

Abstract
Functional protein microarrays were developed as a high‐throughput tool to overcome the limitations of DNA microarrays and to provide a versatile platform for protein functional analyses. Recent years have witnessed tremendous growth in the use of protein microarrays, particularly functional protein microarrays, to address important questions in the field of clinical proteomics. In this review, we will summarize some of the most innovative and exciting recent applications of protein microarrays in clinical proteomics, including biomarker identification, pathogen–host interactions, and cancer biology.
Keywords: Biomarker, Cancer, Pathogen‐host interactions, Protein microarray, Serum profiling
Abbreviations
- AIH
autoimmune hepatitis
- BMV
brome mosaic virus
- CAM
clamped adenine motif
- CD
Crohn's disease
- DDR
DNA damage response
- EBV
Epstein‐Barr virus
- HIV‐1
human immunodeficiency virus type 1
- IBD
inflammatory bowel disease
- KSHV
Kaposi's sarcoma‐associated herpesvirus
- mAbs
monoclonal antibodies
- MME
membrane metallo‐endopeptidase
- PSA
prostate‐specific antigen
- RPA
replication protein A
- SARS‐CoV
SARS coronavirus
- SIM
SUMO interaction motif
- TIP60
Tat‐interactive protein 60
- UC
ulcerative colitis
1. Introduction
The concept of microarrays was developed from an earlier concept termed ambient analyte immunoassay, first introduced by Roger Ekins in 1989. In the following decade, microarrays were first successfully realized as DNA or oligonucleotide microarrays, which allowed the quantification of the mRNA expression levels of thousands of genes in parallel. This technology has changed many aspects of biological research. Though extremely successful, the chemistry of DNA hybridization precludes its application for studying proteins, which are considered the major driving force in cells. Consistent with this view, mRNA profiles do not always correlate with protein expression as reported in many recent mass spectrometry studies 1, 2, 3. Therefore, protein microarrays were developed as a high‐throughput tool to overcome the limitations of DNA microarrays, and to provide a versatile platform for protein functional analyses 4, 5, 6.
At the beginning of the development of protein array technology, bacterial strains of a cDNA expression library were gridded and grown on nylon membranes, followed by lysis of the bacteria, and immobilization of the total protein complement 7, 8. However, these early attempts only had limited success, because (i) heterologous proteins (e.g. human proteins) were expressed in bacteria, yielding proteins that lacked critical eukaryotic PTMs; (ii) denaturing conditions were used to lyse the bacterial host, resulting in improperly folded proteins; (iii) proteins of interest were not purified away from thousands of unwanted bacterial proteins; and (iv) the density of the array was low. Before long, other research groups began to report their efforts to fabricate high‐density protein microarrays with purified proteins or antibodies 9, 10, 11, 12. In order to improve protein stability and preserve the native conformation of purified proteins, many research groups developed a variety of surface features to keep proteins hydrated during protein microarray fabrication. These efforts included reports on the 3D gel‐pad chips 13, nanowell chips 11, and plasma membrane‐coated chips 14, to name a few.
The real breakthrough was a 2001 report on the fabrication of a yeast proteome microarray by the Snyder group 15. In this study, approximately 5800 full‐length yeast ORFs were individually expressed in yeast and their protein products purified as N‐terminal glutathione S‐transferase (GST) fusion proteins. Then, each purified protein was robotically spotted on a single glass slide in duplicate at high‐ ensity to form the first “proteome” microarray, as it covered more than 75% of the yeast proteome. More recently, proteome microarrays have been fabricated from the proteomes of viruses, bacteria, plants, and humans 4, 16, 17, 18, 19, 20, 21.
On the basis of their applications, protein microarrays can be divided into two classes: analytical and functional protein microarrays 22. Unlike antibody arrays (analytical microarrays), functional protein microarrays are made by spotting purified proteins on solid surfaces and are therefore useful for direct characterization of protein functions, such as protein‐binding properties, PTMs, enzyme–substrate relationships, and immune responses 5, 22. More recently, a reverse‐phase array was developed in which tissue or cell lysates, as opposed to antibodies, are used to construct the array 23.
Meanwhile, we and others have developed various types of biochemical assays that can be conducted using protein microarrays to characterize protein‐binding properties, including protein–protein, protein–DNA, protein–RNA, and, protein–lipid interactions, and to identify substrates of various types of enzymes, such as protein kinases, acetyltransferases, and ubiquitin and SUMO E3 ligases via covalent reactions 9, 15, 24, 25, 26, 27, 28, 29, 30, 31 (Table 1). These efforts clearly demonstrate the versatility and power of protein microarray technology as a systems biology and proteomics tool 6, 32. In this review, we will summarize recent applications of protein microarrays in clinical proteomics, including biomarker identification, pathogen–host interactions, and cancer biology (Table 2).
Table 1.
Protein microarray studies by PTM
| PTM | Substrate | Enzyme | Reference |
|---|---|---|---|
| Phosphorylation | Yeast | 87 yeast kinases | 11, 26 |
| Human | Human CDK5, CKII | 33, 34 | |
| Four herpesvirus kinases | 35 | ||
| Arabidopsis | Arabidopsis MAPKs | 18, 19 | |
| Herpesvirus | EBV BGLF4 | 36 | |
| Ubiquitylation | Yeast | HECT‐domain E3 Rsp5 | 28, 37 |
| Human | HECT‐domain E3 Nedd4 and Nedd4L | 38 | |
| Acetylation | Yeast | NuA4 complex | 27, 29 |
| E. coli | PAT | 39 | |
| SUMOylation | Human | E3 RanGAP1 | 30 |
| S‐nitrosylation | Yeast and human | N/A | 40 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 2.
Protein microarray studies in clinical proteomics
| Disease type | Disease | Detection methods | Ref |
|---|---|---|---|
| Infectious | SARS infection | Serum antibodies detected on SARS‐CoV arrays comprised of approximately 60 purified proteins of five coronaviruses | 16 |
| B‐cell lymphoma or AIDS‐related Kaposi's lymphoma | Serum antibodies detected on herpesvirus array comprised of approximately 80 purified EBV and KSHV proteins | 41 | |
| Rabbit model of Plague | Rabbit serum antibodies detected on Yersinia pestis arrays comprised of 149 proteins | 42 | |
| Brucellosis | Serum antibodies detected on Brucella melitensis arrays comprised of 3046 proteins expressed in lysates | 43 | |
| Cervical carcinomas or precursor lesions | Serum antibodies detected on papillomavirus arrays comprised of 154 proteins of 13 viruses | 44 | |
| Streptococcus infection | Human proteins detected on Streptococcal surface protein arrays comprised of 201 purified proteins from two pathogenic strains | 45 | |
| Autoimmune | Inflammatory bowel disease (CD and UC) | Serum antibodies detected on E. coli K12 arrays comprised of purified 4179 proteins | 46 |
| Autoimmune hepatitis | Serum autoantibodies detected on human arrays comprised of 5011 purified proteins | 47 | |
| Primary biliary cirrhosis | Serum autoantibodies detected on human arrays comprised of approximately 17 000 purified proteins | 48 | |
| Sjögren's syndrome | Saliva autoantibody detected on human arrays comprised of approximately 8000 purified proteins | 49 | |
| Cancer | Breast cancer | Serum autoantibodies detected on human arrays comprised of 4988 candidate tumor antigens | 50 |
| Cancer stem‐like cell glycan signature identified using array of 94 lectins | 51 | ||
| Bladder cancer | Serum autoantibodies detected on human arrays comprised of approximately 8000 purified proteins | 52 | |
| Rhabdomyosarcoma | Phosphorylation status of 27 proteins detected on human arrays spotted with cancer cell lysates | 53 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
2. Biomarker identification
One of the rapidly growing applications of protein microarray technology in the field of clinical proteomics is biomarker identification. This application for protein microarrays stemmed from traditional serology studies, which focus on the diagnostic identification of antibodies in patient serum samples. These antibodies can be produced as part of an immune response to an infection, against a foreign protein, or even against a person's own protein. When proteins on a protein microarray are viewed as potential antigens, researchers can use it as a platform to identify autoantibodies that show statistically significant association with an infection or with a disease of interest. In general, the following approach is used: first, patient sera are diluted (e.g. 1000‐fold) and incubated on a preblocked antigen microarray (i.e. protein microarray), followed by a stringent washing step. Then, positive signals are detected using anti‐human IgG, IgM, or IgA antibodies coupled with various fluorophores for detection (Fig. 1). Compared with traditional serology techniques, such as ELISA, agglutination, precipitation, complement‐fixation, and fluorescent antibodies, protein microarray‐based serum profiling is much more sensitive and can be performed at a much higher throughput. Another significant advantage is that it offers an unbiased platform for novel biomarker identification. In this section, we will review four studies to illustrate the history and development of protein microarrays in biomarker identification.
Figure 1.
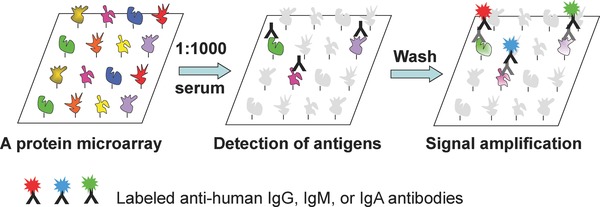
Principle of serum profiling assays performed on a functional protein microarray. A functional protein microarray, composed of hundreds of thousands of individually purified proteins, is first blocked with BSA in PBS buffer. Then, a diluted serum sample is incubated on the microarray typically at RT for 1 h. After extensive washes, bound antibodies (e.g. human IgG, IgA, or IgM) can be detected with anti‐human immunoglobulin antibodies, followed by a signal amplification step with fluorescently labeled secondary antibodies. Detection of immunoglobulin isotypes can be multiplexed with different fluorophores as illustrated.
2.1. SARS‐CoV diagnosis
In 2003, Zhu et al. fabricated the first viral proteome microarray composed of every full‐length protein and protein fragment encoded by SARS coronavirus (SARS‐CoV), as well as proteins from five additional mammalian coronaviruses 16. These microarrays were then used to screen 400 Canadian serum samples collected during the 2002 SARS outbreak, including samples from confirmed SARS‐CoV cases, respiratory illness patients, and healthcare professionals. Antibody response was quantified by the application of both anti‐human IgG and IgM antibodies each coupled to different fluorophores, followed by measurement of fluorescence signal intensity (Fig. 2). To identify potential biomarkers, serum samples were first clustered according to the relative signal intensities of all of the coronavirus proteins in an unsupervised fashion (See Data Analysis section). The serum samples fell into two major groups, which upon subsequent comparison with clinical data were largely correlated with either SARS‐positive or SARS‐negative sera. In the cluster of markers, five fragments of the SARS N protein associated tightly with SARS infection, while SARS sera also exhibited statistically significant binding to one spike protein fragment. However, a few proteins encoded by other coronaviruses also showed significant correlation. To determine the best classifiers and classification model, two different supervised analysis approaches, k nearest neighbor and logistics regression were applied, and the N protein of SARS‐CoV, as well as the spike protein S from both SARS‐CoV and HCoV‐229E, were identified as the best classifiers. A useful feature of a serum test relative to a nucleic acid diagnostic test is that anti‐pathogen antibodies can potentially be detected long after infection. Taking advantage of this, serum samples collected from SARS patients, who recovered from respiratory disease (∼320 days after diagnosis), were used to probe the microarray and positive signals were detected with both anti‐human IgG and IgM antibodies (Fig. 2; middle panel). These results clearly showed that SARS‐CoV N proteins could be readily recognized by human IgG antibodies and importantly, not by IgM antibodies, as expected. However, serum samples collected from the Chinese patients immediately after fever was detected showed much stronger signals both in the IgG and IgM profiling (Fig. 2; left panel). These results indicated that the protein microarray approach is capable of detecting anti‐pathogen antibodies in serum samples long after infection, as well as detecting infection at early stages of infection as demonstrated by anti‐human IgM profiling. The approach developed here is potentially applicable to all viruses and expected to have a great impact on epidemiological studies and possibly in clinical diagnoses.
Figure 2.

Examples of IgG and IgM profiles obtained with serum samples of SARS‐CoV‐infected patients. Sample FP B0352 was collected immediately following detection of fever in a patient in Beijing; Sample DP C08 was collected from a recovered SARS patient in Toronto. Signals in the upper panel and the BSA control were detected with anti‐human IgGs, while signals in the lower panel of the two patient samples were detected with anti‐human IgMs.
2.2. Humoral immune responses to herpesviruses
A similar approach has been applied to profile humoral immune responses to two human herpesviruses, Epstein‐Barr virus (EBV) and Kaposi's sarcoma‐associated herpesvirus (KSHV). EBV is an ubiquitous human herpesvirus, while KSHV has a restricted seroprevalence. Both viruses are associated with malignancies and show an increased frequency in individuals who are co‐infected with human immunodeficiency virus type 1 (HIV‐1). The Zhu and Hayward groups generated a protein microarray consisting of 174 EBV and KSHV full‐length proteins that were individually expressed and purified from yeast 36, 41. Instead of sera, plasma antibody responses to EBV and KSHV were examined from healthy volunteers and patients with B‐cell lymphoma or with AIDS‐related Kaposi's sarcoma or lymphoma. These experiments detected IgG responses to known antigens, as well as the tegument proteins ORF38 (KSHV), BBRF (EBV), BGLF2 (EBV), and BNRF1 (EBV), and to the EBV early lytic proteins BRRF1 and BORF2. Because IgA responses to EBV EBNA1 and viral capsid antigens have long been used as a diagnostic tool for nasopharyngeal carcinoma, they also found IgA responses in healthy and HIV‐infected patients. IgA responses to VCA and to EBNA1 were found to be frequently elevated in lymphoma patients and in individuals who were HIV‐1 positive. Comparison between the IgG and IgA responses indicated that IgA responses were much higher against BCRF1, BRRF2, and LMP2A. Therefore, this study demonstrated that plasma can be used for biomarker identification; immunoglobulin responses of other isotypes, such as IgA, are therefore also worth testing.
2.3. Escherichia coli proteome microarrays for IBD diagnosis
To demonstrate that protein microarrays could also be used to identify new biomarkers in autoimmune diseases, Chen et al. decided to apply an E. coli K12 proteome microarray 17 to profile serum samples collected from Crohn's disease (CD) and ulcerative colitis (UC) patients 46. CD and UC are chronic, idiopathic, and clinically heterogeneous intestinal disorders collectively known as inflammatory bowel disease (IBD). Although IBDs have been suggested to be autoimmune diseases, anti‐microbial antibodies are present in the sera of IBD patients, and some of these antigens have proven to be valuable serological biomarkers for diagnosis and/or prognosis of the disease. In this study, a protein microarray, including 4256 proteins encoded by a commensal K12 strain, was screened using individual serum from healthy controls (n = 39) and clinically well‐characterized patients with IBD (66 CD and 29 UC). Surprisingly, among the 417 E. coli proteins that were differentially recognized by serum antibodies from healthy controls and either CD or UC patients, 169 proteins were identified as highly immunogenic in healthy controls, 186 proteins were identified as highly immunogenic in CD patients, and only 19 proteins were identified as highly immunogenic in UC patients. Using several statistical tools, they identified two sets of serum antibodies as novel biomarkers for specifically distinguishing CD from healthy controls (accuracy, 86 ± 4%; p < 0.01) and CD from UC (accuracy, 80 ± 2%; p < 0.01). This study was the first demonstration of using high‐density, high‐content proteome microarrays to discover novel serological biomarkers. It was also the first effort to examine human immune responses to the entire proteome of a microbial species in a disease context.
2.4. Autoantigen discovery for AIH
A protein microarray composed of individually purified human proteins would be an ideal tool for discovery of novel autoantigens associated with an autoimmune disease. Take autoimmune hepatitis (AIH) as an example: AIH is a chronic necro‐inflammatory disease of human liver with little known etiology. Detection of nonorgan‐specific and liver‐related autoantibodies using immunoserological approaches has been widely used for diagnosis and prognosis. However, these traditional autoantigens, such as anti‐SMA (smooth muscle autoantibodies) and anti‐ANA (antinuclear autoantibodies) are often mixtures of complex biological materials. Unambiguous and accurate detection of the disease demands identification and characterization of these autoantigens. Therefore, Song et al. fabricated a human protein microarray of 5011 nonredundant proteins that were expressed and purified as GST fusions in yeast 45. There are several advantages associated with producing human proteins in yeast rather than bacteria: (i) higher solubility, (ii) higher yields of large proteins (e.g. > 50 kD), (iii) better preserved conformation of proteins, and (iv) proteins are less immunogenic when produced in yeast than in E. coli 17, 21, 25. However, unlike a viral or bacterial protein microarray, a significant obstacle to the use of a human protein microarray of high content is its high cost. For example, a human protein array of 9000 proteins can exceed $1000 per array. In order to reduce this cost, Song et al. developed a two‐phase strategy to identify new biomarkers in AIH. Phase I is designed for rapid selection of candidate biomarkers, which are then validated in Phase II (Fig. 3). In Phase I, 30 AIH and 30 control serum samples were selected and individually used to probe the human protein microarrays at a 1000‐fold dilution, followed by detection of bound human autoantibodies using a Cy‐5‐conjugated anti‐human IgG antibody. Statistical analysis revealed 11 candidate autoantigens. To validate these candidates and to avoid a potential overfitting problem (see below), which is especially likely when dealing with a small sample size, the 11 proteins and three positive controls were re‐purified to build a large number of low‐cost small arrays for Phase II validation. These arrays were then sequentially probed with serum samples used in Phase I and serum samples obtained from an additional 22 AIH, 50 primary biliary cirrhosis, 43 hepatitis B, 41 hepatitis C, 11 system lupus erythematosus, 11 primary Sjögren's syndrome, and two rheumatoid arthritis patients. As negative controls, they also included 26 serum samples from patients suffering from other types of severe disease and 50 samples from healthy subjects. Three new antigens, RPS20, Alb2‐like, and dUTPase, were identified as highly AIH‐specific biomarkers with sensitivities of 47.5% (RPS20), 45.5% (Alba‐like), and 22.7% (dUTPase), which were further validated with additional AIH samples in a double‐blind design. Finally, they demonstrated that these new biomarkers could be readily applied to ELISA‐based assays for clinical diagnosis and prognosis.
Figure 3.
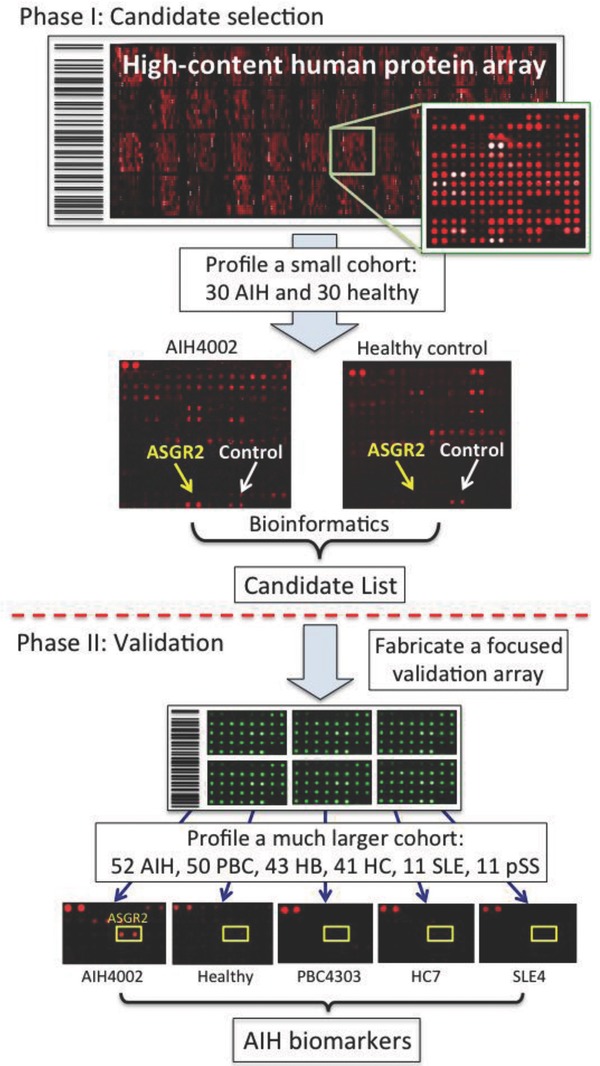
Scheme of the two‐phase strategy for biomarker identification in human autoimmune diseases. In Phase I, a small cohort is used to rapidly identify a group of candidate biomarkers via serum profiling assays on a human protein microarray of high cost. Because a small number of microarrays are needed, cost of the experiments is relatively low. In Phase II, a focused protein microarray of low cost is fabricated by spotting down purified candidate proteins. A much larger cohort is then assayed on these arrays in a double blind fashion to validate the candidates identified in Phase I.
This study represents a new paradigm in biomarker identification using protein microarrays for three reasons. First, a manageable number of candidate biomarkers can be rapidly identified at low cost because fewer expensive protein microarrays of high content are needed in the first phase of this two‐phase strategy. Second, by using small arrays comprised selected candidate proteins, the validation step can be rapidly carried out with a much larger cohort at lower cost. This validation step is extremely important for avoiding the overfitting problem associated with statistical analysis in biomarker or classifier identification, especially when dealing with a small cohort (e.g. <40). Overfitting is a problem in which a statistical model describes random error or noise instead of the underlying relationship. It generally occurs in biomarker identification when the system is excessively complex, such as having too many individual‐to‐individual variations relative to the number of samples used. As a result, biomarkers that have been overfit generally have poor predictive performance. Therefore, testing an additional, larger cohort in a double‐blind design is an effective way to rule out overfit biomarkers. Third, the author developed ELISA‐based assays to examine the performance of the validated biomarkers with additional samples. These newly identified biomarkers serve as a translational step toward clinical practice.
In addition, there have been a series of studies that employed pathogen protein microarrays to profile serological responses following infection. For example, protein microarrays have been developed in bacteria and viruses for biomarker identification in various infectious diseases 43, 44, 47, 54. These studies have clearly demonstrated the power of protein microarrays in identification of potential biomarkers; however, several shortcomings are repeatedly seen in these studies. For instance, many of these arrays were fabricated using proteins translated in E. coli lysates without purification 43, 44, 47, 54. Because these proteins are contaminated with unwanted E. coli proteins, sensitivity of the assay is likely reduced due to their high immunogenicity 41. As a result, E. coli lysates had to be used as a blocking reagent to alleviate this problem. Also problematic is that in many of these studies, identified biomarkers were not validated with additional cohorts and therefore, the possibility of overfitting was not completely ruled out.
3. Pathogen–host interactions
An emerging application of protein microarrays in the field of clinical proteomics is an unbiased, proteome‐wide survey of important players involved in pathogen–host interactions. The identified factors, encoded by either a pathogen or a host, have the potential to be developed into novel therapeutic targets. Protein microarrays can serve as an ideal platform for such purposes: Once a protein microarray is fabricated from a host or pathogen, it can be used to identify direct pathogen–host interactions. This strategy is particularly useful for investigating virus–host interactions because after entering the host cells, the viral genome and encoded proteins are in direct physical contact with the host's biological materials. As we will discuss in this section, such interactions can be investigated at multiple levels, such as RNA–protein interactions, enzyme–substrate relationships, and protein–protein interactions.
3.1. BMV RNA and host proteome interactions
In 2007, Zhu et al. described the first study using a yeast proteome microarray to identify host factors that can affect replication of brome mosaic virus (BMV), a plant‐infecting RNA virus that can also replicate in Saccharomyces cerevisiae 25. Previous studies have shown that this positive‐stranded RNA virus encodes a tRNA‐like structure at the 3′‐end of its RNA genome, in which a clamped adenine motif (CAM) is required for packaging its genome into the capsid. To identify crucial host proteins that can interfere with the viral packaging process, a Cy3‐labeled CAM‐containing RNA stem‐loop structure was incubated on the yeast proteome microarray in the presence of an equal amount of a Cy5‐labeled mutated CAM hairpin. Using Cy3‐to‐Cy5 fluorescence signal intensity ratios, the top hits were identified and validated using an in vitro gel‐shift assay. Two validated candidate proteins, pseudouridine synthase 4 and actin patch protein 1, were selected for further characterization in tobacco plants. Both proteins modestly reduced BMV genomic plus‐strand RNA accumulation, but dramatically inhibited BMV systemic spread in plants. Pseudouridine synthase 4 also prevented the encapsidation of the BMV RNAs in plants and the reassembly of BMV virions in vitro.
This work is significant because it established the first RNA‐binding assay on a proteome microarray and demonstrated the utility of protein microarrays for identifying important players involved in pathogen–host interactions.
3.2. Host phosphorylome of virus‐encoded kinases
In the course of evolution, viruses have been very successful at exploiting the host via development of their own arsenals, some of which were hijacked from the host in the form of both DNA and proteins. To develop more effective antivirals, one must understand the molecular mechanisms by which viruses exploit the host machineries for their own use. The human α, β, and γ herpesviruses infect different tissues and cause distinct diseases, ranging from mild cold sores to pneumonitis, birth defects, and cancers 55. However, they each confront many of the same challenges in infecting their hosts, including reprogramming cellular gene expression, sensing cell‐cycle phase, and modifying cell‐cycle progression, and reactivating the lytic life cycle to produce new virions and spread infection. On the other hand, many lytic cycle genes involved in replication of the viral genomes (e.g. the orthologous serine/threonine protein kinases) are highly conserved across the herpesvirus family. Therefore, it became an attractive hypothesis that the shared substrates targeted by these orthologous viral kinases would reveal host pathways that are critical for replication across the herpesvirus family.
To test the above hypothesis, Li et al. employed a human protein microarray 56. The authors purified four orthologous kinases encoded by EBV, KSHV, human cytomegalovirus (HCMV), and HSV‐1, performed kinase reactions on a human protein microarray described previously 4, and identified 110 shared substrates. Like every large‐scale screen, the next challenge was to select candidates that would be worth pursuing. To do so, the authors then applied Gene Ontology and STRING analyses (http://string-db.org/, a database of known and predicted protein–protein interactions) to these candidates and found a highly connected cluster of 15 proteins. Strikingly, these proteins were all known to be involved in the DNA damage response (DDR) (Fig. 4). The host DDR has been known to be important to many viruses, including human herpesviruses, and is relevant to virus‐induced tumorigenesis 35. To narrow down this list to a single candidate for in‐depth characterization, the authors reasoned that the viruses are likely to target an upstream master regulator, which triggers the DNA damage response. On the basis of a literature search and the structure of this cluster, Tat‐interactive protein 60 (TIP60) emerged as an excellent candidate for follow‐up, because (i) TIP60 is further upstream in the DDR pathway than any of the other candidates in the cluster; (ii) it serves as a master regulator in DDR via activation of ATM autophosphorylation activity by acetylation; (iii) it regulates chromatin dynamics via histone acetylation; and (iv) its importance has been shown in other viruses. Indeed, the authors observed that when TIP60 was knocked down in EBV‐infected B cells, EBV's lytic replication was greatly reduced. Next, the authors applied a series of cell‐based assays and showed that during EBV replication, TIP60 activation by the BGLF4 kinase triggers EBV‐induced DDR and also mediates induction of viral lytic gene expression. Finally, the authors demonstrated that TIP60 was also required for efficient lytic replication in HCMV, KSHV, and HSV‐1.
Figure 4.
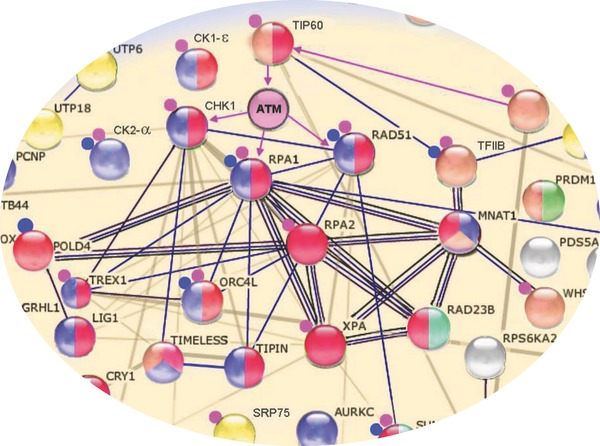
Identification of most relevant candidate for in‐depth in vivo studies. Assisted by GO analysis, 110 shared substrates of conserved herpesvirus kinases were plugged into the STRING database. A highly connected cluster of 15 proteins was revealed, all of which are known to play a role in DDR. Based on the literature and topology of the cluster, TIP60 emerged as the most promising candidate. Protein nodes are color coded by functional class, with proteins involved in DDR colored red. Small blue circles adjacent to protein nodes indicate that the protein is herpesvirus associated, while small pink circles indicate the proteins are associated with other viruses. Edges between the proteins represent known or predicted connections, such as protein–protein interactions, catalytic reactions, and enzyme–substrate relationships.
This work illustrates the value of high‐throughput, unbiased approaches for the discovery of conserved viral targets in the host that have the potential to be developed into novel therapeutic targets for antivirals. Currently, there are few drugs available to treat herpesvirus infections, and viral escape mutants develop as a result of extensive use of this limited repertoire. The herpesvirus protein kinases are attractive antiviral drug targets. However, developing broadly effective drugs targeting protein kinases requires knowledge of their common cellular substrates. The information provided by common substrate identification will assist in the design of assays for new and broadly effective anti‐herpesvirus therapeutics.
3.3. LANA interactome analysis reveals a role in telomere shortening
Protein microarrays can also serve as a convenient tool for profiling protein–protein interactomes between a pathogen and a host. In a recent example, Hayward et al. surveyed the interactome between a KSHV‐encoded virulent factor, LANA, and the human host using human protein microarrays, in order to identify host proteins that can be recognized by LANA 57. LANA functions in latently infected cells as an essential participant in KSHV genome replication and as a driver of dysregulated cell growth. Although yeast two‐hybrid screens, GST affinity, immunoprecipitation assays, and chromatography coupled with MS have been applied to the identification of LANA binding proteins, each approach has its strengths and weaknesses, and each tend to identify different sets of proteins. In this study, the authors used purified FLAG‐tagged LANA applied to human protein microarrays to identify 61 potential binding partners, many of which were previously unknown. Eight out of nine proteins were validated by co‐immunoprecipitation, including TIP60, protein phosphatase 2A, replication protein A (RPA) and XPA. Although human papillomavirus E6, HIV‐1 TAT, and HCMV pUL27 interact with TIP60 and induce TIP60 degradation, LANA‐associated 42 retained acetyltransferase activity and showed increased stability. This observation is in line with the study described in the previous section that showed that TIP60 plays a positive role in KSHV lytic replication. On the other hand, identification of RPA as a LANA interacting partner suggested that LANA may play a role in regulating the length of host telomeres, because RPA1 and RPA2 are known to be essential in the replication of cellular telomeric DNA. To test this hypothesis, the authors performed ChIP assays with anti‐RPA1 and ‐RPA2 antibodies using primers specific to the telomere regions and found that the presence of LANA drastically reduced the recruitment of both RPA1 and RPA2 to the host telomeres, while it had no impact on the protein level of the RPA complex. This observation raised the possibility that LANA might have an impact on telomere length. Using Southern blot analysis of terminal restriction fragments, the standard method for quantifying telomere length, the authors demonstrated that the average length of telomeres was shortened by at least 50% in both LANA‐expressing endothelial cells and KSHV‐infected primary effusion lymphoma cells. Many interesting questions remain to be answered. How does LANA block the RPA complex recruitment to the telomeres? Is it achieved via direct competition since LANA is also an ssDNA‐binding protein? Or, does LANA serve as a kinase sink for the RPA complex and regulate RPA recruitment via phosphorylation?
3.4. SUMO‐EBV interactome revealed a new mechanism of EBV lytic replication
On the flip side, a human factor of interest can be used to survey a virus protein microarray to identify important viral factors. Similar to the ubiquitylation pathway, SUMOylation involves a series of sequential enzymatic reactions that conjugate SUMO to lysine residues on substrate proteins. Previous studies have shown that both latent and lytic EBV proteins interact with the SUMO system. Noncovalent SUMO‐EBV protein interactions can occur via a SUMO interaction motif (SIM) in the target proteins. To comprehensively identify additional EBV proteins that bind to SUMO, Li et al. performed a protein‐binding assay with human SUMO2 58 on a previously described EBV proteome microarray 41 and identified a total of 11 proteins, including the conserved viral kinase BGLF4. The mutation of potential SIMs in BGLF4 at both N‐ and C‐termini changed the intracellular localization of BGLF4 from nuclear to cytoplasmic, while BGLF4 mutated in the N‐terminal SIM remained predominantly nuclear. The mutation of the C‐terminal SIM yielded an intermediate phenotype with nuclear and cytoplasmic staining. The authors also found that BGLF4 abolished the SUMOylation of the EBV lytic cycle transactivator ZTA, and that this inhibitory effect on ZTA SUMOylation was dependent on both BGLF4 SUMO binding and BGLF4 kinase activity. The global profile of protein SUMOylation was also suppressed by BGLF4 but not by the SIM or kinase‐dead BGLF4 mutant. Furthermore, BGLF's interaction with SUMO was required to induce the cellular DNA damage response and to enhance the production of extracellular virus during EBV lytic replication.
3.5. Identification of novel streptococcal proteins that bind human ligands
The identification of pathogen proteins that interact with human factors has also been applied to understanding the mechanisms of bacterial infection. Margarit et al. harnessed the power of protein microarrays to identify proteins expressed by two species of the streptococcus gram‐positive bacteria, Streptococcus pyogenes, and S. agalactiae, that interact with human factors known to mediate pathogenesis 59. Rather than developing whole‐proteome arrays, they used a bioinformatics approach to predict those proteins present on the cellular surface—and thus most likely to play a role in infection, and used this list of 200 proteins to develop their arrays. They also carefully considered the human probes that they would use, choosing three human ligands: fibronectin, fibrinogen, and C4‐binding protein, all known to play important roles in the colonization and infection processes. Binding experiments conducted using the streptococcal arrays and human protein probes identified 17 of the 20 known interactions previously reported as well as eight newly identified streptococcal proteins, many of which they confirmed by far Western blot analysis. These novel proteins included proteins of unknown function as well as three related proteins that they termed the fib proteins. They then used domain mapping to identify regions of the fib proteins required for their interaction with the human ligands. Interestingly, sera samples from patients with S. agalactiae infections show high titers of fib‐specific antibodies, indicating that these proteins are highly expressed during infection. Further work will determine the role of these proteins in infection and whether they will emerge as suitable drug targets to fight pathogenic Streptococcus infections.
In summary, the above studies have demonstrated the power of protein microarrays in the discovery of novel molecular mechanisms underlying host–pathogen interactions at various levels. In recent years, other high‐throughput approaches, such as shotgun MS 60, genome‐wide RNAi screens 61, 62, and yeast two‐hybrid 63, 64 have been applied to understand host–pathogen interactions; however, the protein microarray approach provides a more versatile platform than any of these single approaches for identifying multiple types of direct interactions between a pathogen and host, including protein–protein interactions 57, 58, 59, RNA–protein interactions 25, and enzyme–substrate interactions 35.
4. Cancer biology
Over the past 5 years, rapid development of genome‐wide sequencing technologies (i.e. next‐gen sequencing) has revealed the heterogeneous nature of tumors 65, 66. However, clinical diagnosis of tumors is largely still dependent on morphologic patterns. The fact that tumors with indistinguishable morphology can have vastly different clinical outcomes suggests that the molecular heterogeneity of each patient's tumor cells have to be better understood before more effective therapies can be developed. Therefore, the future of cancer treatment is tailored molecular therapy specific for each individual, which will require a new class of proteomic profiling technologies. As a widely adopted technology, protein microarrays can meet this need for the profiling of the functional state of tumors and for cancer biomarker identification.
4.1. Reverse phase protein microarrays in cancer biology
A widely adopted approach to determining the status of signaling pathways in tumor cells is based on immunoblot analysis with antibodies that can recognize phosphorylated proteins. To transform the low‐throughput immunoblot to a high‐throughput format, Haab et al. first reported the development of antibody microarray technology, in which individual commercial antibodies were spotted on glass at high density 12. This technology allows for simultaneous detection of multiple antigens presented in a complex biological sample, such as cells, tissues, and body fluids 67, 68.
The term “reverse phase protein microarray” was proposed by Liotta, Petricoin et al. in 2001 69 as an array in which lysates of cells or tissues are immobilized on the array surface rather than antibodies. Using phosphoprotein‐specific antibodies, these arrays can then be used to interrogate the phosphorylation state of proteins present in these mixtures as a proxy of signaling status in tumors. There are several ongoing clinical trials currently utilizing the reverse phase protein microarray 70. An obvious advantage of this approach is that it allows to evaluate the state of multiple components of a signaling pathway, even though the cell is lysed. Because fabrication of such microarrays requires a minimum amount of sample, multiple clinical samples, such as biopsy samples, tumors, and body fluids, can be printed on a series of identical arrays and analyzed in parallel using commercially available anti‐phosphoprotein or other specific antibodies.
For example, Petricoin et al. used cancer cell lysates, representing 59 patient samples obtained from the Children's Oncology Group Intergroup Rhabdomyosarcoma Study (IRS), to fabricate reverse phase microarrays 53. Rhabodymyosarcoma is a rare childhood cancer that arises from undifferentiated muscle progenitor cells. Although current treatments can yield as high as a 67% disease‐free survival rate, the reasons for treatment failure in the remaining one‐third of patients are unknown. The identification of biomarkers to allow distinguishing these patients from those that respond to traditional therapies would help identify those patients best suited for alternative therapies, and also potential drug targets. These reverse phase arrays were therefore used as platforms to detect the phosphorylation status of proteins thought to underlie whether rhabdomyosarcoma subtypes were responsive to standard chemotherapy regimens. Using phosphosite‐specific antibodies, the authors identified higher phosphorylation levels in four Akt/mTOR pathway components in patients with poor survival outcomes. Network analysis based on this data and known pathway information was used to find that, on the other hand, patients with good treatment outcomes showed mTOR pathway suppression. Together, these findings suggested that pharmacologically inducing mTOR pathway suppression could result in improved outcomes for patients that failed to respond to standard chemotherapy regimens. The authors proceeded to test this hypothesis using a mouse xenograft model with rhabdoymyosarcoma cell lines and a known mTOR pathway inhibitor. They found that treatment with the inhibitor resulted in reduced phosphorylation of the protein 4E‐BP1 that had been identified by the protein microarray studies, as well as inhibition of tumor growth. These results suggest that mTOR pathway inhibitors may be a potential way to improve treatment outcomes for patients that fail to respond to standard chemotherapy regimens. Additionally, they demonstrate the power of tumor cell lysate arrays in (i) identifying specific patient subpopulations that could benefit from tailored therapies, as well as (ii) identifying the specific molecules that should be targeted in developing and testing these therapies. However, there are several major problems with the reverse phase array approach. First, well‐characterized antibodies are not available for the great majority of human proteins 71, 72. Second, several recent studies have suggested that many commercially available monoclonal antibodies (mAbs) may not even recognize their purported targets, and cross‐react extensively with other cellular antigens 73. Third, antibody cross‐reactivity is an even more pressing problem in diagnostic and therapeutic applications, as underlined by the recent withdrawal of several mAb‐based pharmaceuticals from the market 74, 75. Finally, this approach requires prior knowledge as to which phosphoproteins should be evaluated and as a result, is not an ideal platform for the discovery of novel biomarkers.
4.2. Identification of autoantibody biomarkers for the early detection of breast cancer
Current screening for breast cancer using mammograms detects only 70% of breast cancers, and false‐positive mammograms lead to unnecessary biopsies. The identification of biomarkers that would allow early detection of breast cancer could provide a noninvasive, low cost method that could improve patient outcomes. One promising category of cancer biomarkers is autoantibodies to tumor antigens that offer better stability, specificity, ease of purification, and ease of detection compared to other serum proteins. In order to identify autoantibodies to tumor antigens associated with breast cancer, Anderson et al. used protein arrays containing candidate tumor antigens and applied breast cancer patient and control serum samples to identify differences in the human antibody repertoire that could be used as biomarkers 50. These custom protein arrays, termed “NAPPA” arrays (Nucleic Acid Protein Programmable Array), are fabricated by the spotting of cDNAs that encode the target proteins at each feature of the array. Proteins are transcribed and translated by a cell‐free system and immobilized by encoded epitope tags, thus bypassing the protein purification process. Additionally, the authors used a three‐phase screening approach to home in on the best candidate breast cancer biomarkers. In the first phase, they used arrays with the full set of 4988 tumor antigens in order to eliminate uninformative autoantibodies that were present at similar levels in both early breast cancer patients and healthy women. Subtracting these antigens, the protein set was reduced to 761, allowing them to fabricate smaller arrays for the next phase that offer the benefits of reduced cost and fewer false positives. In the second phase, sera from patients with invasive early breast cancer and benign breast disease were compared, in order to identify antigens specific to early breast cancer but absent from benign breast disease, resulting in 119 antigens. In the third phase, they set out to validate this antigen list, finding 28 antigens that maintained high levels of specificity in a blinded validation assay, including the protein ATP6AP1, a known autoantigen. They then focused on this protein and went on to show high expression of ATP6AP1 in four breast cancer cell lines by Western blot, as well as significantly higher ATP6AP1 autoantibody levels in approximately 13% of early breast cancer serum cases compared to controls. Although only a first step, this work demonstrates the power of protein microarrays, in particular programmable protein microarrays, in identifying biomarkers for the early detection of breast cancer.
4.3. Finding autoantibody biomarkers in bladder cancer
An important goal of identifying cancer biomarkers is to define new strategies for early diagnosis that can allow early intervention with current therapies to improve patient survival rates. Additionally, since cancer‐associated autoantibodies often target proteins that are mutated, modified, or aberrantly expressed in tumor cells, they could also be considered immunologic reporters that could uncover molecular events underlying tumorigenesis 52. The molecular players in these events, in turn, may be the best place to start in efforts to develop novel therapies. In order to identify autoantibody biomarkers that could act as indicators of bladder cancer, as well as the underlying molecular pathology contributing to disease, Orenes‐Pinero turned to a protein array strategy using the Invitrogen Protoarray containing approximately 8000 purified human proteins to identify antibodies to tumor‐associated antigens in serum 52. Comparing serum samples collected from 12 patients with bladder cancer and ten control patients without bladder cancer, they identified 171 differentially expressed proteins. Among these, they selected clusterin and dynamin for validation based in part on their known role in cancer biology. Using immunohistochemistry on a custom tissue microarray comprised bladder cancer tumor samples, they found reduced expression levels of clusterin in muscle invasive bladder tumors as compared to nonmuscle invasive tumors. On the other hand, they found that low‐protein expression of dynamin was associated with increased tumor stage and grade, higher recurrence rate after surgery, as well as shorter survival. Paradoxically, their follow‐up tests revealed lower expression levels of dynamin and clusterin associated with disease, in contrast to their protein array results that showed increased autoantibody levels to these proteins among bladder cancer patients compared to controls. Despite these contradictory findings, the authors demonstrated significant associations between dynamin and clusterin expression levels and bladder cancer disease progression that could potentially allow them to use these as informative biomarkers in the clinic as well as potential drug targets. This work demonstrates the power of protein microarrays for the identification of autoantibodies to tumor‐associated antigens and its application to the discovery of cancer biomarkers.
4.4. Application of lectin microarrays in cancers
Cell surfaces, especially mammalian cell surfaces, are heavily coated with complex poly‐ and oligosaccharides, and these glycans have been implicated in many functions, such as cell‐to‐cell communication, host–pathogen interactions, and cell matrix interactions. Aberrations of glycosylation are usually indicative of the onset of specific diseases, such as cancer. There are a handful of tools that can be used to study glycosylation, such as LC 76, 77, MC 78, CE 79, 80, and flow cytometry 81, 82.
To take advantage of the specific glycan‐binding properties of lectins and the parallel analysis capability of microarrays 83, we and others have developed the lectin microarray technology for profiling glycans in a high‐throughput fashion 51, 84, 85, 86, 87. Although lectin microarrays in early reports were only composed of a small number of lectins, in a later report a high‐content lectin microarray composed of 94 unique commercial lectins was fabricated and used to profile accessible surface glycans of mammalian cells 51. A total of 24 human cell lines were labeled and applied to this lectin microarray. A binary algorithm was developed to generate a “glycan signature” for each cell line, resulting in a hierarchical cluster based on their accessible glycan composition. By comparing the glycan profiles of a breast cancer cell line and its cancer stem‐like cell derivatives, three lectins, namely LEL, AAL, and WGA, were found to specifically recognize MCF7 cells but not the derivatives. To confirm this result, the authors first employed LEL‐conjugated beads to purify away the normal MCF7 cells from the cancer stem‐like cells (estimated as ∼0.1% in the cell population) in an MCF7 cell population as a means to enrich cancer stem‐like cells. Next, using a mouse model to test the enrichment of the cancer stem‐like cells, they showed that 2 weeks following injection of the LEL‐depleted cancer stem‐like cell enriched cultures, average tumor size was greater than twofold bigger than the control group injected with a similar number of regular MCF7 cells. This study demonstrated the utility of a lectin microarray in the identification of novel cell surface markers on cancer stem‐like cells that subsequently allowed enrichment for cancer stem‐like cells.
Because the affinity of lectins is usually low (Kd is in the range of 10−3–10−6 M) 84, in processing a cell‐binding assay on a lectin microarray, an analyte of low affinity may be washed away from the immobilized lectins, especially when dealing with live cells. In order to overcome these problems, researchers have modified lectin microarray technology in many ways 88. One example is antibody‐assisted lectin profiling that was developed for detecting glycoproteins at low concentrations. Kuno et al. used this method to analyze the glycan structures of a protein hPod, which has been proposed to enhance the metastatic potential of glioblastoma cells 86. The hPod protein complex was first IP enriched with the appropriate antibody and then incubated on a lectin array to identify its associated glycans. An additional modification of the lectin microarray platform is the use of the evanescent‐field activated fluorescence detection system, which allows for label‐free, real‐time detection. Since the evanescent field is generated within 200 nm, the background signals are so low that washing steps are not necessary. It has been reported that this detection system has by far the highest sensitivity among the lectin microarray detection systems, with a reported detection limit in the 100 pM range 89. Finally, Li et al. reported a two‐phase discovery and validation approach to improve the sensitivity of the lectin microarray technology in cancer biomarker discovery for prostate cancer 90. First, pooled tissue samples for each group were generated by mixing equal amounts of tissue proteins (50 μg) from the four cases in the normal, nonaggressive cancer, aggressive cancer, and metastasis groups. In the first phase, or discovery phase, prostate‐specific antigen (PSA) and membrane metallo‐endopeptidase (MME) proteins were extracted from each tissue group using anti‐total PSA antibody and anti‐MME mAbs, respectively. After incubating the IPed PSA or MME proteins on a lectin microarray, captured PSA or MME proteins were detected with anti‐PSA or anti‐MME mAbs. Comparison of signals between each group of pooled tissue revealed that the fraction of PSA that is O‐glycosylated (as recognized by jacalin) or Neu5Ac conjugated (as detected by SNA), as well as the fraction of MME that was modified by either GalNAc or GlcNac was highly elevated in aggressive prostate cancer and metastatic prostate cancer groups. These results were confirmed with immunosorbent assay in phase II, in which PSA and MME were first captured on an ECL plate coated with anti‐PSA and anti‐MME mAbs, followed by detection with biotinylated lectins. These studies demonstrate the power and adaptability of protein microarrays for detection of even low‐affinity interactions such as those between lectins and glycans.
5. Outlook
Recent years have witnessed tremendous growth in the use of protein microarrays to address important questions in the field of clinical proteomics. In the area of biomarker identification, most of the recent research has been focused on either infections or autoimmune diseases. We believe that protein microarrays, especially functional protein microarrays, will be widely used for identification of cancer biomarkers in the near future. Indeed, recent advances in immunoproteomics and high‐throughput technologies have suggested that the autoantibody repertoire in cancer patients might be quite different as compared with that in healthy subjects, leading to the hypothesis that autoantigens might be identified as biomarkers for cancer diagnosis, as well as cancer prognosis 91. Ideally, a human protein microarray developed for such a purpose should cover the entire human proteome, in order to enable a comprehensive screen for the autoantigens. To our knowledge, we have fabricated a human proteome microarray of the best coverage (>70%) 21. However, when hundreds, if not thousands, of serum samples are needed to screen for biomarkers, the cost of using these human proteome microarrays accumulates very rapidly. An effective strategy to overcome this obstacle is to apply the two‐phase strategy as described in the AIH study 45. We expect that this strategy will become popular in the near future. Finally, we expect that functional protein microarrays will be used as a readout to obtain reaction profiles of the collected activities of various types of enzymes, such as kinases, acetyltransferases, ubiquitin, and SUMO E3 ligases in cancerous tissues. Comparing PTM profiles obtained from cancer and healthy tissues will allow us to identify biomarkers and to gain new insights into the molecular mechanisms of disease.
ACKNOWLEDGMENTS
This work is supported in part by the NIH (RR020839, DK082840, RO1GM076102, CA125807, CA160036, and HG006434 to HZ; R01EY017589 to JQ, and an AHA Predoctoral Fellowship (10PRE3040000) to EC).
The authors have declared no conflicts of interest.
Colour Online: See the article online to view Figs. 1–4 in colour.
6 References
- 1. Gygi, S. P. , Rochon, Y. , Franza, B. R. , Aebersold, R. , Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 1999, 19, 1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kopf, E. , Zharhary, D. , Antibody arrays–an emerging tool in cancer proteomics. Int. J. Biochem. Cell Biol. 2007, 39, 1305–1317. [DOI] [PubMed] [Google Scholar]
- 3. Zhu, H. , Snyder, M. , Protein arrays and microarrays. Curr. Opin. Chem. Biol. 2001, 5, 40–45. [DOI] [PubMed] [Google Scholar]
- 4. Hu, S. , Xie, Z. , Onishi, A. , Yu, X. et al., Profiling the human protein‐DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 2009, 139, 610–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith, M. , Jona, G. , Ptacek, J. , Devgan, J. et al., Global analysis of protein function using protein function using protein microarrays. Mech. Age Dev. 2005, 126, 171–175. [DOI] [PubMed] [Google Scholar]
- 6. Xie, Z. , Hu, S. , Qian, J. , Blackshaw, S. et al., Systematic characterization of protein‐DNA interactions. Cell Mol. Life Sci. 2011, 68, 1657–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bussow, K. , Cahill, D. , Nietfeld, W. , Bancroft, D. et al., A method for global protein expression and antibody screening on high‐density filters of an arrayed cDNA library. Nucleic Acids Res. 1998, 26, 5007–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ge, H. , UPA, a universal protein array system for quantitative detection of protein‐protein, protein‐DNA, protein‐RNA and protein‐ligand interactions. Nucleic Acids Res. 2000, 28, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. MacBeath, G. , Schreiber, S. L. , Printing proteins as microarrays for high‐throughput function determination. Science 2000, 289, 1760–1763. [DOI] [PubMed] [Google Scholar]
- 10. Schweitzer, B. , Wiltshire, S. , Lambert, J. , O'Malley, S. et al., Immunoassays with rolling circle DNA amplification: a versatile platform for ultrasensitive antigen detection. Proc. Natl. Acad. Sci. USA 2000, 97, 10113–10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu, H. , Klemic, J. F. , Chang, S. , Bertone, P. et al., Analysis of yeast protein kinases using protein chips. Nat. Genet. 2000, 26, 283–289. [DOI] [PubMed] [Google Scholar]
- 12. Haab, B. B. , Dunham, M. J. , Brown, P. O. , Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001, 2, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arenkov, P. , Kukhtin, A. , Gemmell, A. , Voloshchuk, S. et al., Protein microchips: use for immunoassay and enzymatic reactions. Anal. Biochem. 2000, 278, 123–131. [DOI] [PubMed] [Google Scholar]
- 14. Bieri, C. , Ernst, O. P. , Heyse, S. , Hofmann, K. P. et al., Micropatterned immobilization of a G protein‐coupled receptor and direct detection of G protein activation. Nat. Biotechnol. 1999, 17, 1105–1108. [DOI] [PubMed] [Google Scholar]
- 15. Zhu, H. , Bilgin, M. , Bangham, R. , Hall, D. et al., Global analysis of protein activities using proteome chips. Science 2001, 293, 2101–2105. [DOI] [PubMed] [Google Scholar]
- 16. Zhu, H. , Hu, S. , Jona, G. , Zhu, X. et al., Severe acute respiratory syndrome diagnostics using a coronavirus protein microarray. Proc. Natl. Acad. Sci. USA 2006, 103, 4011–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen, C. S. , Korobkova, E. , Chen, H. , Zhu, J. et al., A proteome chip approach reveals new DNA damage recognition activities in Escherichia coli . Nat. Methods 2008, 5, 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Popescu, S. C. , Popescu, G. V. , Bachan, S. , Zhang, Z. et al., Differential binding of calmodulin‐related proteins to their targets revealed through high‐density Arabidopsis protein microarrays. Proc. Natl. Acad. Sci. USA 2007, 104, 4730–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feilner, T. , Hultschig, C. , Lee, J. , Meyer, S. et al., High throughput identification of potential Arabidopsis mitogen‐activated protein kinases substrates. Mol. Cell. Proteomics 2005, 4, 1558–1568. [DOI] [PubMed] [Google Scholar]
- 20. Lueking, A. , Possling, A. , Huber, O. , Beveridge, A. et al., A nonredundant human protein chip for antibody screening and serum profiling. Mol. Cell. Proteomics 2003, 2, 1342–1349. [DOI] [PubMed] [Google Scholar]
- 21. Jeong, J. S. , Jiang, L. , Albino, E. , Marrero, J. et al., Rapid identification of monospecific monoclonal antibodies using a human proteome microarray. Mol. Cell. Proteomics 2012, 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen, C. S. , Zhu, H. , Protein microarrays. BioTechniques 2006, 40, 423–429. [DOI] [PubMed] [Google Scholar]
- 23. Poetz, O. , Schwenk, J. M. , Kramer, S. , Stoll, D. et al., Protein microarrays: catching the proteome. Mech. Ageing Dev. 2005, 126, 161–170. [DOI] [PubMed] [Google Scholar]
- 24. Hall, D. , Zhu, H. , Zhu, X. , Royce, T. et al., Regulation of gene expression by a metabolic enzyme. Science 2004, 306, 482–484. [DOI] [PubMed] [Google Scholar]
- 25. Zhu, J. , Gopinath, K. , Murali, A. , Yi, G. et al., RNA‐binding proteins that inhibit RNA virus infection. Proc. Natl. Acad. Sci. USA 2007, 104, 3129–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ptacek, J. , Devgan, G. , Michaud, G. , Zhu, H. et al., Global analysis of protein phosphorylation in yeast. Nature 2005, 438, 679–684. [DOI] [PubMed] [Google Scholar]
- 27. Lin, Y. Y. , Lu, J. Y. , Zhang, J. , Walter, W. et al., Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 2009, 136, 1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu, J. , Lin, Y. , Qian, J. , Tao, S. et al., Functional dissection of an HECT ubiquitin E3 ligase. Mol. Cell. Proteomics 2007, 7, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu, J. Y. , Lin, Y. Y. , Sheu, J. C. , Wu, J. T. et al., Acetylation of yeast AMPK controls intrinsic aging independently of caloric restriction. Cell 2011, 146, 969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oh, Y. H. , Hong, M. Y. , Jin, Z. , Lee, T. et al., Chip‐based analysis of SUMO (small ubiquitin‐like modifier) conjugation to a target protein. Biosens. Bioelectr. 2007, 22, 1260–1267. [DOI] [PubMed] [Google Scholar]
- 31. Jeong, J. S. , Rho, H. S. , Zhu, H. , A functional protein microarray approach to characterizing posttranslational modifications on lysine residues. Methods Mol. Biol. 2011, 723, 213–223. [DOI] [PubMed] [Google Scholar]
- 32. Hu, S. , Xie, Z. , Qian, J. , Blackshaw, S. et al., Functional protein microarray technology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schnack, C. , Hengerer, B. , Gillardon, F. , Identification of novel substrates for Cdk5 and new targets for Cdk5 inhibitors using high‐density protein microarrays. Proteomics 2008, 8, 1980–1986. [DOI] [PubMed] [Google Scholar]
- 34. Tarrant, M. K. , Rho, H. S. , Xie, Z. , Jiang, Y. L. et al., Regulation of CK2 by phosphorylation and O‐GlcNAcylation revealed by semisynthesis. Nat. Chem. Biol. 2012, 8, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nikitin, P. A. , Luftig, M. A. , The DNA damage response in viral‐induced cellular transformation. Br. J. Cancer 2012, 106, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu, J. , Liao, G. , Shan, L. , Zhang, J. et al., Protein array identification of substrates of the Epstein‐Barr virus protein kinase BGLF4. J. Virol. 2009, 83, 5219–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gupta, R. , Kus, B. , Fladd, C. , Wasmuth, J. et al., Ubiquitination screen using protein microarrays for comprehensive identification of Rsp5 substrates in yeast. Mol. Syst. Biol. 2007, 3, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Persaud, A. , Alberts, P. , Amsen, E. M. , Xiong, X. et al., Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4–2 using proteome arrays. Mol. Syst. Biol. 2009, 5, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thao, S. , Chen, C. S. , Zhu, H. , Escalante‐Semerena, J. , Nε‐lysine acetylation of a bacterial transcription factor inhibits its DNA‐binding activity. PLoS ONE 2010, 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Foster, M. W. , Forrester, M. T. , Stamler, J. S. , A protein microarray‐based analysis of S‐nitrosylation. Proc. Natl. Acad. Sci. USA 2009, 106, 18948–18953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zheng, D. , Wan, J. , Cho, Y. G. , Wang, L. et al., Comparison of humoral immune responses to Epstein‐Barr virus and Kaposi's sarcoma‐associated herpesvirus using a viral proteome microarray. J. Infect. Dis. 2011, 204, 1683–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li, B. , Jiang, L. , Song, Q. , Yang, J. et al., Protein microarray for profiling antibody responses to Yersinia pestis live vaccine. Infect. Immun. 2005, 73, 3734–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vigil, A. , Chen, C. , Jain, A. , Nakajima‐Sasaki, R. et al., Profiling the humoral immune response of acute and chronic Q fever by protein microarray. Mol. Cell. Proteomics 2011, 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Doolan, D. L. , Mu, Y. , Unal, B. , Sundaresh, S. et al., Profiling humoral immune responses to P. falciparum infection with protein microarrays. Proteomics 2008, 8, 4680–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Song, Q. , Liu, G. , Hu, S. , Zhang, Y. et al., Novel autoimmune hepatitis‐specific autoantigens identified using protein microarray technology. J. Proteome Res. 2010, 9, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen, C. S. , Sullivan, S. , Anderson, T. , Tan, A. C. et al., Identification of novel serological biomarkers for inflammatory bowel disease using Escherichia coli proteome chip. Mol. Cell. Proteomics 2009, 8, 1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liang, L. , Tan, X. , Juarez, S. , Villaverde, H. et al., Systems biology approach predicts antibody signature associated with Brucella melitensis infection in humans. J. Proteome Res. 2011, 10, 4813–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hu, C. J. , Song, G. , Huang, W. , Liu, G. Z. et al., Identification of new autoantigens for primary biliary cirrhosis using human proteome microarrays. Mol. Cell. Proteomics 2012, 11, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hu, S. , Vissink, A. , Arellano, M. , Roozendaal, C. et al., Identification of autoantibody biomarkers for primary Sjogren's syndrome using protein microarrays. Proteomics 2011, 11, 1499–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anderson, K. S. , Sibani, S. , Wallstrom, G. , Qiu, J. et al., Protein microarray signature of autoantibody biomarkers for the early detection of breast cancer. J. Proteome Res. 2011, 10, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tao, S. C. , Li, Y. , Zhou, J. , Qian, J. et al., Lectin microarrays identify cell‐specific and functionally significant cell surface glycan markers. Glycobiology 2008, 18, 761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Orenes‐Pinero, E. , Barderas, R. , Rico, D. , Casal, J. I. et al., Serum and tissue profiling in bladder cancer combining protein and tissue arrays. J. Proteome Res. 2010, 9, 164–173. [DOI] [PubMed] [Google Scholar]
- 53. Petricoin, E. F. , Espina, V. , Araujo, R. P. , Midura, B. et al., Phosphoprotein pathway mapping: Akt/mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res. 2007, 67, 3431–3440. [DOI] [PubMed] [Google Scholar]
- 54. Luevano, M. , Bernard, H. U. , Barrera‐Saldana, H. A. , Trevino, V. et al., High‐throughput profiling of the humoral immune responses against thirteen human papillomavirus types by proteome microarrays. Virology 2010, 405, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Arvin, A. , Campadelli‐Fiume, G. , Mocarski, E. , Moore, P. S. et al., Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge University Press, Cambridge: 2007. [PubMed] [Google Scholar]
- 56. Li, R. , Zhu, J. , Xie, Z. , Liao, G. et al., Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell. Host Microbe 2011, 10, 390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shamay, M. , Liu, J. , Li, R. , Liao, G. et al., A protein array screen for Kaposi's sarcoma‐associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity, and telomere shortening. J. Virol. 2012, 86, 5179–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li, R. , Wang, L. , Liao, G. , Guzzo, C. M. et al., SUMO binding by the Epstein‐Barr virus protein kinase BGLF4 is crucial for BGLF4 function. J. Virol. 2012, 86, 5412–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Margarit, I. , Bonacci, S. , Pietrocola, G. , Rindi, S. et al., Capturing host‐pathogen interactions by protein microarrays: identification of novel streptococcal proteins binding to human fibronectin, fibrinogen, and C4BP . FASEB J. 2009, 23, 3100–3112. [DOI] [PubMed] [Google Scholar]
- 60. Komarova, A. V. , Combredet, C. , Meyniel‐Schicklin, L. , Chapelle, M. et al., Proteomic analysis of virus‐host interactions in an infectious context using recombinant viruses. Mol. Cell. Proteomics 2011, 10, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Krishnan, M. N. , Ng, A. , Sukumaran, B. , Gilfoy, F. D. et al., RNA interference screen for human genes associated with West Nile virus infection. Nature 2008, 455, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Brass, A. L. , Huang, I. C. , Benita, Y. , John, S. P. et al., The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Uetz, P. , Dong, Y. A. , Zeretzke, C. , Atzler, C. et al., Herpesviral protein networks and their interaction with the human proteome. Science 2006, 311, 239–242. [DOI] [PubMed] [Google Scholar]
- 64. Pfefferle, S. , Schopf, J. , Kogl, M. , Friedel, C. C. et al., The SARS‐coronavirus‐host interactome: identification of cyclophilins as target for pan‐coronavirus inhibitors. PLoS Pathog. 2011, 7, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Parsons, D. W. , Li, M. , Zhang, X. , Jones, S. et al., The genetic landscape of the childhood cancer medulloblastoma. Science 2011, 331, 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gerlinger, M. , Rowan, A. J. , Horswell, S. , Larkin, J. et al., Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Borrebaeck, C. A. , Wingren, C. , Design of high‐density antibody microarrays for disease proteomics: key technological issues. J. Proteomics 2009, 72, 928–935. [DOI] [PubMed] [Google Scholar]
- 68. Haab, B. B. , Methods and applications of antibody microarrays in cancer research. Proteomics 2003, 3, 2116–2122. [DOI] [PubMed] [Google Scholar]
- 69. Paweletz, C. P. , Charboneau, L. , Bichsel, V. E. , Simone, N. L. et al., Reverse phase protein microarrays which capture disease progression show activation of pro‐survival pathways at the cancer invasion front. Oncogene 2001, 20, 1981–1989. [DOI] [PubMed] [Google Scholar]
- 70. Mueller, C. , Liotta, L. A. , Espina, V. , Reverse phase protein microarrays advance to use in clinical trials. Mol. Oncol. 2010, 4, 461–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kalyuzhny, A. E. , The dark side of the immunohistochemical moon: industry. J. Histochem. Cytochem. 2009, 57, 1099–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Couchman, J. R. , Commercial antibodies: the good, bad, and really ugly. J. Histochem. Cytochem. 2009, 57, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jensen, B. C. , Swigart, P. M. , Simpson, P. C. , Ten commercial antibodies for alpha‐1‐adrenergic receptor subtypes are nonspecific. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 379, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hughes, B. , Antibody‐drug conjugates for cancer: poised to deliver? Nat. Rev. Drug Discov. 2010, 9, 665–667. [DOI] [PubMed] [Google Scholar]
- 75. Berger, J. R. , Houff, S. A. , Major, E. O. , Monoclonal antibodies and progressive multifocal leukoencephalopathy. MAbs 2009, 1, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tomiya, N. , Awaya, J. , Kurono, M. , Endo, S. et al., Analyses of N‐linked oligosaccharides using a two‐dimensional mapping technique. Anal. Biochem. 1988, 171, 73–90. [DOI] [PubMed] [Google Scholar]
- 77. Hase, S. , Ikenaka, T. , Matsushima, Y. , Structure analyses of oligosaccharides by tagging of the reducing end sugars with a fluorescent compound. Biochem. Biophys. Res. Commun. 1978, 85, 257–263. [DOI] [PubMed] [Google Scholar]
- 78. Kameyama, A. , Kikuchi, N. , Nakaya, S. , Ito, H. et al., A strategy for identification of oligosaccharide structures using observational multistage mass spectral library. Anal. Chem. 2005, 77, 4719–4725. [DOI] [PubMed] [Google Scholar]
- 79. Kamoda, S. , Nakanishi, Y. , Kinoshita, M. , Ishikawa, R. et al., Analysis of glycoprotein‐derived oligosaccharides in glycoproteins detected on two‐dimensional gel by capillary electrophoresis using on‐line concentration method. J. Chromatogr. A 2006, 1106, 67–74. [DOI] [PubMed] [Google Scholar]
- 80. Kamoda, S. , Kakehi, K. , Capillary electrophoresis for the analysis of glycoprotein pharmaceuticals. Electrophoresis 2006, 27, 2495–2504. [DOI] [PubMed] [Google Scholar]
- 81. Poulain, S. , Lepelley, P. , Cambier, N. , Cosson, A. et al., Assessment of P‐glycoprotein expression by immunocytochemistry and flow cytometry using two different monoclonal antibodies coupled with functional efflux analysis in 34 patients with acute myeloid leukemia. Adv. Exp. Med. Biol. 1999, 457, 57–63. [DOI] [PubMed] [Google Scholar]
- 82. Frojmovic, M. , Wong, T. , Dynamic measurements of the platelet membrane glycoprotein IIb‐IIIa receptor for fibrinogen by flow cytometry. II. Platelet size‐dependent subpopulations. Biophys. J. 1991, 59, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kung, L. A. , Tao, S. C. , Qian, J. , Smith, M. G. et al., Global analysis of the glycoproteome in Saccharomyces cerevisiae reveals new roles for protein glycosylation in eukaryotes. Mol. Syst. Biol. 2009, 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pilobello, K. T. , Krishnamoorthy, L. , Slawek, D. , Mahal, L. K. , Development of a lectin microarray for the rapid analysis of protein glycopatterns. Chembiochem. 2005, 6, 985–989. [DOI] [PubMed] [Google Scholar]
- 85. Angeloni, S. , Ridet, J. L. , Kusy, N. , Gao, H. et al., Glycoprofiling with micro‐arrays of glycoconjugates and lectins. Glycobiology 2005, 15, 31–41. [DOI] [PubMed] [Google Scholar]
- 86. Kuno, A. , Kato, Y. , Matsuda, A. , Kaneko, M. K. et al., Focused differential glycan analysis with the platform antibody‐assisted lectin profiling for glycan‐related biomarker verification. Mol. Cell. Proteomics 2009, 8, 99–108. [DOI] [PubMed] [Google Scholar]
- 87. Zheng, T. , Peelen, D. , Smith, L. M. , Lectin arrays for profiling cell surface carbohydrate expression. J. Am. Chem. Soc. 2005, 127, 9982–9983. [DOI] [PubMed] [Google Scholar]
- 88. Zhou, S. M. , Cheng, L. , Guo, S. J. , Zhu, H. et al., Lectin microarrays: a powerful tool for glycan‐based biomarker discovery. Comb. Chem. High Throughput Screen. 2011, 14, 711–719. [DOI] [PubMed] [Google Scholar]
- 89. Uchiyama, N. , Kuno, A. , Koseki‐Kuno, S. , Ebe, Y. et al., Development of a lectin microarray based on an evanescent‐field fluorescence principle. Methods Enzymol. 2006, 415, 341–351. [DOI] [PubMed] [Google Scholar]
- 90. Li, Y. , Tao, S. C. , Bova, G. S. , Liu, A. Y. et al., Detection and verification of glycosylation patterns of glycoproteins from clinical specimens using lectin microarrays and lectin‐based immunosorbent assays. Anal. Chem. 2011, 83, 8509–8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Murphy, M. A. , O'Leary, J. J. , Cahill, D. J. , Assessment of the humoral immune response to cancer. J. Proteomics 2012, 75, 4573–4579. [DOI] [PubMed] [Google Scholar]