

Abstract
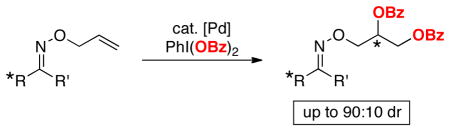
A Pd-catalyzed asymmetric alkene 1,2-dioxygenation reaction is described. The diastereoselectivity of the reaction is controlled by tethering a chiral oxime ether directing group to the alkene substrate. The best selectivities are obtained with 8-substituted menthone-derived oxime ether auxiliaries.
Alkene difunctionalization reactions are synthetically valuable methods that generate two new bonds and up to two stereocenters in a single operation. These transformations are frequently catalyzed by transition metals, with osmium-based catalysts being particularly effective at controlling the relative and absolute stereochemistry of the products.1 Recently, a number of exciting reports have demonstrated the feasibility of high-oxidation state Pd-catalyzed alkene difunctionalization.2,3 These Pd-catalyzed reactions are of particular interest because they are mechanistically distinct from the corresponding Os systems. Whereas Os promotes concerted syn addition of two heteroatoms across the alkene, high valent Pd catalysis involves the formation of each new carbon–X bond in a discrete step.2a As a result, this mechanistic manifold offers the potential for greater reaction diversity, with the possibility of adding two different functional groups across a carbon carbon double bond in a highly selective fashion.2,3
Over the past 10 years, numerous high-valent Pd-catalyzed alkene difunctionalization reactions have been developed.4–12 These include methods for alkene dioxygenation,4 aminooxygenation,5 diamination,7 amino-halogenation,8,9 and aryl halogenation.10 These transformations often proceed with selectivity for either syn or anti addition of the vicinal groups. However, the stereoselective formation of non-racemic products in these reactions has remained largely elusive.13
One appealing approach for achieving asymmetric Pd-catalyzed alkene difunctionalization would involve the use of a chiral directing group. A related strategy has been successfully employed by Yu to achieve asymmetric high valent Pd-catalyzed ligand-directed C–H bond oxidation.14 In a similar fashion, we envisioned that the chiral directing group in substrate 1 (Scheme 1) could relay stereochemical information to the stereocenter formed in the product (3) via palladacycle intermediate 2. This approach should also enable more facile difunctionalization, because the directing group would bring the Pd proximal to the target olefin.
Scheme 1.
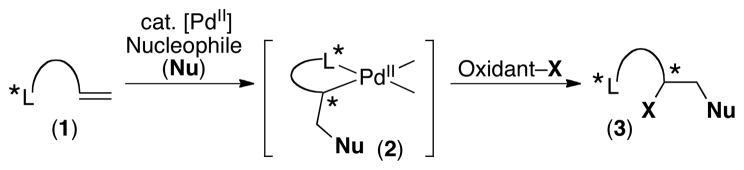
Chiral Ligand-Directed Approach to Asymmetric Alkene Difunctionalization
Our initial investigations have focused on chiral oxime ethers as directing groups for asymmetric alkene dioxygenation. These directing groups were selected based on three key criteria: (1) oxime ethers are known to be effective directing ligands for other Pd-catalyzed reactions (particularly C–H functionalization),15 (2) the substrates can easily be prepared from an allyl alcohol and a chiral ketone, and (3) diverse chiral non-racemic ketones are readily available.
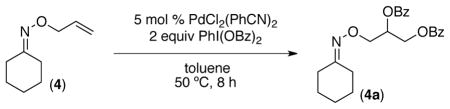
We first examined the Pd-catalyzed dioxygenation of achiral oxime ether substrate 4 with PhI(O2CPh)2.4 Using PdCl2(PhCN)2 as the catalyst, this transformation proceeded in good yield under mild conditions (8 h at 50 °C in toluene) (Table 1, entry 1).16 Importantly, no dioxygenation was observed in the absence of Pd under these conditions, even upon the addition of 5–10 mol % of TfOH or BF3•OEt2.17 Although allyl propyl ether (5) and oxime ether 4 both contain an olefin allylic to an oxygen atom, 5 was completely unreactive under the optimized conditions. This result demonstrates that the oxime ether moiety in 4 is necessary to facilitate the dioxygenation reaction under these mild conditions.18
Table 1.
Dibenzoylation of 4 and Control Reactionsa
| |||||
|---|---|---|---|---|---|
| entry | substrate | Pd (mol %) | additive | product | % yield |
| 1 | 4 | 5 | none | 4a | 59b |
| 2 | 0 | none | <1c | ||
| 3 | 0 | 5 mol % TfOH | <1c | ||
| 4 | 0 | 10 mol % BF3•OEt2 | <1c | ||
| 5 |
|
5 | none |
|
<1c |
Conditions: substrate (1 equiv), PdCl2(PhCN)2 (0.05 equiv), PhI(OBz)2 (2 equiv), dry toluene (0.12 M in substrate), 50 °C, 8 h.
Isolated yield.
Product was not detected by NMR spectroscopic analysis of the crude reaction mixture.
Literature precedent suggests that this transformation proceeds via a mechanism involving oxime ether coordination, oxypalladation, oxidation from PdII to PdIV and then C–O bond forming reductive elimination (Scheme 2). Within the context of this general manifold, there are several unresolved details that are critical for the stereochemical outcome of the reaction. In particular, the initial step could proceed via either trans (paths a and b) or cis oxypalladation (paths c and d);2,4 furthermore, the final reductive elimination could proceed by direct (paths a and c) or SN2-type (paths b and d) mechanisms.2,4,5 In an achiral system, these four pathways would lead to two different products (overall syn or anti addition).
Scheme 2.
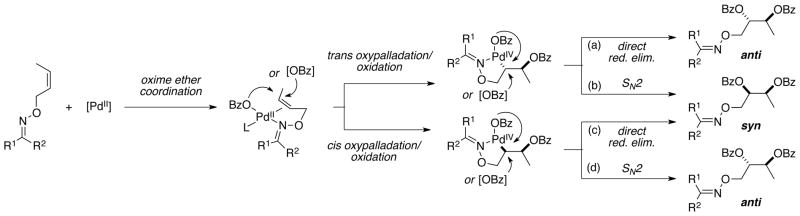
Possible Mechanistic Pathways for Oxime-Directed Dibenzoylation
To probe these possibilities, we examined the stereochemical outcome of Pd-catalyzed dibenzoylation with cis- and trans-7 as substrates. Under our optimal conditions, dioxygenation of both alkenes proceeded with high (≥10:1) selectivity for the syn addition product (Scheme 3).19 The syn selectivity suggests that the primary operative mechanism is either (b) or (c) in Scheme 2. The observation of high diastereoselectivity in this transformation is critical for our goal of achieving asymmetric dioxygenation, as it indicates that the stereocenter-forming steps proceed with high stereochemical fidelity.
Scheme 3.
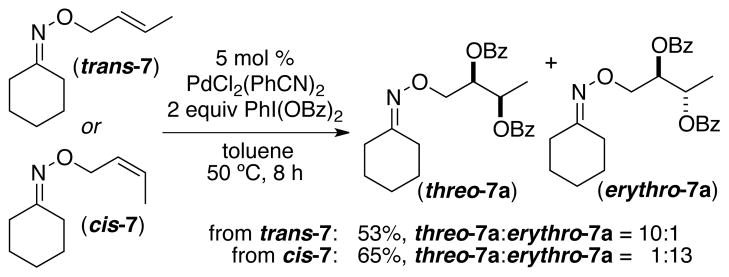
Syn Diastereoselectivity in the Dibenzoylation of trans and cis Internal Alkenes 7
We next examined a series of chiral allyl oxime ethers derived from commercially available chiral ketones (8–12, Table 2). Notably, the E and Z oxime isomers of the same parent ketone present significantly different steric environments to a coordinated Pd center. Therefore, the E and Z diastereomers were separated and studied individually when possible (substrates 8 and 10).
Table 2.
Alkene Dioxygenation with Chiral Auxiliaries Derived from Commercial Ketonesa
| |||
|---|---|---|---|
| substrate | product | yieldb | drc |
 (E-8) |
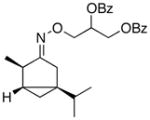 (E-8a) |
64% | 63:37 |
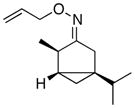 (Z-8) |
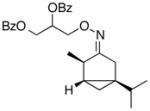 (Z-8a) |
63% | 52:48 |
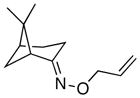 (9) |
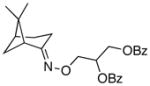 (9a) |
68% | 59:41 |
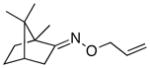 (E-10) |
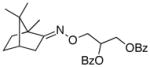 (E-10a) |
25% | 75:25 |
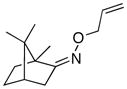 (Z-10) |
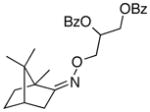 (Z-10a) |
67% | 66:34 |
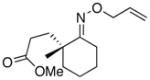 (11) |
 (11a) |
<1%d | -- |
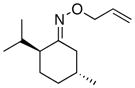 (12) |
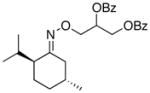 (12a) |
75% | 74:26 |
Conditions: substrate (1 equiv), PdCl2(PhCN)2 (0.05 equiv), PhI(OBz)2 (2 equiv), dry toluene (0.12 M in substrate), 50 °C, 8 h.
Isolated yield.
Determined by relative integrations of at least 2 pairs of peaks in the 13C and/or 1H NMR spectra; see Supporting Information for details.
Product was not detected by NMR spectroscopic analysis of the crude reaction mixture.
Oxime stereoisomers E-8 and Z-8 underwent Pd-catalyzed dibenzoylation to afford two spectroscopically different sets of products. This result demonstrates that the oxime ether is configurationally stable under the mild reaction conditions. The stereocenter α to the oxime will be in closer proximity to the coordinated Pd center in E-8 relative to Z-8. Thus, as expected, the former afforded the dibenzoylated product in higher dr than the latter (63:37 versus 52:48, respectively).
In keeping with the trend observed with E-8 and Z-8, the dr’s obtained with substrates derived from other commercial chiral ketones tracked with the steric environment at the α-position of the oxime. For example, the diastereoselectivity of the reaction of 9 was lower than that seen with E-8. This can be rationalized based on the fact that the 3°-α stereocenter of 9 is tied back into a bicyclic scaffold. Improved diastereoselectivity (75:25) was observed in E-10, which contains a 4°-α stereocenter. However, the overall yield was low, likely due to the highly congested steric environment proximal to the oxime. In keeping with this hypothesis, the opposite oxime isomer (Z-10) afforded lower dr (66:33) but better reactivity. Furthermore, the use of substrate 11, which contains an even more hindered 4°-α stereocenter, resulted in dramatically decreased reactivity (no dioxygenated product was observed in this case). Among the substrates in Table 2, the best balance between reactivity and selectivity was obtained with menthone-derived substrate 12, which features β-branching adjacent to a 3°-α stereocenter.
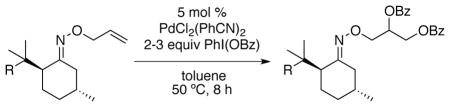
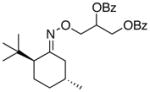
To improve further on the selectivity seen with 12, we prepared a number of 8-substituted menthone auxiliaries. Interestingly, the addition of a third methyl group to the β-position of the ketone (13) provided a substantial improvement in stereoselectivity (dr = 86:14 for 13a, compared to 74:26 for 12a). The selectivity remained unchanged upon additional mono-substitution at the γ position. However, a further improvement in dr (to 90:10) was observed with 17, which contains a 3°-carbon center at the γ position. Notably, as shown in Table 3, a decline in reaction yield was seen as the steric bulk of the auxiliary increased (compare 17a and 15a to 13a). Overall, auxilliaries 13 and 17 provide particularly promising results and have potential for further applications in this field.
Table 3.
Alkene Dioxygenation with Chiral Auxiliaries Derived from 8-Substituted Menthonea
| |||
|---|---|---|---|
| substrate | product | yieldb | drc |
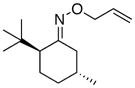 (13) |
 (13a) |
69% | 86:14 |
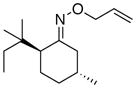 (14) |
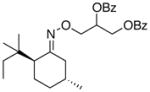 (14a) |
70% | 86:14 |
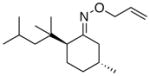 (15) |
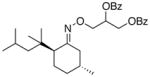 (15a) |
54% | 86:14 |
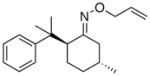 (16) |
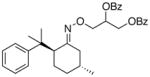 (16a) |
44% | 85:15 |
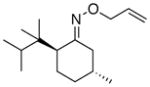 (17) |
 (17a) |
37% | 90:10 |
Conditions: substrate (1 equiv), PdCl2(PhCN)2 (0.05 equiv), PhI(OBz)2 (2–3 equiv), dry toluene (0.12 M in substrate), 50 °C, 8 h.
Isolated yield.
Determined by relative integrations of at least 2 pairs of peaks in the 13C and/or 1H NMR spectra; see Supporting Information for details.
In summary, the results presented herein demonstrate that a chiral oxime ether directing group can be used to control the stereochemical outcome of high valent Pd-catalyzed alkene dioxygenation. Readily accessible menthone derivatives are particularly effective auxiliaries for these transformations. Ongoing investigations are focused on broadening the scope of this work to a more diverse set of nucleophiles and alkene substrates. In addition, current studies seek to expand this strategy to the use of related chiral auxiliaries as catalysts via reversible in situ formation of oxime ether and/or imine derivatives.
Supplementary Material
Acknowledgments
This work was supported by the NIH NIGMS (GM073836).
Footnotes
Supporting Information Available: Experimental details and spectroscopic and analytical data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Reviews: Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483.Johnson RA, Sharpless KB. In: Catalytic Asymmetric Synthesis. 2. Ojima I, editor. Wiley-VCH; New York: 2000. p. 357.Bolm C, Hildebrand JP, Muñiz K. In: Catalytic Asymmetric Synthesis. 2. Ojima I, editor. Wiley-VCH; New York: 2000. p. 399.
- 2.Reviews on Pd-catalyzed alkene difunctionalization: Jensen KH, Sigman MS. Org Biomol Chem. 2008;6:4083. doi: 10.1039/b813246a.Wolfe JP. Synlett. 2008:2913.Jacques B, Muñiz K. In: Catalyzed Carbon-Heteroatom Bond Formation. Yudin AK, editor. Wiley-VCH; Weinhem, Germany: 2010. p. 119.McDonald RI, Liu G, Stahl SS. Chem Rev. 2011;111:2981. doi: 10.1021/cr100371y.
- 3.Reviews on high valent Pd: Muñiz K. Angew Chem Int Ed. 2009;48:9412. doi: 10.1002/anie.200903671.Canty AJ. Dalton Trans. 2009;47:10409. doi: 10.1039/b914080h.Xu LM, Li BJ, Yang Z, Shi ZJ. Chem Soc Rev. 2010;39:712. doi: 10.1039/b809912j.Hickman AJ, Sanford MS. Nature. 2012;484:177. doi: 10.1038/nature11008.
- 4.Dioxygenation: Li Y, Song D, Dong VM. J Am Chem Soc. 2008;130:2962. doi: 10.1021/ja711029u.Wang A, Jiang H, Chen H. J Am Chem Soc. 2009;131:3846. doi: 10.1021/ja900213d.Wang W, Wang F, Shi M. Organometallics. 2010;29:928.Park CP, Lee JH, Yoo KS, Jung KW. Org Lett. 2010;12:2450. doi: 10.1021/ol1001686.
- 5.Aminooxygenation: Alexanian EJ, Lee C, Sorensen EJ. J Am Chem Soc. 2005;127:7690. doi: 10.1021/ja051406k.Liu G, Stahl SS. J Am Chem Soc. 2006;128:7179. doi: 10.1021/ja061706h.Desai LV, Sanford MS. Angew Chem Int Ed. 2007;46:5737. doi: 10.1002/anie.200701454.
- 6.Aminoarylation: Rosewall CF, Sibbald PA, Liskin DV, Michael FE. J Am Chem Soc. 2009;131:9488. doi: 10.1021/ja9031659.Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J Am Chem Soc. 2009;131:15945. doi: 10.1021/ja906915w.
- 7.Diamination: Streuff J, Hövelmann CH, Nieger M, Muñiz K. J Am Chem Soc. 2005;127:14586. doi: 10.1021/ja055190y.Muñiz K. J Am Chem Soc. 2007;129:14542. doi: 10.1021/ja075655f.Muñiz K, Hövelmann CH, Streuff J. J Am Chem Soc. 2008;130:763. doi: 10.1021/ja075041a.Sibbald PA, Michael FE. Org Lett. 2009;11:1147. doi: 10.1021/ol9000087.Iglesias Á, Pérez EG, Muñiz K. Angew Chem Int Ed. 2010;49:8109. doi: 10.1002/anie.201003653.Muñiz K, Kirsch J, Chávez P. Adv Synth Catal. 2011;353:689.Martínez C, Muñiz K. Angew Chem Int Ed. 2012;51:7031. doi: 10.1002/anie.201201719.
- 8.Aminofluorination: Wu T, Yin G, Liu G. J Am Chem Soc. 2009;131:16354. doi: 10.1021/ja9076588.Qiu S, Xu T, Zhou J, Guo Y, Liu G. J Am Chem Soc. 2010;132:2856. doi: 10.1021/ja909716k.
- 9.Chloroamination: Michael FE, Sibbald PA, Cochran BM. Org Lett. 2008;10:793. doi: 10.1021/ol702922c.Yin G, Wu T, Liu G. Chem Eur J. 2012;18:451. doi: 10.1002/chem.201102776.
- 10.Arylhalogenation: Kalyani D, Sanford MS. J Am Chem Soc. 2008;130:2150. doi: 10.1021/ja0782798.Kalyani D, Satterfield AD, Sanford MS. J Am Chem Soc. 2010;132:8419. doi: 10.1021/ja101851v.
- 11.Cyclopropanation: Tong X, Beller M, Tse MK. J Am Chem Soc. 2007;129:4906. doi: 10.1021/ja070919j.Welbes LL, Lyons TW, Cychosz KA, Sanford MS. J Am Chem Soc. 2007;129:5836. doi: 10.1021/ja071204j.Lyons TW, Sanford MS. Tetrahedron. 2009;65:3211. doi: 10.1016/j.tet.2008.10.107.Tsujihara T, Takenaka K, Onitsuka K, Hatanaka M, Sasai H. J Am Chem Soc. 2009;131:3452. doi: 10.1021/ja809965e.
- 12.Aryltrifluoromethylation: Mu X, Wu T, Wang H-y, Guo Y-l, Liu G. J Am Chem Soc. 2012;134:878. doi: 10.1021/ja210614y.
- 13.For an example of asymmetric alkene difunctionalization via high valent Pd catalysis that utilizes chiral SPRIX ligands, see ref 11d.
- 14.(a) Giri R, Chen X, Yu JQ. Angew Chem Int Ed. 2005;44:2112. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]; (b) Giri R, Liang J, Lei JG, Li JJ, Wang DH, Chen X, Naggar IC, Guo C, Foxman BM, Yu JQ. Angew Chem Int Ed. 2005;44:7420. doi: 10.1002/anie.200502767. [DOI] [PubMed] [Google Scholar]; (c) Giri R, Chen X, Hao XS, Li JJ, Liang J, Fan ZP, Yu JQ. Tetrahedron: Asymmetry. 2005;16:3502. [Google Scholar]; (d) Giri R, Lan Y, Liu P, Houk KN, Yu JQ. J Am Chem Soc. 2012;134:14118. doi: 10.1021/ja304643e. [DOI] [PubMed] [Google Scholar]
- 15.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e.and references therein. Chan CW, Zhou Z, Chan ASC, Yu WY. Org Lett. 2010;12:3926. doi: 10.1021/ol101618u.Neufeldt SR, Sanford MS. Org Lett. 2010;12:532. doi: 10.1021/ol902720d.Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J Am Chem Soc. 2011;133:18566. doi: 10.1021/ja208068w.
- 16.See Supporting Information for full details of the optimization of this transformation.
- 17.For TfOH- or BF3•OEt2-catalyzed alkene dioxygenation with PhI(OAc)2 see Kang YB, Gade LH. J Am Chem Soc. 2011;133:3658. doi: 10.1021/ja110805b.Zhong W, Yang J, Meng X, Li Z. J Org Chem. 2011;76:9997. doi: 10.1021/jo201752y.
-
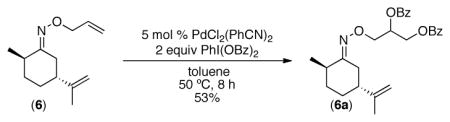
18.A remote alkene was also tolerated:
- 19.Relative stereochemistry was determined by deprotection of each product to the diol and spectroscopic comparison to an authentic sample of the erythro diol prepared by dihydroxylation of cis-7 with AD-mix-α; see Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.