individual mutations (singletons) and mutational clusters (doublets, triplets, etc.). The objective is to deduce a scanning mechanism for AID using a one-dimensional random walk model to simulate the clonal data [17]. The computer-simulated clones were compared with the data based on attaining a best match for the numbers of mutational singletons, doublets, triplets, and so on, for the ensemble of individual clones, and for the distances between mutated motifs.
Fig. 5.

Stochastic AID-catalyzed deamination. a Diverse AID deamination patterns on individual substrates containing 30 AAC (red dot) and AGC (orange dot) hot deamination motifs. AID deamination at the target motifs is shown as T [17]. The representative clones contain 2–10 mutations, distributed as singletons or clusters (doublets, triplets, etc.). b Random walk model depicting AID scanning and catalyzing inefficient deamination of C to U on ssDNA [17]. AID binds to ssDNA randomly and slides in both direction, while catalyzing C deaminations processively. The sliding/hopping distance is determined by a geometric distribution. A good fit of the model to the data occurs when AID slides/hops for average distance of 10 motifs (30 nt), but only rarely deaminates C to U, at about 3 % efficiency for even “hottest” AAC motifs, thus ensuring mutational diversity [17]
A deamination efficiency of 1–7 % provided an excellent fit to the data [17]. A model-independent experimental measurement placed an upper bound of 10 % (Fig. 5b) on the efficiency of AID-catalyzed deamination during transcription of dsDNA by T7 RNA polymerase in vitro [17]. The inefficient catalytic action when encountering its favored motifs suggests that AID is just barely an enzyme, which seems ideal for ensuring IgV hypermutational diversity. The “mechanism” for 1D scanning involves AID moving in either direction, by sliding or making short hops, with an excursion length determined by a geometric distribution (i.e., the discrete statistical analog of an exponential distribution). This means that AID tends to scan ssDNA in short slides or hops, over an average distance of roughly 30 nt (10 motifs), giving a good fit to the data [17]. The low deamination efficiency ensures that most deaminations are isolated. Clusters of deaminations arise from AID revisiting the same motifs over and over again. An animation depicting a model simulation originally published in [17] is included as Supplemental material.
During transcription of dsDNA, AID binds presumably to the non-transcribed strand within a small (~9 nt) transcription bubble. Deamination clusters (doublets and triplets) are observed during transcription by T7 RNA pol in the presence of AID in vitro [17]. These short clusters could arise from constrained back and forth scanning of AID within stalled transcription bubbles, favoring deamination of closely spaced motifs [17, 200]. The clustered mutations found to occur in Ig V- and S-regions in B cells [196] may reflect the processive action of AID targeted at transcriptional stall sites with V and S. If either the sequence context of the V regions or some epigenetic change were to increase the likelihood of entry or activation of AID in the complementarity determining regions, then locally restricted scanning would enrich for mutations in the parts of the antibody that contact antigen.
Watching as Apo3G scans and deaminates ssDNA in real-time
Single-molecule (SM) techniques are in vogue to study protein–DNA interaction dynamics (reviewed in [174, 201–203]). Total internal reflectance microscopy (TIRF) smFRET was recently used to visualize Apo3G scanning and catalysis on ssDNA [176] (Fig. 6a). By placing a fluorescent donor dye at the 5′ or 3′ end of ssDNA attached to a microscope slide, with an acceptor dye affixed to Apo3G free in solution and thus able to bind to the DNA, the motion of Apo3G relative to one end of the DNA can be observed by the increase or decrease in Förster (fluorescence) resonance energy transfer as the enzyme moves closer to or further from the labeled DNA end (Fig. 6, Scanning trajectories). Apo3G scans the entire length of ssDNA in slides or short hops, and also contracts (i.e., bends) ssDNA while scanning [176], which may serve to reposition the enzyme on ssDNA, perhaps by a longer hopping or intersegmental transfer process [18, 19, 29, 169, 172].
Fig. 6.
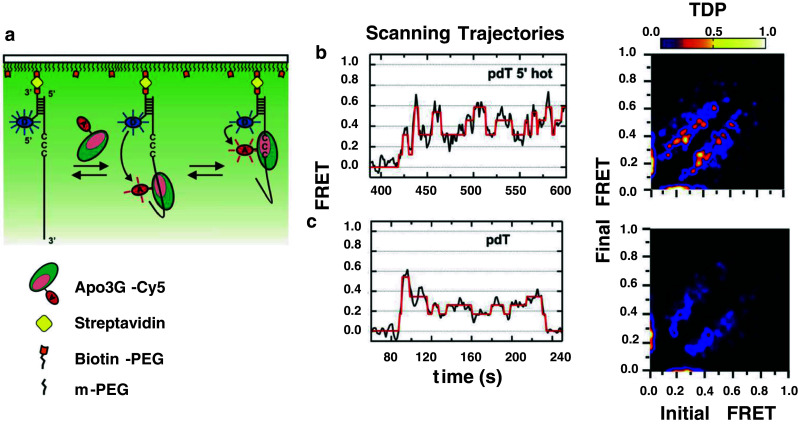
Single-molecule microscopy to visualize Apo3G scanning on ssDNA. a Total internal reflectance fluorescence (TIRF) microscope set up for analyzing Apo3G motion along ssDNA. ssDNA (92 nt) is annealed to a Cy3-labeled (D donor), surface immobilized anchor DNA. Binding and scanning of Cy5-labeled (A acceptor) Apo3G results in FRET changes. b, c The representative smFRET Scanning Trajectories for poly dT substrate with a 5′ hot CCC motif (pdT 5′ hot) or poly dT without C (pdT) show FRET traces (black) with a hidden Markov Model fit (red). Apo3G motion toward 5′- and 3′-directions is observed as increases and decreases in FRET, respectively. The transition density plot (TDP) showing the relative frequency of FRET transitions observed between initial and final FRET states (low frequency = blue, high frequency = yellow). Movements toward the 5′- and 3′-ends appear as peaks above and below the diagonal, respectively. The transitions are symmetric in both directions, indicating that Apo3G scans ssDNA without directional preference. The presence of bright yellow spots in the TDP plot for the pdT 5′ hot motif, but not for pdT, arise from a large excess of FRET transitions caused by quasi-localized scanning, i.e., “hovering”, of Apo3G in the vicinity of the 5′ hot motif [176]
Bulk solution studies with Apo3G and AID provide deamination spectra and clonal deamination patterns ([16–18, 176]; see, e.g., Fig. 5a), along with ssDNA binding constants and lifetimes (i.e., dissociation-rates) [19, 20, 176]. The smFRET data provide some of the same information, enabling one to compare bulk with SM protein–DNA interactions. Interaction lifetimes measured in solution showed that Apo3G binds to ssDNA in two modes, half as short binders (<30 s), half as long binders (30 s–10 min) [19, 20, 176]. The SM data showed the same ~50:50 distribution of short- and long-binders; for the scanning trajectory shown in Fig. 6b, Apo3G moves back and forth on the DNA while remaining bound ≥3 min.
An SM analysis can offer surprising and important new insights that cannot be provided by bulk solution measurements. A transition density plot (TDP) extracts the data from individual scanning trajectories (typically 100 or more trajectories) enabling the combined data to be analyzed with a hidden Markov model [204]. The TDP (Fig. 6b, c) contains symmetrical oblong lobes showing that Apo3G moves from high-to-low and low-to-high FRET states with equal probabilities. Although Apo3G favors deamination in a 3′ → 5′ direction [18–20, 159] (Fig. 4a), the enzyme moves equally in both directions, as it must in the absence of an energy source.
An unexpected observation is the obvious presence of relatively intense bright spots within the TDP lobes at a FRET value of ~0.4, corresponding to the 5′ location of the CCC hot motif [176] (Fig. 6b, TDP). These brighter spots arise from a large excess of FRET transitions caused by quasi-localized scanning, i.e., “hovering”, of Apo3G in the vicinity of the 5′ hot motif. Notably, hovering is not observed in the absence of a CCC motif (Fig. 6c, TDP). TDPs reported for 5′ cold motifs and 3′ hot motifs showed that hovering occurs with 5′ hot >5′ cold >3′ hot [176]. Thus, the time spent by Apo3G in different locations on the DNA corresponds to the polarity favoring deamination toward the 5′-direction, with the polarity “strength” able to override the hot over cold motif preference. Apo3G-catalyzed deaminations can be detected by including Cy5-labeled Pfu DNA polymerase in the reaction in the presence of unlabeled Apo3G. Pfu pol has the remarkably useful property of binding avidly and selectively to U on ssDNA [205, 206]. Therefore, upon Apo3G-catalyzed C → U deamination, Pfu pol binds rapidly to U and FRETs with Cy3-labeled ssDNA. The SM data show that deaminations occur with 5′ hot > 5′ cold > 3′ hot > dead zone hot [176], in accord with the asymmetric binding model (Fig. 4b).
Glancing ahead
AID functions in conjunction with proteins involved in MMR, BER, and perhaps most importantly factors participating in transcription. Steady progress has been made in identifying proteins needed by AID to perform its programmed roles in antibody diversity, the deamination of C → U in Ig V- and S-regions that initiate SHM and CSR [16, 35–40]. Recent efforts have focused on an important non-programmed property of AID, its unfortunate ability to deaminate C at many non-Ig genetic loci, resulting in human disease. To investigate the regulation and targeting of programmed and non-programmed AID–protein interactions will likely require some type of cell-free human or mouse RNA pol II transcription assay.
Although the T7 RNA pol transcription model was useful in showing that AID is biochemically active on a moving transcription bubble [16, 37, 40, 200], it is probably unlikely to be effective in studying AID targeting. If luck would have it, and AID were to act principally at transcription stall sites in vivo, then maybe a biochemical model using a transcription bubble substrate might suffice for some “simple” studies using human RNA Pol II and Spt5, associated with a stalled Pol II, but surely not for studies on proteins that differentially target AID to Ig and non-Ig actively transcribed regions, or for proteins that may prevent AID from binding to actively transcribed Ig constant regions. But it is important to keep in mind an even more immediate roadblock: active AID itself is difficult to isolate in high purity and yield. Not only that, but AID can exist as monomers, dimers, and perhaps larger oligomers in solution [167, 168], and while AID is thought to function as a dimer in vivo [168], it is just not clear what AID is structurally. What seems clear, however, is that the integrity of every region of AID is required for its biological function. A total of 23 missense mutants have been identified in hyper IgM-2 syndrome, and although this is an exceedingly rare human disease, these single amino acid changes span virtually the entire AID gene, each having a marked biological consequence [22, 34, 103].
The APOBEC family of proteins contains 11 members, and here we have focused on AID and Apo3G, which have received the most biological and biochemical attention. These two dC deaminases offer a study in contrasts and similarities. AID and Apo3G bind to ssDNA for a remarkably long time, remaining bound often for several minutes during which multiple deaminations occur [16–18, 176]. The ability to perform multiple deaminations makes sense for Apo3G by providing an effective way to inactivate HIV-1, or to prevent retroviral transposition. Stable binding and high processivity would also be useful for AID, by keeping it bound to a moving transcription bubble or to cause multiple mutations within a stalled bubble. Multiple deaminations could serve as foci for double-strand breaks in Ig switch regions during CSR, but a potential role for multiple deaminations during SHM is not evident because hypermutating B cells typically accumulate just one or at most a few new IgV mutations during a single cell division in vivo [196].
A crystal structure for AID or for a two-domain Apo3G would be a “holy grail”, and a co-crystal containing an ssDNA substrate an even holier grail. What we have instead is a crystal structure of Apo2, a “Monty Python” holy grail. Based on its basic structural features, including a zinc hydroxylation site, Apo2 should be functional catalytically, but it is not, having no measurable C deamination activity on DNA or RNA. Even so, Apo2 shares strong homology with AID and Apo3G. By serving as a surrogate for AID and Apo3G, until now Apo2 has played a pivotal role in relating structure to biochemical function. Mutations made in AID and Apo3G, based on the “equivalent” sites in Apo2, have allowed one to make accurate predictions about biochemical activity and protein structure. The structural predictions may in the future lead to a breakthrough strategy to obtain crystals for both proteins, but especially for Apo3G. Full-length Apo3G is obtained in high yield when expressed in insect cells and has been purified to apparent homogeneity with excellent recovery [18–20]. The problem is that native Apo3G is present in aqueous solution as an equilibrium mix of monomers, dimers, and large oligomers, not an ideal way to try to grow crystals [20]. However, one can obtain a homogeneous, catalytically active Apo3G monomer by mutating Apo3G amino acid residues corresponding to the dimer and tetramer interfaces predicted from the Apo2 structure [19]. Whether or not an Apo2 surrogate strategy will lead to an Apo3G monomer mutant crystal structure in the near-term is anyone’s guess, but it is surely a way forward.
As a “purely intellectual” exercise, an investigation of enzymes that scan ssDNA is new. The use of laser-based single-molecule microscopy may prove especially valuable since it is possible to watch as the enzymes move along the DNA, no longer a novelty for dsDNA scanning enzymes, but as of now a novelty for ssDNA scanning. An ssDNA scanning experiment with Apo3G provided the unexpected mechanistic finding that enzyme hovering occurs proximal to a deamination motif, dependent on the motif sequence and location [176]. This type of quasi-localized scanning motion could only have been obtained at single-molecule resolution. Although back-and-forth scanning along the ssDNA is readily observed, the current resolution times (~30 ms) are too “slow” to distinguish between continuous sliding and short hopping movements, but that is soon likely to change.
As another intellectual exercise, a recent one-dimensional random walk model to analyze clonal deamination data for individual and clustered deaminations enables one to derive scanning distances and deamination efficiencies for AID [17]. Scanning distances predicted by the model can be either verified or disproved using single-molecule microscopy, which joins the theoretical modeling to the single-molecule microscopy. But of course, these are not solely interesting intellectual exercises but do have biological meaning. The stochastic modeling of the deamination patterns provide pretty solid evidence that AID is an extremely poor catalyst—when encountering a favored hot motif it catalyzes C deamination less than 1 out of 10 times. That has profound biological significance because deamination inefficiency breeds mutational variety, a hallmark of antibody diversity.
We conclude by mentioning an entirely new biological role for AID in DNA demethylation. DNA demethylation in mammals occurs after fertilization in preimplantation embryos and during development of primordial germ cells (PGCs) [207, 208]. The loss of methylated C (meC) can either occur by a passive mechanism, when DNA is replicated in the absence of maintenance by methyltransferases, or by an active mechanism that culminates in the removal of meC in the absence of replication [207, 208]. Biochemical studies showed that, beyond its mutagenic role at unmethylated Cs, AID could also deaminate meC → T [194, 209, 210], and studies in zebrafish embryos showed that AID-dependent demethylation could occur in exogenous and endogenous meC [211].
The observation that AID is located in the genome close to pluripotency genes, such as NANOG, reinforced the idea that it might be involved in early development [209]. Although AID-deficient mice and humans have no obvious developmental defects, and they can give rise to apparently normal offspring [7, 34], changes in litter size and abnormal demethylation of germ cells suggest an epigenetic role for AID during the first stages of development [212]. Demethylation of the promoters of OCT4 and NANOG during reprogramming of differentiated cells towards pluripotency requires AID [213], and it has been postulated that AID’s epigenetic role might play a deleterious role during tumorigenesis [214]. Thus, as expanded roles for AID in normal and malignant cells are discovered, it becomes even more important to understand how it works biochemically and to reconstitute these processes in vitro.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by National Institutes of Health grants ES13192 and GM21422 (M.F.G) and CA72649 and CA102705 (M.D.S.).
References
- 1.Milstein C. Diversity and the genesis of high affinity antibodies. Biochem Soc Trans. 1987;15:779–787. doi: 10.1042/bst0150779. [DOI] [PubMed] [Google Scholar]
- 2.Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu Rev Genet. 2011;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- 3.Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 4.Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, Scharff MD. The biochemistry of somatic hypermutation. Annu Rev Immunol. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- 5.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 7.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/S0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 8.Reynaud CA, Delbos F, Faili A, Gueranger Q, Aoufouchi S, Weill JC. Competitive repair pathways in immunoglobulin gene hypermutation. Philos Trans R Soc Lond B. 2009;364:613–619. doi: 10.1098/rstb.2008.0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teng G, Papavasiliou FN. Immunoglobulin somatic hypermutation. Annu Rev Genet. 2007;41:107–120. doi: 10.1146/annurev.genet.41.110306.130340. [DOI] [PubMed] [Google Scholar]
- 10.Chahwan R, Edelmann W, Scharff MD, Roa S. AIDing antibody diversity by error-prone mismatch repair. Semin Immunol. 2012;24:293–300. doi: 10.1016/j.smim.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol. 2012;12:517–531. doi: 10.1038/nri3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prochnow C, Bransteitter R, Chen XS. APOBEC deaminases-mutases with defensive roles for immunity. Sci China C Life Sci. 2009;52:893–902. doi: 10.1007/s11427-009-0133-1. [DOI] [PubMed] [Google Scholar]
- 13.Chelico L, Pham P, Petruska J, Goodman MF. Biochemical basis of immunological and retroviral responses to DNA-targeted cytosine deamination by activation-induced cytidine deaminase and APOBEC3G. J Biol Chem. 2009;284:27761–27765. doi: 10.1074/jbc.R109.052449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 15.Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- 16.Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424:103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- 17.Pham P, Calabrese P, Park SJ, Goodman MF. Analysis of a single-stranded DNA-scanning process in which activation-induced deoxycytidine deaminase (AID) deaminates C to U haphazardly and inefficiently to ensure mutational diversity. J Biol Chem. 2011;286:24931–24942. doi: 10.1074/jbc.M111.241208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chelico L, Pham P, Calabrese P, Goodman MF. APOBEC3G DNA deaminase acts processively 3′ → 5′ on single-stranded DNA. Nat Struct Mol Biol. 2006;13:392–399. doi: 10.1038/nsmb1086. [DOI] [PubMed] [Google Scholar]
- 19.Chelico L, Prochnow C, Erie DA, Chen XS, Goodman MF. Structural model for deoxycytidine deamination mechanisms of the HIV-1 inactivation enzyme APOBEC3G. J Biol Chem. 2010;285:16195–16205. doi: 10.1074/jbc.M110.107987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chelico L, Sacho EJ, Erie DA, Goodman MF. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J Biol Chem. 2008;283:13780–13791. doi: 10.1074/jbc.M801004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conticello SG, Langlois MA, Yang Z, Neuberger MS. DNA deamination in immunity: aID in the context of its APOBEC relatives. Adv Immunol. 2007;94:37–73. doi: 10.1016/S0065-2776(06)94002-4. [DOI] [PubMed] [Google Scholar]
- 22.Durandy A. Immunoglobulin class switch recombination: study through human natural mutants. Philos Trans R Soc Lond B. 2009;364:577–582. doi: 10.1098/rstb.2008.0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorman J, Chowdhury A, Surtees JA, Shimada J, Reichman DR, Alani E, Greene EC. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol Cell. 2007;28:359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blainey PC, Luo G, Kou SC, Mangel WF, Verdine GL, Bagchi B, Xie XS. Nonspecifically bound proteins spin while diffusing along DNA. Nat Struct Mol Biol. 2009;16:1224–1229. doi: 10.1038/nsmb.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc Natl Acad Sci USA. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porecha RH, Stivers JT. Uracil DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proc Natl Acad Sci USA. 2008;105:10791–10796. doi: 10.1073/pnas.0801612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennett SE, Sanderson RJ, Mosbaugh DW. Processivity of Escherichia coli and rat liver mitochondrial uracil-DNA glycosylase is affected by NaCl concentration. Biochemistry. 1995;34:6109–6119. doi: 10.1021/bi00018a014. [DOI] [PubMed] [Google Scholar]
- 28.Schonhoft JD, Stivers JT. Timing facilitated site transfer of an enzyme on DNA. Nat Chem Biol. 2012;8:205–210. doi: 10.1038/nchembio.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedman JI, Stivers JT. Detection of damaged DNA bases by DNA glycosylase enzymes. Biochemistry. 2010;49:4957–4967. doi: 10.1021/bi100593a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeltsch A, Pingoud A. Kinetic characterization of linear diffusion of the restriction endonuclease EcoRV on DNA. Biochemistry. 1998;37:2160–2169. doi: 10.1021/bi9719206. [DOI] [PubMed] [Google Scholar]
- 31.Gowers DM, Wilson GG, Halford SE. Measurement of the contributions of 1D and 3D pathways to the translocation of a protein along DNA. Proc Natl Acad Sci USA. 2005;102:15883–15888. doi: 10.1073/pnas.0505378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonnet I, et al. Sliding and jumping of single EcoRV restriction enzymes on non-cognate DNA. Nucleic Acids Res. 2008;36:4118–4127. doi: 10.1093/nar/gkn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van den Broek B, Lomholt MA, Kalisch S-MJ, Metzler R, Wiuite GJL. How DNA coiling enhances target localization by proteins. Proc Natl Acad Sci USA. 2008;105:15738–15742. doi: 10.1073/pnas.0804248105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Revy P, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/S0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 35.Storb U, et al. Molecular aspects of somatic hypermutation of immunoglobulin genes. Cold Spring Harb Symp Quant Biol. 1999;64:227–234. doi: 10.1101/sqb.1999.64.227. [DOI] [PubMed] [Google Scholar]
- 36.Yoshikawa K, Okazaki IM, Eto T, Kinoshita K, Muramatsu M, Nagaoka H, Honjo T. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 2002;296:2033–2036. doi: 10.1126/science.1071556. [DOI] [PubMed] [Google Scholar]
- 37.Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- 38.Sohail A, Klapacz J, Samaranayake M, Ullah A, Bhagwat AS. Human activation-induced cytidine deaminase causes transcription-dependent, strand-biased C to U deaminations. Nucl Acids Res. 2003;31:2990–2994. doi: 10.1093/nar/gkg464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol. 2003;4:452–456. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- 40.Shen HM, Ratnam S, Storb U. Targeting of the activation-induced cytosine deaminase is strongly influenced by the sequence and structure of the targeted DNA. Mol Cell Biol. 2005;25:10815–10821. doi: 10.1128/MCB.25.24.10815-10821.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 42.Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 43.Longerich S, Tanaka A, Bozek G, Nicolae D, Storb U. The very 5′ end and the constant region of Ig genes are spared from somatic mutation because AID does not access these regions. J Exp Med. 2005;202:1443–1454. doi: 10.1084/jem.20051604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xue K, Rada C, Neuberger MS. The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2-/- ung-/- mice. J Exp Med. 2006;203:2085–2094. doi: 10.1084/jem.20061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Z, et al. Examination of Msh6- and Msh3-deficient mice in class switching reveals overlapping and distinct roles of mutS homologues in antibody diversification. J Exp Med. 2004;200:47–59. doi: 10.1084/jem.20040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen HM, Tanaka A, Bozek G, Nicolae D, Storb U. Somatic hypermutation and class switch recombination in Msh6(−/−)Ung(−/−) double-knockout mice. J Immunol. 2006;177:5386–5392. doi: 10.4049/jimmunol.177.8.5386. [DOI] [PubMed] [Google Scholar]
- 47.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 48.Gordon MS, Kanegai CM, Doerr JR, Wall R. Somatic hypermutation of the B cell receptor genes B29 (Igbeta, CD79b) and mb1 (Igalpha, CD79a) Proc Natl Acad Sci USA. 2003;100:4126–4131. doi: 10.1073/pnas.0735266100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muschen M, Re D, Jungnickel B, Diehl V, Rajewsky K, Kuppers R. Somatic mutation of the CD95 gene in human B cells as a side-effect of the germinal center reaction. J Exp Med. 2000;192:1833–1840. doi: 10.1084/jem.192.12.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pasqualucci L, et al. BCL-6 Mutations in Normal Germinal Center B Cells: evidence of Somatic Hypermutation Acting Outside Ig Loci. Proc Natl Acad Sci USA. 1998;95:11816–11821. doi: 10.1073/pnas.95.20.11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shen HM, Peters A, Baron B, Zhu X, Storb U. Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science. 1998;280:1750–1752. doi: 10.1126/science.280.5370.1750. [DOI] [PubMed] [Google Scholar]
- 52.Kotani A, Okazaki IM, Muramatsu M, Kinoshita K, Begum NA, Nakajima T, Saito H, Honjo T. A target selection of somatic hypermutations is regulated similarly between T and B cells upon activation-induced cytidine deaminase expression. Proc Natl Acad Sci USA. 2005;102:4506–4511. doi: 10.1073/pnas.0500830102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamane A, et al. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2011;12:62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hogenbirk MA, Velds A, Kerkhoven RM, Jacobs H. Reassessing genomic targeting of AID. Nat Immunol. 2012;13:797–798. doi: 10.1038/ni.2367. [DOI] [PubMed] [Google Scholar]
- 55.Pavri R, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamane A, Resch W, Nussezweig M, Casellas R. Reply to “Reassessing genomic targeting of AID”. Nat Immunol. 2012;13:798–800. doi: 10.1038/ni.2368. [DOI] [PubMed] [Google Scholar]
- 57.Peron S, et al. AID-driven deletion causes immunoglobulin heavy chain locus suicide recombination in B cells. Science. 2012;336:931–934. doi: 10.1126/science.1218692. [DOI] [PubMed] [Google Scholar]
- 58.Klein IA, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang Y, Soong TD, Wang L, Melnick AM, Elemento O. Genome-wide detection of genes targeted by non-Ig somatic hypermutation in lymphoma. PLoS ONE. 2012;7:e40332. doi: 10.1371/journal.pone.0040332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kato L, et al. Nonimmunoglobulin target loci of activation-induced cytidine deaminase (AID) share unique features with immunoglobulin genes. Proc Natl Acad Sci USA. 2012;109:2479–2484. doi: 10.1073/pnas.1120791109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Betz AG, Milstein C, Gonzalez-Fernandez A, Pannell R, Larson T, Neuberger MS. Elements regulating somatic hypermutation of an immunoglobulin kappa gene: critical role for the intron enhancer/matrix attachment region. Cell. 1994;77:239–248. doi: 10.1016/0092-8674(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 62.Lebecque SG, Gearhart PJ. Boundaries of somatic mutation in rearranged immunoglobulin genes: 5′ boundary is near the promoter, and 3′ boundary is approximately 1 kb from V(D)J gene. J Exp Med. 1990;172:1717–1727. doi: 10.1084/jem.172.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rada C, Gonzalez-Fernandez A, Jarvis JM, Milstein C. The 5′ boundary of somatic hypermutation in a V kappa gene is in the leader intron. Eur J Immunol. 1994;24:1453–1457. doi: 10.1002/eji.1830240632. [DOI] [PubMed] [Google Scholar]
- 64.Hackett JJ, Rogerson BJ, O’Brien RL, Storb U. Analysis of somatic mutations in kappa transgenes. J Exp Med. 1990;172:131–137. doi: 10.1084/jem.172.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weber JS, Berry J, Manser T, Claflin JL. Position of the rearranged V kappa and its 5′ flanking sequences determines the location of somatic mutations in the J kappa locus. J Immunol. 1991;146:3652–3655. [PubMed] [Google Scholar]
- 66.Storb U, Peters A, Klotz E, Kim N, Shen HM, Kage K, Rogerson B, Martin TE. Somatic hypermutation of immunoglobulin genes is linked to transcription. Curr Top Microbiol Immunol. 1998;229:11–19. doi: 10.1007/978-3-642-71984-4_2. [DOI] [PubMed] [Google Scholar]
- 67.Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4:57–65. doi: 10.1016/S1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- 68.Tumas-Brundage K, Manser T. The transcriptional promoter regulates hypermutation of the antibody heavy chain locus. J Exp Med. 1997;185:239–250. doi: 10.1084/jem.185.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goyenechea B, Klix N, Yélamos J, Williams GT, Riddell A, Neuberger MS, Milstein C. Cells highly expressing Igk transgenes show clonal recruitment of hypermutation: a role for both MAR and the enhancers. EMBO J. 1997;16:3987–3994. doi: 10.1093/emboj/16.13.3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fukita Y, Jacobs H, Rajewsky K. Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity. 1998;9:105–114. doi: 10.1016/S1074-7613(00)80592-0. [DOI] [PubMed] [Google Scholar]
- 71.Bachl J, Carlson C, Gray-Schopfer V, Dessing M, Olsson C. Increased transcription levels induce higher mutation rates in a hypermutating cell line. J Immunol. 2001;166:5051–5057. doi: 10.4049/jimmunol.166.8.5051. [DOI] [PubMed] [Google Scholar]
- 72.Ronai D, Iglesias-Ussel MD, Fan M, Li Z, Martin A, Scharff MD. Detection of chromatin-associated single-stranded DNA in regions targeted for somatic hypermutation. J Exp Med. 2007;204:181–190. doi: 10.1084/jem.20062032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perlot T, Li G, Alt FW. Antisense transcripts from immunoglobulin heavy-chain locus V(D)J and switch regions. Proc Natl Acad Sci USA. 2008;105:3843–3848. doi: 10.1073/pnas.0712291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Basu U, et al. The RNA Exosome Targets the AID Cytidine Deaminase to Both Strands of Transcribed Duplex DNA Substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shen HM, Storb U. Activation-induced cytidine deaminase (AID) can target both DNA strands when the DNA is supercoiled. Proc Natl Acad Sci USA. 2004;101:12997–13002. doi: 10.1073/pnas.0404974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shockett P, Stavnezer J. Effect of cytokines on switching to IgA and alpha germline transcripts in the B lymphoma I.29 mu. Transforming growth factor-beta activates transcription of the unrearranged C alpha gene. J Immunol. 1991;147:4374–4383. [PubMed] [Google Scholar]
- 77.Lutzker S, Rothman P, Pollock R, Coffman R, Alt FW. Mitogen- and IL-4-regulated expression of germ-line Ig gamma 2b transcripts: evidence for directed heavy chain class switching. Cell. 1988;53:177–184. doi: 10.1016/0092-8674(88)90379-0. [DOI] [PubMed] [Google Scholar]
- 78.Snapper CM, Paul WE. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science. 1987;236:944–947. doi: 10.1126/science.3107127. [DOI] [PubMed] [Google Scholar]
- 79.Jung S, Rajewsky K, Radbruch A. Shutdown of class switch recombination by deletion of a switch region control element. Science. 1993;259:984–987. doi: 10.1126/science.8438159. [DOI] [PubMed] [Google Scholar]
- 80.Zhang J, Bottaro A, Li S, Stewart V, Alt FW. A selective defect in IgG2b switching as a result of targeted mutation of the I gamma 2b promoter and exon. EMBO J. 1993;12:3529–3537. doi: 10.1002/j.1460-2075.1993.tb06027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang FT, Yu K, Balter BB, Selsing E, Oruc Z, Khamlichi AA, Hsieh CL, Lieber MR. Sequence dependence of chromosomal R-loops at the immunoglobulin heavy-chain Smu class switch region. Mol Cell Biol. 2007;27:5921–5932. doi: 10.1128/MCB.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol. 2003;4:442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- 83.Zarrin AA, Alt FW, Chaudhuri J, Stokes N, Kaushal D, Du Pasquier L, Tian M. An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nat Immunol. 2004;5:1275–1281. doi: 10.1038/ni1137. [DOI] [PubMed] [Google Scholar]
- 84.Yelamos J, et al. Targeting of non-Ig sequences in place of the V segment by somatic hypermutation. Nature. 1995;376:225–229. doi: 10.1038/376225a0. [DOI] [PubMed] [Google Scholar]
- 85.MacCarthy T, Kalis SL, Roa S, Pham P, Goodman MF, Scharff MD, Bergman A. V-region mutation in vitro, in vivo, and in silico reveal the importance of the enzymatic properties of AID and the sequence environment. Proc Natl Acad Sci USA. 2009;106:8629–8634. doi: 10.1073/pnas.0903803106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Luby TM, Schrader CE, Stavnezer J, Selsing E. The MU switch region tandem repeats are important, but not required, for antibody class switch recombination. J Exp Med. 2001;193:159–168. doi: 10.1084/jem.193.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Khamlichi AA, Glaudet F, Oruc Z, Denis V, Le Bert M, Cogne M. Immunoglobulin class-switch recombination in mice devoid of any Smicro tandem repeat. Blood. 2004;103:3828–3836. doi: 10.1182/blood-2003-10-3470. [DOI] [PubMed] [Google Scholar]
- 88.Xu Z, et al. 14–3-3 adaptor proteins recruit AID to 5′-AGCT-3′-rich switch regions for class switch recombination. Nat Struct Mol Biol. 2010;17:1124–1135. doi: 10.1038/nsmb.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Maizels N. Immunoglobulin gene diversification. Annu Rev Genet. 2005;39:23–46. doi: 10.1146/annurev.genet.39.073003.110544. [DOI] [PubMed] [Google Scholar]
- 90.Klotz EL, Storb U. Somatic hypermutation of a lambda 2 transgene under the control of the lambda enhancer or the heavy chain intron enhancer. J Immunol. 1996;157:4458–4463. [PubMed] [Google Scholar]
- 91.Bachl J, Olsson C, Chitkara N, Wabl M. The Ig mutator is dependent on the presence, position, and orientation of the large intron enhancer. Proc Natl Acad Sci USA. 1998;95:2396–2399. doi: 10.1073/pnas.95.5.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Komori A, Xu Z, Wu X, Zan H, Casali P. Biased dA/dT somatic hypermutation as regulated by the heavy chain intronic iEmu enhancer and 3′Ealpha enhancers in human lymphoblastoid B cells. Mol Immunol. 2006;43:1817–1826. doi: 10.1016/j.molimm.2005.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ronai D, Iglesias-Ussel MD, Fan M, Shulman MJ, Scharff MD. Complex regulation of somatic hypermutation by cis-acting sequences in the endogenous IgH gene in hybridoma cells. Proc Natl Acad Sci USA. 2005;102:11829–11834. doi: 10.1073/pnas.0505449102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Morvan CL, Pinaud E, Decourt C, Cuvillier A, Cogne M. The immunoglobulin heavy-chain locus hs3b and hs4 3′ enhancers are dispensable for VDJ assembly and somatic hypermutation. Blood. 2003;102:1421–1427. doi: 10.1182/blood-2002-12-3827. [DOI] [PubMed] [Google Scholar]
- 95.Pinaud E, Marquet M, Fiancette R, Peron S, Vincent-Fabert C, Denizot Y, Cogne M. The IgH locus 3′ regulatory region: pulling the strings from behind. Adv Immunol. 2011;110:27–70. doi: 10.1016/B978-0-12-387663-8.00002-8. [DOI] [PubMed] [Google Scholar]
- 96.Michael N, Shen HM, Longerich S, Kim N, Longacre A, Storb U. The E box motif CAGGTG enhances somatic hypermutation without enhancing transcription. Immunity. 2003;19:235–242. doi: 10.1016/S1074-7613(03)00204-8. [DOI] [PubMed] [Google Scholar]
- 97.Tanaka A, Shen HM, Ratnam S, Kodgire P, Storb U. Attracting AID to targets of somatic hypermutation. J Exp Med. 2010;207:405–415. doi: 10.1084/jem.20090821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Conlon TM, Meyer KB. The chicken Ig light chain 3′-enhancer is essential for gene expression and regulates gene conversion via the transcription factor E2A. Eur J Immunol. 2006;36:139–148. doi: 10.1002/eji.200535219. [DOI] [PubMed] [Google Scholar]
- 99.Schoetz U, Cervelli M, Wang YD, Fiedler P, Buerstedde JM. E2A expression stimulates Ig hypermutation. J Immunol. 2006;177:395–400. doi: 10.4049/jimmunol.177.1.395. [DOI] [PubMed] [Google Scholar]
- 100.Okazaki IM, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, Honjo T. Constitutive expression of AID leads to tumorigenesis. J Exp Med. 2003;197:1173–1181. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Martin A, Scharff MD. Somatic hypermutation of the AID transgene in B and non-B cells. Proc Natl Acad Sci USA. 2002;99:12304–12308. doi: 10.1073/pnas.192442899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Larijani M, Petrov AP, Kolenchenko O, Berru M, Krylov SN, Martin A. AID associates with single-stranded DNA with high affinity and a long complex half-life in a sequence-independent manner. Mol Cell Biol. 2007;27:20–30. doi: 10.1128/MCB.00824-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mu Y, Prochnow C, Pham P, Chen XS, Goodman MF. A structural basis for the biochemical behavior of activation-induced deoxycytidine deaminase class-switch recombination-defective hyper-IgM-2 mutants. J Biol Chem. 2012;287:28007–28016. doi: 10.1074/jbc.M112.370189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nambu Y, Sugai M, Gonda H, Lee C, Katakai T, Agata Y, Yokota Y, Shimizu A. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 2003;302:2137–2140. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- 105.Jeevan-Raj BP, et al. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med. 2011;208:1649–1660. doi: 10.1084/jem.20110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ranjit S, Khair L, Linehan EK, Ucher AJ, Chakrabarti M, Schrader CE, Stavnezer J. AID binds cooperatively with UNG and Msh2-Msh6 to Ig switch regions dependent upon the AID C terminus. J Immunol. 2011;187:2464–2475. doi: 10.4049/jimmunol.1101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Conticello SG, Ganesh K, Xue K, Lu M, Rada C, Neuberger MS. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol Cell. 2008;31:474–484. doi: 10.1016/j.molcel.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 108.Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol. 2010;12:160–166. doi: 10.1038/ni.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kanehiro Y, et al. Activation-induced cytidine deaminase (AID)-dependent somatic hypermutation requires a splice isoform of the serine/arginine-rich (SR) protein SRSF1. Proc Natl Acad Sci USA. 2012;109:1216–1221. doi: 10.1073/pnas.1120368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maeda K, Singh SK, Eda K, Kitabatake M, Pham P, Goodman MF, Sakaguchi N. GANP-mediated recruitment of activation-induced cytidine deaminase to cell nuclei and to immunoglobulin variable region DNA. J Biol Chem. 2010;285:23945–23953. doi: 10.1074/jbc.M110.131441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- 112.Wu X, Geraldes P, Platt JL, Cascalho M. The double-edged sword of activation-induced cytidine deaminase. J Immunol. 2005;174:934–941. doi: 10.4049/jimmunol.174.2.934. [DOI] [PubMed] [Google Scholar]
- 113.Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS, Chaudhuri J. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nat Immunol. 2009;10:420–426. doi: 10.1038/ni.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kuang FL, Luo Z, Scharff MD. H3 trimethyl K9 and H3 acetyl K9 chromatin modifications are associated with class switch recombination. Proc Natl Acad Sci USA. 2009;106:5288–5293. doi: 10.1073/pnas.0901368106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kodgire P, Mukkawar P, North JA, Poirier MG, Storb U. Nucleosome stability dramatically impacts the targeting of somatic hypermutation. Mol Cell Biol. 2012;32:2030–2040. doi: 10.1128/MCB.06722-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 117.Alce TM, Popik W. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J Biol Chem. 2004;279:34083–34086. doi: 10.1074/jbc.C400235200. [DOI] [PubMed] [Google Scholar]
- 118.Bach D, Peddi S, Mangeat B, Lakkaraju A, Strub K, Trono D. Characterization of APOBEC3G binding to 7SL RNA. Retrovirology. 2008;5:54. doi: 10.1186/1742-4690-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bogerd HP, Cullen BR. Single-stranded RNA facilitates nucleocapsid: aPOBEC3G complex formation. RNA. 2008;14:1228–1236. doi: 10.1261/rna.964708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Burnett A, Spearman P. APOBEC3G multimers are recruited to the plasma membrane for packaging into human immunodeficiency virus type 1 virus-like particles in an RNA-dependent process requiring the NC basic linker. J Virol. 2007;81:5000–5013. doi: 10.1128/JVI.02237-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cen S, Guo F, Niu M, Saadatmand J, Deflassieux J, Kleiman L. The interaction between HIV-1 Gag and APOBEC3G. J Biol Chem. 2004;279:33177–33184. doi: 10.1074/jbc.M402062200. [DOI] [PubMed] [Google Scholar]
- 122.Douaisi M, Dussart S, Courcoul M, Bessou G, Vigne R, Decroly E. HIV-1 and MLV Gag proteins are sufficient to recruit APOBEC3G into virus-like particles. Biochem Biophys Res Commun. 2004;321:566–573. doi: 10.1016/j.bbrc.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 123.Khan MA, et al. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J Virol. 2005;79:5870–5874. doi: 10.1128/JVI.79.9.5870-5874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Luo K, Liu B, Xiao Z, Yu Y, Yu X, Gorelick R, Yu XF. Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J Virol. 2004;78:11841–11852. doi: 10.1128/JVI.78.21.11841-11852.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schafer A, Bogerd HP, Cullen BR. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology. 2004;328:163–168. doi: 10.1016/j.virol.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 126.Strebel K, Khan MA. APOBEC3G encapsidation into HIV-1 virions: which RNA is it? Retrovirology. 2008;5:55. doi: 10.1186/1742-4690-5-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Svarovskaia ES, et al. Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J Biol Chem. 2004;279:35822–35828. doi: 10.1074/jbc.M405761200. [DOI] [PubMed] [Google Scholar]
- 128.Hache G, Liddament MT, Harris RS. The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J Biol Chem. 2005;280:10920–10924. doi: 10.1074/jbc.M500382200. [DOI] [PubMed] [Google Scholar]
- 129.Iwatani Y, Takeuchi H, Strebel K, Levin JG. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J Virol. 2006;80:5992–6002. doi: 10.1128/JVI.02680-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Navarro F, Bollman B, Chen H, Konig R, Yu Q, Chiles K, Landau NR. Complementary function of the two catalytic domains of APOBEC3G. Virology. 2005;333:374–386. doi: 10.1016/j.virol.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 131.Suspene R, et al. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 2004;32:2421–2429. doi: 10.1093/nar/gkh554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 133.Yu Q, et al. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol. 2004;11:435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- 134.Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the VIF protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 135.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/S0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 137.Yang B, Chen K, Zhang C, Huang S, Zhang H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J Biol Chem. 2007;282:11667–11675. doi: 10.1074/jbc.M606864200. [DOI] [PubMed] [Google Scholar]
- 138.Kaiser SM, Emerman M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J Virol. 2006;80:875–882. doi: 10.1128/JVI.80.2.875-882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Langlois MA, Neuberger MS. Human APOBEC3G can restrict retroviral infection in avian cells and acts independently of both UNG and SMUG1. J Virol. 2008;82:4660–4664. doi: 10.1128/JVI.02469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Guenzel CA, Herate C, Le Rouzic E, Maidou-Peindara P, Sadler HA, Rouyez MC, Mansky LM, Benichou S. Recruitment of the nuclear form of uracil DNA glycosylase into virus particles participates in the full infectivity of HIV-1. J Virol. 2012;86:2533–2544. doi: 10.1128/JVI.05163-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li XY, Guo F, Zhang L, Kleiman L, Cen S. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J Biol Chem. 2007;282:32065–32074. doi: 10.1074/jbc.M703423200. [DOI] [PubMed] [Google Scholar]
- 142.Iwatani Y, et al. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007;35:7096–7108. doi: 10.1093/nar/gkm750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Browne EP, Allers C, Landau NR. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology. 2009;387:313–321. doi: 10.1016/j.virol.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Miyagi E, Opi S, Takeuchi H, Khan M, Goila-Gaur R, Kao S, Strebel K. Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. J Virol. 2007;81:13346–13353. doi: 10.1128/JVI.01361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schumacher AJ, Hache G, Macduff DA, Brown WL, Harris RS. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD and HIV-1 restriction. J Virol. 2008;82(6):2652–2660. doi: 10.1128/JVI.02391-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ding Q, et al. APOBEC3G promotes liver metastasis in an orthotopic mouse model of colorectal cancer and predicts human hepatic metastasis. J Clin Invest. 2011;121:4526–4536. doi: 10.1172/JCI45008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nik-Zainal S, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- 149.Pham P, Bransteitter R, Goodman MF. Reward versus risk: DNA cytidine deaminases triggering immunity and disease. Biochemistry. 2005;44:2703–2715. doi: 10.1021/bi047481+. [DOI] [PubMed] [Google Scholar]
- 150.Holden LG, et al. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature. 2008;456:121–124. doi: 10.1038/nature07357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Prochnow C, Bransteitter R, Klein MG, Goodman MF, Chen XS. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature. 2007;445:447–451. doi: 10.1038/nature05492. [DOI] [PubMed] [Google Scholar]
- 152.Shandilya SM, et al. Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure. 2010;18:28–38. doi: 10.1016/j.str.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kitamura S, et al. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat Struct Mol Biol. 2012;19:1005–1010. doi: 10.1038/nsmb.2378. [DOI] [PubMed] [Google Scholar]
- 154.Chen KM, Harjes E, Gross PJ, Fahmy A, Lu Y, Shindo K, Harris RS, Matsuo H. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature. 2008;452:116–119. doi: 10.1038/nature06638. [DOI] [PubMed] [Google Scholar]
- 155.Furukawa A, et al. Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 2009;28:440–451. doi: 10.1038/emboj.2008.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Harjes E, et al. An extended structure of the APOBEC3G catalytic domain suggests a unique holoenzyme model. J Mol Biol. 2009;389:819–832. doi: 10.1016/j.jmb.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bransteitter R, Prochnow C, Chen XS. The current structural and functional understanding of APOBEC deaminases. Cell Mol Life Sci. 2009;66:3137–3147. doi: 10.1007/s00018-009-0070-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Autore F, Bergeron JR, Malim MH, Fraternali F, Huthoff H. Rationalisation of the differences between APOBEC3G structures from crystallography and NMR studies by molecular dynamics simulations. PLoS One. 2010;5:e11515. doi: 10.1371/journal.pone.0011515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chelico L, Pham P, Goodman MF. Stochastic properties of processive cytidine DNA deaminases AID and APOBEC3G. Philos Trans R Soc Lond B. 2009;364:583–593. doi: 10.1098/rstb.2008.0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Conticello SG. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008;9:229. doi: 10.1186/gb-2008-9-6-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med. 2003;197:1291–1296. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen KM, Martemyanova N, Lu Y, Shindo K, Matsuo H, Harris RS. Extensive mutagenesis experiments corroborate a structural model for the DNA deaminase domain of APOBEC3G. FEBS Lett. 2007;581:4761–4766. doi: 10.1016/j.febslet.2007.08.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kohli RM, Abrams SR, Gajula KS, Maul RW, Gearhart PJ, Stivers JT. A portable hotspot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. J Biol Chem. 2009;284:22898–22904. doi: 10.1074/jbc.M109.025536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kohli RM, et al. Local sequence targeting in the AID/APOBEC family differentially impacts retroviral restriction and antibody diversification. J Biol Chem. 2010;285:40956–40964. doi: 10.1074/jbc.M110.177402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang M, Rada C, Neuberger MS. Altering the spectrum of immunoglobulin V gene somatic hypermutation by modifying the active site of AID. J Exp Med. 2010;207:141–153. doi: 10.1084/jem.20092238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Iwatani Y, Rosen AE, Guo J, Musier-Forsyth K, Levin JG. Efficient initiation of HIV-1 reverse transcription in vitro. Requirement for RNA sequences downstream of the primer binding site abrogated by nucleocapsid protein-dependent primer-template interactions. J Biol Chem. 2003;278:14185–14195. doi: 10.1074/jbc.M211618200. [DOI] [PubMed] [Google Scholar]
- 167.Brar SS, Sacho EJ, Tessmer I, Croteau DL, Erie DA, Diaz M. Activation-induced deaminase, AID, is catalytically active as a monomer on single-stranded DNA. DNA Repair (Amst) 2008;7:77–87. doi: 10.1016/j.dnarep.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ta VT, et al. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat Immunol. 2003;4:843–848. doi: 10.1038/ni964. [DOI] [PubMed] [Google Scholar]
- 169.Feng Y, Chelico L. Intensity of deoxycytidine deamination of HIV-1 proviral DNA by the retroviral restriction factor APOBEC3G is mediated by the noncatalytic domain. J Biol Chem. 2011;286:11415–11426. doi: 10.1074/jbc.M110.199604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Shlyakhtenko LS, Lushnikov AY, Miyagi A, Li M, Harris RS, Lyubchenko YL. Nanoscale structure and dynamics of ABOBEC3G complexes with single-stranded DNA. Biochemistry. 2012;51:6432–6440. doi: 10.1021/bi300733d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–133. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 172.Nowarski R, Britan-Rosich E, Shiloach T, Kotler M. Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat Struct Mol Biol. 2008;15:1059–1066. doi: 10.1038/nsmb.1495. [DOI] [PubMed] [Google Scholar]
- 173.Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gorman J, Greene EC. Visualizing one-dimensional diffusion of proteins along DNA. Nat Struct Mol Biol. 2008;15:768–774. doi: 10.1038/nsmb.1441. [DOI] [PubMed] [Google Scholar]
- 175.Kad NM, Wang H, Kennedy GG, Warshaw DM, Van Houten B. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single-molecule imaging of quantum-dot-labeled proteins. Mol Cell. 2010;37:702–713. doi: 10.1016/j.molcel.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Senavirathne G, Jaszczur M, Auerbach PA, Upton TG, Chelico L, Goodman MF, Rueda D. Single-stranded DNA scanning and deamination by APOBEC3G cytidine deaminase at single-molecule resolution. J Biol Chem. 2012;287:15826–15835. doi: 10.1074/jbc.M112.342790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lee WI, Torgerson TR, Schumacher MJ, Yel L, Zhu Q, Ochs HD. Molecular analysis of a large cohort of patients with the hyper immunoglobulin M (IgM) syndrome. Blood. 2005;105:1881–1890. doi: 10.1182/blood-2003-12-4420. [DOI] [PubMed] [Google Scholar]
- 178.Razanajaona D, et al. Somatic mutations in human Ig variable genes correlate with a partially functional CD40-ligand in the X-linked hyper-IgM syndrome. J Immunol. 1996;157:1492–1498. [PubMed] [Google Scholar]
- 179.Aruffo A, et al. The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked hyper-IgM syndrome. Cell. 1993;72:291–300. doi: 10.1016/0092-8674(93)90668-G. [DOI] [PubMed] [Google Scholar]
- 180.DiSanto JP, Bonnefoy JY, Gauchat JF, Fischer A, de Saint Basile G. CD40 ligand mutations in x-linked immunodeficiency with hyper-IgM. Nature. 1993;361:541–543. doi: 10.1038/361541a0. [DOI] [PubMed] [Google Scholar]
- 181.Korthauer U, et al. Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. 1993;361:539–541. doi: 10.1038/361539a0. [DOI] [PubMed] [Google Scholar]
- 182.Fuleihan R, Ramesh N, Geha RS. Role of CD40-CD40-ligand interaction in Ig-isotype switching. Curr Opin Immunol. 1993;5:963–967. doi: 10.1016/0952-7915(93)90113-7. [DOI] [PubMed] [Google Scholar]
- 183.Durandy A, Peron S, Taubenheim N, Fischer A. Activation-induced cytidine deaminase: structure-function relationship as based on the study of mutants. Hum Mutat. 2006;27:1185–1191. doi: 10.1002/humu.20414. [DOI] [PubMed] [Google Scholar]
- 184.Durandy A, Taubenheim N, Peron S, Fischer A. Pathophysiology of B cell intrinsic immunoglobulin class switch recombination deficiencies. Adv Immunol. 2007;94:275–306. doi: 10.1016/S0065-2776(06)94009-7. [DOI] [PubMed] [Google Scholar]
- 185.Kracker S, Durandy A. Insights into the B cell specific process of immunoglobulin class switch recombination. Immunol Lett. 2011;138:97–103. doi: 10.1016/j.imlet.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 186.Minegishi Y, et al. Mutations in activation-induced cytidine deaminase in patients with hyper IgM syndrome. Clin Immunol. 2000;97:203–210. doi: 10.1006/clim.2000.4956. [DOI] [PubMed] [Google Scholar]
- 187.Zhu Y, Nonoyama S, Morio T, Muramatsu M, Honjo T, Mizutani S. Type two hyper-IgM syndrome caused by mutation in activation-induced cytidine deaminase. J Med Dent Sci. 2003;50:41–46. [PubMed] [Google Scholar]
- 188.Imai K, Zhu Y, Revy P, Morio T, Mizutani S, Fischer A, Nonoyama S, Durandy A. Analysis of class switch recombination and somatic hypermutation in patients affected with autosomal dominant hyper-IgM syndrome type 2. Clin Immunol. 2005;115:277–285. doi: 10.1016/j.clim.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 189.Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol Cell. 2003;12:501–508. doi: 10.1016/S1097-2765(03)00309-5. [DOI] [PubMed] [Google Scholar]
- 190.Wang M, Yang Z, Rada C, Neuberger MS. AID upmutants isolated using a high-throughput screen highlight the immunity/cancer balance limiting DNA deaminase activity. Nat Struct Mol Biol. 2009;16:769–776. doi: 10.1038/nsmb.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bransteitter R, Pham P, Calabrese P, Goodman MF. Biochemical analysis of hypermutational targeting by wild type and mutant activation-induced cytidine deaminase. J Biol Chem. 2004;279:51612–51621. doi: 10.1074/jbc.M408135200. [DOI] [PubMed] [Google Scholar]
- 192.Suspene R, Rusniok C, Vartanian JP, Wain-Hobson S. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 2006;34:4677–4684. doi: 10.1093/nar/gkl555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Beale RC, Petersen-Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol. 2004;337:585–596. doi: 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 194.Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci USA. 2003;100:4102–4107. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Shen HM, Poirier MG, Allen MJ, North J, Lal R, Widom J, Storb U. The activation-induced cytidine deaminase (AID) efficiently targets DNA in nucleosomes but only during transcription. J Exp Med. 2009;206:1057–1071. doi: 10.1084/jem.20082678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Storb U, Shen HM, Nicolae D. Somatic hypermutation: processivity of the cytosine deaminase AID and error-free repair of the resulting uracils. Cell Cycle. 2009;8:3097–3101. doi: 10.4161/cc.8.19.9658. [DOI] [PubMed] [Google Scholar]
- 197.Zaheen A, Boulianne B, Parsa JY, Ramachandran S, Gommerman JL, Martin A. AID constrains germinal center size by rendering B cells susceptible to apoptosis. Blood. 2009;114:547–554. doi: 10.1182/blood-2009-03-211763. [DOI] [PubMed] [Google Scholar]
- 198.Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 199.Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 200.Canugovi C, Samaranayake M, Bhagwat AS. Transcriptional pausing and stalling causes multiple clustered mutations by human activation-induced deaminase. FASEB J. 2009;23:34–44. doi: 10.1096/fj.08-115352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.van Oijen AM. Single-molecule approaches to characterizing kinetics of biomolecular interactions. Curr Opin Biotechnol. 2011;22:75–80. doi: 10.1016/j.copbio.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 202.Myong S, Ha T. Stepwise translocation of nucleic acid motors. Curr Opin Struct Biol. 2010;20:121–127. doi: 10.1016/j.sbi.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.McKinney SA, Joo C, Ha T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys J. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lasken RS, Schuster DM, Rashtchian A. Archaebacterial DNA polymerases tightly bind uracil-containing DNA. J Biol Chem. 1996;271:17692–17696. doi: 10.1074/jbc.271.30.17692. [DOI] [PubMed] [Google Scholar]
- 206.Fogg MJ, Pearl LH, Connolly BA. Structural basis for uracil recognition by archaeal family B DNA polymerases. Nat Struct Biol. 2002;9:922–927. doi: 10.1038/nsb867. [DOI] [PubMed] [Google Scholar]
- 207.Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330:622–627. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 210.Larijani M, Frieder D, Sonbuchner TM, Bransteitter R, Goodman MF, Bouhassira EE, Scharff MD, Martin A. Methylation protects cytidines from AID-mediated deamination. Mol Immunol. 2005;42:599–604. doi: 10.1016/j.molimm.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 211.Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Chahwan R, Wontakal SN, Roa S. Crosstalk between genetic and epigenetic information through cytosine deamination. Trends Genet. 2010;26:443–448. doi: 10.1016/j.tig.2010.07.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.