

Abstract
Although targeting oncogenic mutations in the BRAF serine/threonine kinase with small molecule inhibitors can lead to significant clinical responses in melanoma, it fails to eradicate tumors in nearly all patients. Successful therapy will be aided by identification of intrinsic mechanisms that protect tumor cells from death. Here, we used a bioinformatics approach to identify drug-able, “driver” oncogenes restricted to tumor versus normal tissues. Applying this method to 88 short-term melanoma cell cultures, we show that the antiapoptotic BCL2 family member BCL2A1 is recurrently amplified in ∼30% of melanomas and is necessary for melanoma growth. BCL2A1 overexpression also promotes melanomagenesis of BRAF-immortalized melanocytes. We find that high-level expression of BCL2A1 is restricted to melanoma due to direct transcriptional control by the melanoma oncogene MITF. Although BRAF inhibitors lead to cell cycle arrest and modest apoptosis, we find that apoptosis is significantly enhanced by suppression of BCL2A1 in melanomas with BCL2A1 or MITF amplification. Moreover, we find that BCL2A1 expression is associated with poorer clinical responses to BRAF pathway inhibitors in melanoma patients. Cotreatment of melanomas with BRAF inhibitors and obatoclax, an inhibitor of BCL2A1 and other BCL2 family members, overcomes intrinsic resistance to BRAF inhibitors in BCL2A1-amplified cells in vitro and in vivo. These studies identify MITF-BCL2A1 as a lineage-specific oncogenic pathway in melanoma and underscore its role for improved response to BRAF-directed therapy.
High-resolution somatic copy number and genome sequencing of cancer have identified key driver mutations that form the basis for rationally targeted therapeutics. In melanoma, the most commonly mutated molecule, the protein kinase BRAF gene, is mutated in ∼50% of cases. The majority of BRAF mutations result in the substitution of valine by glutamic acid at position 600 (termed V600E), leading to a ∼500-fold increase in its kinase activity (1). BRAF(V600E) promotes oncogenesis through activation of the MEK1/2 kinases and the MAPK signal transduction cascade. BRAF has been shown by overexpression and knockdown experiments to be a critical mediator of melanomagenesis. Introduction of mutated BRAF into immortalized melanocytes leads to anchorage-independent growth and tumors in mice. However, oncogenesis induced by BRAF requires other genetic alterations, because oncogenic BRAF induces cellular senescence in primary melanocytes. In mice, dysregulation of BRAF, in cooperation with inactivation of the tumor suppressors PTEN or INK4A, leads to development of melanoma with short latency (2, 3).
Conversely, suppression of BRAF by RNA interference or small molecule inhibitors leads to cell cycle arrest and apoptosis in preclinical models (4–7). BRAF mutations generally predict response to the BRAF inhibitor vemurafenib (PLX4032), yet some BRAF mutant melanoma cell lines are relatively resistant (8–10). Treatment of most patients whose tumors have the BRAF(V600E) mutation also leads to tumor regression and improved survival (11). However, the duration of such responses is highly variable and virtually all patients eventually relapse (11–13), indicating that resistance mechanisms limit both the magnitude and duration of clinical response.
Here we undertook an integrated bioinformatic and functional analysis to identify genomically amplified therapeutic targets in melanoma and other malignancies. We identify the antiapoptotic factor BCL2A1 as a unique melanoma oncogene located on chromosome 15q. This region is significantly amplified in ∼30–40% of melanomas by large-scale copy number analyses and was previously observed to correlate with resistance of melanomas to chemotherapy (14). Unexpectedly, we find that high-level expression of BCL2A1 is largely restricted to melanomas compared with other tumor types. The lineage-specific expression was attributable to its direct regulation by the melanoma oncogene MITF. BCL2A1 is essential for survival in those melanomas in which it is amplified, and its overexpression is shown to promote tumorigenesis in cooperation with BRAF(V600E). Although BRAF inhibitors lead to cell cycle arrest and modest apoptosis, apoptosis is significantly enhanced by suppression of BCL2A1 in melanomas harboring BCL2A1 or MITF amplification. Finally, the combination of a BRAF inhibitor and obatoclax, an inhibitor of BCL2 family members including BCL2A1 currently in clinical trials, enhances apoptosis and tumor regression in vitro and in vivo.
Results
Bioinformatic Analysis Identifies Targets of Genomic Amplification.
High-resolution somatic copy number amplifications combined with gene expression profiles have been previously applied to identify causal oncogenes in a variety of malignancies (15–21). However, considerable obstacles exist to translation of these analyses to the clinic. Reasoning that the ability to identify amplified genes that are restricted to tumor cells compared with host tissues could aid the development of targeted therapy with decreased risk of toxicity, we performed a bioinformatics screen for candidate oncogenes in several tumor types, including breast, glioblastoma, colon, and melanoma for which comprehensive genomic datasets were publically available. For each tumor type, we compared gene expression to a pooled set of 72 normal tissues and identified genes whose expression was significantly higher in cancer compared with normal tissues [at false discovery rate (FDR) = 0] and that were significantly genomically amplified by the GISTIC algorithm (22). We considered genomic copy number of 3 or more as significant, given that this level of amplification can meaningfully affect protein expression and sensitivity to knockdown (15). The analysis yielded several known oncogenes including ERBB2 (23) and CCND1 in breast cancer (SI Appendix, Table S1). In melanoma, eight genes overlapped between the two analyses (Fig. 1A and SI Appendix, Fig. S1A), including the lineage-specific transcription factor MITF, consistent with its previously described amplification (21). Of particular interest was BCL2A1, one of six antiapoptotic BCL2 family members that has not been previously described as a human oncogene (24). Other BCL2 family members have been described as oncogenes by amplification or translocation, and several approaches to pharmacologically target these proteins, including BCL2A1, are in clinical development (25–29). To confirm these results, we also evaluated an independent microarray dataset (30). BCL2A1 and MITF mRNA were higher in melanoma compared with skin, which predominantly consists of nonmelanocytes (SI Appendix, Fig. S2). To confirm that BCL2A1 genomic amplification occurs within primary tumor specimens, in addition to the early passage melanoma cultures we examined above, we examined data from The Cancer Genome Atlas Project (www.broadinstitute.org/tcga/home). This dataset demonstrated that both BCL2A1 and MITF were amplified significantly only in melanoma and that 30.8% of primary melanoma biopsies had BCL2A1 amplification.
Fig. 1.

Scheme for identification of unique amplified oncogenic targets. (A) Diagram showing intersection of amplified genes that were significantly increased in melanoma compared with pooled normal tissues. (B) Approach for identification of oncogenes located on chromosome 15q. Eighty-eight melanomas with copy number analysis and gene expression were analyzed by GISTIC analysis and filtered for higher expression in 15q-amplified versus nonamplified melanomas (P < 0.05, Wilkcoxon test). (C) Dependence of M14 melanoma cell line on BCL2A1 and other candidate genes located in the 15q amplicon. M14 cells were transfected with siRNA targeting BCL2A1 or control. Colony formation is normalized to control siRNA 72 h after transfection. Data represent an average of at least six independent experiments with reported SE. *P < 0.05 versus siControl.
The genomic region 76–100 Mb of chromosome 15q encompassing BCL2A1 was amplified in 28 of 88 melanoma cell lines (31.8%), early passage tumors, and primary specimens evaluated (GISTIC analysis, q = 6.7 × 10−6) (31, 32) (Fig. 1B). Because SNP arrays are only semiquantitative with respect to copy number, we confirmed BCL2A1 copy number using genomic quantitative PCR (SI Appendix, Fig. S3A). Genomic amplification of BCL2A1 was observed in melanoma and was not seen in other tumor types (SI Appendix, Fig. S1B). Different parts of the amplicon were mainly coamplified, with an 80% overlap among samples amplified at different loci in two independent datasets (SI Appendix, Fig. S1 C and D). After filtering undetectable or weakly expressed genes (SI Appendix, Fig. S1E), only four genes within the 15q amplicon were expressed twofold or greater in amplified versus unamplified cells, including BCL2A1 (Fig. 1B). BCL2A1 mRNA (SI Appendix, Fig. S3B) or protein (SI Appendix, Fig. S3D) correlated well with its amplification level. There was a significant correlation between mRNA expression and protein expression (Pearson correlation R = 0.88, P = 0.03). We evaluated whether BCL2A1 or other candidate genes were required for proliferation of an amplified melanoma cell line, M14. Knockdown of BCL2A1 by siRNA significantly reduced proliferation, whereas knockdown of each of the other genes did not affect growth (Fig. 1C), despite >80% knockdown of mRNA (SI Appendix, Fig. S4A) or protein (SI Appendix, Fig. S4B). Similar results were seen in an additional 15q-amplified cell line (SI Appendix, Fig. S4C). We were unable to reliably detect Cathepsin H (CTSH) by validated antibodies, although mRNA was significantly suppressed at 72 h. Thus, we cannot exclude the possibility that CTSH is functionally required for the growth of 15q-amplified melanoma cells, in addition to BCL2A1, despite the absence of a phenotype under conditions where its mRNA levels are significantly suppressed. We did not detect any relationship between BCL2A1 amplification and MITF amplification (Fisher’s exact test, P = 0.06) or BRAF mutations (Wilcoxon test, P = 0.30), although MITF amplification correlated with higher BCL2A1 expression (see below).
BCL2A1 Is Dysregulated in Melanoma.
To examine whether BCL2A1 expression was increased in melanomas compared with normal melanocytes (and not simply a marker of the melanocytic lineage), we stained primary human melanomas with a BCL2A1-specific monoclonal antibody (SI Appendix, Fig. S5). In a representative case, we observed that BCL2A1 was highly expressed in the melanoma but had much lower expression in normal melanocytes of adjacent skin (Fig. 2A). Using a tumor progression tissue array consisting of benign nevi, primary cutaneous melanomas, and melanoma metastases, we observed no or low expression in 70% of nevi (n = 27), and no nevi had robust (3+) staining (Fig. 2B and Materials and Methods). In contrast, robust expression was observed in 72% of primary melanomas (n = 60, P < 0.0001 for nevi versus melanoma, Fisher’s exact test). A comparison of an independent set of primary melanoma and benign skin nevi (30) also demonstrated significantly increased expression of BCL2A1 in melanoma (P = 5.4 × 10−15, Wilcoxon rank sum test). We also examined whether expression of BCL2A1 predicted prognosis. We found that in a series of stage III and stage IV melanomas, high BCL2A1 mRNA (Fig. 2C) or protein (Fig. 2D and SI Appendix, Fig. S6) expression were associated with decreased survival after diagnosis. Collectively, these data suggest an important role of BCL2A1 dysregulation in melanoma pathogenesis.
Fig. 2.

BCL2A1 is dysregulated in melanoma. (A) A primary melanoma with overlying attenuated overlying epidermis was stained with anti-BCL2A1. The arrows show examples of intraepidermal melanocytes, which uniformly have weak BCL2A1 staining, whereas most melanoma cells have robust BCL2A1 expression. (B) Melanoma progression array showing correlation of BCL2A1 staining with tumor progression. High BCL2A1 mRNA (C) or staining (D) is associated with poorer overall survival of stage III and stage IV melanoma patients (P = 0.0076 and P = 0.0064, respectively).
BCL2A1 Is a Melanoma Oncogene.
To evaluate the functional requirement for BCL2A1 in melanoma, we suppressed BCL2A1 by siRNA and evaluated growth in a colony formation assay. A breast cancer cell line, MCF7, which does not express BCL2A1, was not sensitive to BCL2A1 knockdown (Fig. 3A). However, melanomas with 15q amplifications were dependent on BCL2A1. In contrast, BCL2A1-nonamplified melanomas were not dependent on BCL2A1, despite efficient knockdown (SI Appendix, Fig. S3C). BCL2A1 knockdown by shRNA (SI Appendix, Fig. S4D) also significantly impaired the tumorigenicity of M14 melanoma cells in mouse xenografts (Fig. 3B). To evaluate whether BCL2A1 can promote transformation, we used a previously described assay using genetically immortalized human melanocytes that exhibit soft-agar clonogenic growth upon oncogenic transformation (21). BCL2A1 expression (Fig. 3C) together with BRAF(V600E) efficiently promoted growth of these cells in soft agar compared with control-infected cells (Fig. 3D).
Fig. 3.

Requirement of BCL2A1 for melanoma growth. (A) Colony formation assay of cell lines 72 h after transfection with siRNA targeting BCL2A1. Results are normalized to control siRNA. Data represent an average of at least three independent experiments with SE. ***P < 0.001 compared with siControl. (B) Effect of BCL2A1 knockdown on growth of M14 melanoma xenotransplants. Volume of tumors was determined 12 d after injection. Representative tumors from mice are shown for each shRNA. (C) Expression of HA-tagged BCL2A1 in pmel*BRAF(V600E) (21) detected by Western blotting. (D) BCL2A1 overexpression promotes soft-agar growth in oncogenic BRAF-transformed melanocytes. Cell number was determined after 2 wk.
Lineage Restricted BCL2A1 Expression in Melanoma Owing to Its Direct Regulation by MITF.
We next evaluated expression of BCL2A1 across a panel of melanoma and other tumor cell lines. The expression of BCL2A1 was strikingly restricted to melanomas compared with tumors from other tissue types (Fig. 4A; P = 6.05 × 10−10, Wilcoxon rank sum test). These results were confirmed in other datasets (Fig. 4B; P = 2.99 × 10−6, t test). Consistent with prior reports, moderate expression was found in some lymphoid malignancies (33) in a larger collection of 319 cancer cell lines, although BCL2A1 was not amplified in these tissues (SI Appendix, Fig. S1B).
Fig. 4.
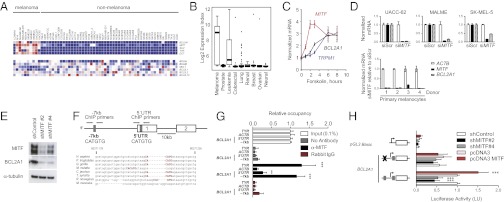
MITF directly regulates BCL2A1 in the melanocytic lineage. Expression of antiapoptotic BCL-2 family members, MITF, and MITF-regulated targets in (A) the NCI-60 tumor panel and (B) an independent dataset of 954 cancer cell lines (GlaxoSmithKline). (C) The cAMP agonist forskolin (20 µM) induces MITF, TRPM1 (a known MITF target), and BCL2A1 mRNA. A representative of three independent experiments using different donors is shown. (D) Knockdown of MITF by siRNA suppresses BCL2A1 expression in melanoma cells and primary melanocytes of different donors. Indicated RNA was quantified 72 h after siRNA transfection. (E) Knockdown of MITF by two independent lentiviral-expressed shMITFs suppresses BCL2A1 in UACC-62 melanoma cells. (F) Genomic structure of BCL2A1 promoter with conserved E-box at −7 kb site and within 5′UTR (gray boxes), showing exons 1 and 2 and location of primers used for chromatin precipitation. Below, alignment of the BCL2A1 promoter among mammalian species at the −7 kb site, based on Feb 2009 Build. (G) Chromatin immunopreciptation of indicated genomic region with no antibody, anti-MITF, or rabbit IgG. Precipitated DNA was amplified using primers surrounding the −7 kb or 5′UTR E-boxes. Results are normalized to input DNA. ***P < 0.001 compared with rabbit IgG control; **P < 0.01. (H) BCL2A1 promoters were cloned upstream of the luciferase gene as indicated. UACC-62 cells were transfected with the indicated promoters and two distinct shRNA hairpins targeting MITF (#2 and #4). Forty-eight hours later, luciferase activity was determined. Results reported are averages of at least three independent experiments, performed in duplicate and normalized for transfection efficiency using Renilla luciferase. All data are normalized to the wild-type BCL2A1 promoter transfected with the control. ***P < 0.001 compared with control.
We identified transcription factors whose expression was associated with BCL2A1 expression (SI Appendix, Table S2). Levels of MITF were strongly correlated with BCL2A1 expression (Pearson correlation 0.56, P = 4.4 × 10−9). Moreover, BCL2A1 was correlated with other genes known to be directly regulated by MITF, such as SILV, TYR, and DCT (34–36), but not other highly expressed genes (SI Appendix, Fig. S7C). MITF itself has been previously identified as a lineage-specific oncogene that regulates melanoma growth and survival (21). Although it is amplified in only ∼15–20% of melanomas, it is essential for the survival of most melanomas. Although MITF directly regulates the BCL2 gene (37), which is functionally related to BCL2A1, MITF deficiency produces a significantly more severe melanocyte defect (embryonic melanocyte lineage loss) than BCL2 deficiency (postnatal melanocyte loss), suggesting that other MITF target genes contribute importantly to MITF’s melanocytic survival phenotype. Unlike other mammalian species, the mouse genome contains four closely related homologs of BCL2A1 (38), precluding straightforward analysis of its genetic contribution to the murine melanocyte lineage.
To evaluate whether MITF is sufficient to activate BCL2A1, we treated primary melanocytes with the cAMP agonist, forskolin, which increased MITF expression within 2–3 h (39) (Fig. 4C). BCL2A1 was also induced by forskolin and was delayed relative to MITF, similar to another direct MITF target, TRPM1. Forced lentiviral overexpression of MITF in melanocytes was also sufficient to induce BCL2A1 mRNA (SI Appendix, Fig. S7A). To evaluate whether MITF is required for BCL2A1 expression we suppressed endogenous MITF by siRNA in primary melanocytes and several melanoma cell lines (Fig. 4D). BCL2A1 mRNA was significantly reduced in all cells examined. Similar results were seen at the protein level using two lentiviral-delivered shRNAs targeting MITF (Fig. 4E). MITF knockdown suppressed BCL2 in some but not all melanocytic cells and did not suppress other antiapoptotic BCL2 family members (BCL2L1, BCL2L2, or MCL1) (SI Appendix, Fig. S7B).
We compared the promoters of BCL2A1 in mammalian species (Fig. 4F) and identified putative MITF binding sites (E-boxes), located 7 kb upstream of the transcriptional start site and within the 5′ untranslated region. These sequences were found in all species examined except Mus musculus, which as stated above has four distinctly encoded BCL2A1 genes (38). To detect in vivo occupancy of MITF at these binding sites, we performed ChIP using primers spanning each region. Strong MITF binding was detected at −7 kb at levels similar to another MITF target, tyrosinase (Fig. 4G). Weak binding was detected at the 5′UTR site. We also evaluated whether BCL2A1 transcription was dependent on the conserved E-boxes and observed that mutation of the E-box at −7 kb suppressed basal activity of the promoter by 44% compared with the wild-type promoter (Fig. 4H). shRNA targeting of MITF reduced BCL2A1 promoter activity by 50% in an E-box–dependent manner. Conversely, overexpressed MITF increased BCL2A1 promoter activity, also in an E-box–dependent fashion.
BCL2A1 Confers Resistance to BRAF Inhibitors.
We evaluated genomic amplification of the prosurvival BCL2 family in melanoma and other cancer types. Both MCL1 and BCL2A1 were genomically amplified in subsets of melanomas (SI Appendix, Fig. S8). Whereas MCL1 is known to be amplified in multiple human tumor types (15), BCL2A1 amplification is seen exclusively in melanoma. Because amplification of BCL2 family members may limit the effectiveness of chemotherapy or targeted therapy, we evaluated whether BCL2A1 might mediate relative intrinsic resistance to BRAF inhibitors (refer to SI Appendix, Table S3 for BRAF mutation and BCL2A1 amplification of cell lines used). In a BCL2A1-unamplified BRAF-mutant cell line, overexpression of BCL2A1 inhibited the antiproliferative effect of PLX4720 (SI Appendix, Fig. S9A) or chemotherapy (SI Appendix, Fig. S10E) and protected from apoptosis (Fig. 5A) but not cell cycle arrest (SI Appendix, Fig. 10 A and B). BCL2A1 did not affect the ability of BRAF pathway inhibitors to suppress ERK activity (Fig. 5B). Conversely, in a BCL2A1-amplified cell line that also contains an oncogenic mutation in BRAF, knockdown of BCL2A1 significantly increased sensitivity to PLX4720 (Fig. 5 C and D and SI Appendix, Fig. S9B). Similar results were obtained with the structurally unrelated MEK inhibitor, GSK1120212, diminishing the likelihood of off-target effects (Fig. 5 A and D and SI Appendix, Fig. S10D). To further minimize the likelihood of off-target effects, we knocked down BCL2A1 using individual, rather than pooled, siRNAs (SI Appendix, Fig. S9F). The sensitization to apoptosis correlated with the degree of BCL2A1 knockdown by each siRNA. The sensitivity to PLX4720 in a cell line carrying wild-type BRAF and no BCL2A1 amplification was not affected by the knockdown of BCL2A1 (Fig. 5E), despite similar knockdown to BCL2A1-amplified M14 cells (SI Appendix, Fig. S9G). PLX4720 only modestly induced apoptosis, consistent with observations that the drug has predominately a cytostatic, noncytocidal effect on many melanoma cell lines in vitro (4–7). Suppression of BCL2A1 significantly enhanced apoptosis in BRAF-mutant BCL2A1- or MITF-expressing cell lines or short-term cultures, but not in melanomas lacking BCL2A1 or a BRAF-mutant colon cancer cell line (Fig. 5F). We also found that suppression of BCL2A1 by shRNA significantly reduced the number of resistant clones after 2 wk of PLX4720 treatment (SI Appendix, Fig. S9C). MITF knockdown also enhanced overall cytotoxicity of BRAF inhibitors (SI Appendix, Fig. S9 D and E).
Fig. 5.

BCL2A1 and MITF mediate resistance to BRAF-directed therapy. (A) BRAF-mutant A375 melanoma cells with overexpressed BCL2A1 were treated with PLX4720 or GSK1120212 and apoptosis was measured by Annexin V staining after 48 h of drug treatment. (B) Effect of BCL2A1 overexpression on ERK signaling after 4 h of treatment with 3 µM PLX4720. (C) Effect of concomitant BCL2A1 suppression by siRNA and PLX4720 in BCL2A1 amplified cell line, M14. Quantification of three independent experiments is shown in D. (E) Effect of siBCL2A1 and PLX4720 in BRAF-wild-type (15q-unamplified) cell line MeWo. (F) Effect of siBCL2A1 and PLX4720 (3 µM) on apoptosis in cell lines and short-term cultures. WDir is a colon cancer cell line with BRAF V600E mutation. (G) Effect of obatoclax (100 nM), ABT-737 (1 µM), and PLX4720 (3 µM) on apoptosis of BCL2A1-amplified cell line 501mel after 48 h of treatment. (H) Effect of PLX4720 alone or in combination with obatoclax in mouse xenotransplants. A375 cells expressing BCL2A1 or control vector (five biological replicates each) were injected into mice. Treatment of mice began 12 d after implantation, and tumor volume was measured at baseline and on the 12th d of treatment. *P < 0.05 compared with control. (I) Box-whisker plot comparing BCL2A1 mRNA expression in melanomas from patients before treatment with vemurafenib or GSK1120212 + GSK2118436 (SI Appendix, Table S4). Normalized expression (arbitrary units) was compared in patients who achieved objective responses by standard RECIST criteria (n = 12) versus those without objective responses (n = 7).
Consistent with these genetic data, treatment with a pan-BCL2 family inhibitor, obatoclax, synergistically induced apoptosis of a BCL2A1-amplified cell line (Fig. 5G and SI Appendix, Fig. S11A), whereas ABT-737 (an inhibitor of BCL2, BCL2L1, and BCL2L2 but not of MCL1 or BCL2A1) did not. To evaluate this combination in vivo, we treated mouse xenotransplants of A375 cells (which do not express appreciable BCL2A1) or derivatives expressing BCL2A1 (Fig. 5A) with PLX4720, obatoclax, or the combination for 2 wk (Fig. 5H). PLX4720 modestly decreased the size of the control tumors over this period, whereas tumors overexpressing BCL2A1 slightly grew in size. Obatoclax alone did not significantly affect the growth of tumors, but in combination with PLX4720 it significantly decreased tumor volume of both BCL2A1-overexpressing and control tumors. Two of five animals treated with the combination had no detectable tumors present after 2 wk. Moreover, we did not observe any overt toxicity of the combination of PLX4720 and obatoclax or significant difference in weight of the animals (SI Appendix, Fig. S11B). These data indicate that the combination of BRAF inhibitors and obatoclax may be an attractive therapeutic combination for melanomas with high expression of BCL2A1, either by genomic amplification or dysregulation of its upstream regulator, MITF.
To evaluate the clinical relevance of BCL2A1 in conferring resistance to BRAF inhibitors, we evaluated 19 melanoma patients for whom we have biopsies before treatment to either vemurafenib or the combination of GSK1120212 (MEK inhibitor) and GSK2118436 (BRAF inhibitor) (refer to SI Appendix, Table S4 for patient details). Tumors from patients with objective RECIST responses exhibited significantly lower levels of BCL2A1 expression (P = 0.03) compared with those that had no objective response (Fig. 5I). Collectively these data suggest that identification of patients with dysregulated BCL2A1 may have poorer clinical outcomes that may be improved by concomitant treatment with BCL2 antagonists such as obatoclax.
Discussion
Small molecule suppression of the BRAF/MEK pathway in melanoma produces clinical responses in a majority of melanoma patients (13), but the responses are variable and all patients eventually relapse. Although recent reports have elucidated several mechanisms by which melanomas acquire resistance to BRAF inhibitors (40–42), it is notable that patients treated with BRAF inhibitors rarely have complete initial responses. We propose that amplification of BCL2A1 or its direct regulator MITF may limit the primary efficacy of BRAF-directed therapy. Although we find a correlation between BCL2A1 expression and sensitivity to BRAF inhibitors in vitro and in patients, larger, prospective trials to evaluate the effect of BCL2A1 dysregulation on BRAF inhibitor efficacy will be necessary. Moreover, it will be necessary to correlate genomic copy number with protein expression in primary melanoma tissues.
Our finding that the well-established melanoma oncogene MITF directly regulates BCL2A1 suggests that it may also contribute to resistance to BRAF inhibitors. MITF is amplified in 15–20% of melanomas, although it is functionally required in a larger group of melanomas, including many that lack MITF amplification (21). Although there are no small molecules that target MITF, targeting downstream pathways such as BCL2A1 may be of clinical utility to this group of melanomas. In contrast, MITF-negative melanomas do not express BCL2A1 and are not sensitive to BCL2A1 suppression.
Previous work has evaluated the role of BCL2 family members in mediating resistance to targeted therapy. In lung cancer, reducing MCL1 expression sensitized epidermal growth factor receptor mutant nonsmall cell lung cancers to MEK inhibitors (43). MEK inhibitors synergized with the BH3 mimetic ABT-737 in BRAF mutant colon cancer cell lines (44). In contrast, we found that ABT-737 did not synergize with PLX4720 in melanoma. These data are consistent with the lack of efficacy of a BCL2 antisense oligonucleotide in patients with melanoma (45). The differences between melanoma and other colon cancer cell lines may be related to the inability of ABT-737 to inhibit BCL2A1 or MCL1. Indeed, BCL2A1 has been previously shown to confer resistance to ABT-737 (46). Whereas we find that BCL2A1 is genomically amplified in 30% of melanomas and is the most abundant prosurvival BCL2 family in the melanocyte lineage, we cannot exclude that other BCL2 family members may contribute to resistance to BRAF inhibitors in some cases. We have also observed that MCL1 is inversely correlated with BCL2A1 expression in melanomas (Fig. 4A). Because its overexpression would also likely suppress apoptosis, pharmacologic inhibition of MCL1 may also be therapeutically attractive, although it may engender greater toxicity risks than BCL2A1, given MCL1’s ubiquitous distribution compared with the melanocyte-restricted expression of BCL2A1. Collectively, our data suggest that dysregulation of antiapoptotic BCL family members in melanoma, particularly BCL2A1, may be important in the management of patients treated with BRAF inhibitors.
Materials and Methods
Cell Lines and Cultures.
Cell lines were from American Type Culture Collection and were maintained as described in SI Appendix, SI Text.
Western Blotting and Immunohistochemistry.
Antibodies used were MITF C5 hybridoma, BCL2A1 (Cell Signaling Technology), α-tubulin (clone DM1A; Sigma), GAPDH (Cell Signaling Technology), BLM (Epitomics), ISG20 (Sigma), and HA (3F10; Roche). For immunohistochemistry, tumor arrays were stained with anti-BCL2A1 (rabbit monoclonal clone 1639–1, 1:50 dilution; Epitomics). Details of tissue microarray, staining, and scoring are described in SI Appendix, SI Text.
RNA Isolation, Chromatin Immunoprecipitation, and Quantitative Real-Time PCR.
Total RNA was isolated using RNeasy RNA kits (Qiagen) and quantitative real-time PCR was performed on an Applied Biosystems 7700 machine. Primers and conditions for PCR are described in SI Appendix, SI Text.
siRNA Delivery and Analysis.
siRNAs SMARTpools (Dharmacon) were transfected into melanomas or primary melanocytes using the lipidoid delivery agent C12-133-B as described in SI Appendix, SI Text. Lentivirus was prepared by transfection in 293T cells as described in SI Appendix, SI Text.
Cell Growth and Soft-Agar Assays.
pLEX HA-MYC BCL2A1 (Open Biosystems) was packaged in 293T cells and infected into indicated cells as described in SI Appendix, SI Text. Infected pmel* BRAFV600E cells were plated onto soft agar as described (21) and cell number was estimated with the CellTiter-Glo luminescence assay after 2 wk. All mouse experiments were done in accordance with Institutional Animal Care and Use Committee (IACUC) approved animal protocols at Massachusetts General Hospital.
Promoter Assays and Luciferase Experiments.
The BCL2A1 promoter was amplified from discarded human foreskin and cloned into the pGL3-Basic vector. Mutagenesis was performed using the QuikChange Mutagenesis Kit (Stratagene). Primer sequences for mutagenesis are indicated in SI Appendix, SI Text. Luciferase readings were normalized to cotransfected pRL-CMV Renilla Control.
Apoptosis and Flow Cytometry Assays.
Cells were reverse-transfected with 25 nM siRNA as above and treated with PLX4720 (Sai Advantium Pharma Limited), GSK1120212 (Active Biochem), ABT-737, or obatoclax (Selleck Chemicals) for 48 h. Apoptosis was measured using the Annexin V Apoptosis Kit (Becton Dickinson). Cell cycle analysis was done 72 h after treatment with drug using propidium iodide. Cell cycle phases were estimated using FlowJo software (Treestar Software).
Supplementary Material
Acknowledgments
We thank Meenhard Herlyn (Wistar Institute) for cell lines, Gideon Bollag and Plexxikon for PLX4720-containing chow, Hans Widlund (Brigham and Women’s Hospital), and members of the Fisher laboratory for discussions and suggestions. We thank Juying Li for advice, Su-Jean Seo (Beth-Israel Deaconess Hospital) for help with microscopy, and Myung-Shin Sims (John Wayne Cancer Institute) for statistical help. This work was supported by the American Skin Association (R.H.); National Institute of Arthritis and Musculoskeletal and Skin Diseases/National Institutes of Health (NIH); the Melanoma Research Alliance; the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (D.L.M., D.S.B.H., and D.E.F.); and Grant-in-Aid 24700971 for Young Scientists (B) (to S.Y.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan. D.G.A. is supported by NIH Grant eb00244. J.S.S. acknowledges support from the PhRMA Foundation, University of California at San Francisco (UCSF), Committee on Research, UCSF Research Allocation Program, and National Cancer Institute Grant R01CA163336. D.E.F. is a Distinguished Clinical Scholar of the Doris Duke Medical Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. A.R. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1205575110/-/DCSupplemental.
References
- 1.Wan PT, et al. Cancer Genome Project Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 2.Dhomen N, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15(4):294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 3.Dankort D, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41(5):544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joseph EW, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci USA. 2010;107(33):14903–14908. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solit DB, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439(7074):358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smalley KS, Flaherty KT. Integrating BRAF/MEK inhibitors into combination therapy for melanoma. Br J Cancer. 2009;100(3):431–435. doi: 10.1038/sj.bjc.6604891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haass NK, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14(1):230–239. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 8.Paraiso KH, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71(7):2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tap WD, et al. Pharmacodynamic characterization of the efficacy signals due to selective BRAF inhibition with PLX4032 in malignant melanoma. Neoplasia. 2010;12(8):637–649. doi: 10.1593/neo.10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Søndergaard JN, et al. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J Transl Med. 2010;8:39. doi: 10.1186/1479-5876-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapman PB, et al. BRIM-3 Study Group Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bollag G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flaherty KT, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nessling M, Kern MA, Schadendorf D, Lichter P. Association of genomic imbalances with resistance to therapeutic drugs in human melanoma cell lines. Cytogenet Cell Genet. 1999;87(3–4):286–290. doi: 10.1159/000015451. [DOI] [PubMed] [Google Scholar]
- 15.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim M, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125(7):1269–1281. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 17.Bass AJ, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41(11):1238–1242. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott KL, et al. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature. 2009;459(7250):1085–1090. doi: 10.1038/nature08109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Firestein R, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455(7212):547–551. doi: 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weir BA, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450(7171):893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garraway LA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436(7047):117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 22.Beroukhim R, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci USA. 2007;104(50):20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Fiore PP, et al. erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science. 1987;237(4811):178–182. doi: 10.1126/science.2885917. [DOI] [PubMed] [Google Scholar]
- 24.Vogler M. BCL2A1: The underdog in the BCL2 family. Cell Death Differ. 2012;19(1):67–74. doi: 10.1038/cdd.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paoluzzi L, et al. Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood. 2008;111(11):5350–5358. doi: 10.1182/blood-2007-12-129833. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen M, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA. 2007;104(49):19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 28.Walensky LD, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305(5689):1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cashman JR, et al. Inhibition of Bfl-1 with N-aryl maleimides. Bioorg Med Chem Lett. 2010;20(22):6560–6564. doi: 10.1016/j.bmcl.2010.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Talantov D, et al. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 2005;11(20):7234–7242. doi: 10.1158/1078-0432.CCR-05-0683. [DOI] [PubMed] [Google Scholar]
- 31.Thomas RK, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39(3):347–351. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 32.Lin WM, et al. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer Res. 2008;68(3):664–673. doi: 10.1158/0008-5472.CAN-07-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karsan A, Yee E, Kaushansky K, Harlan JM. Cloning of human Bcl-2 homologue: Inflammatory cytokines induce human A1 in cultured endothelial cells. Blood. 1996;87(8):3089–3096. [PubMed] [Google Scholar]
- 34.Miller AJ, et al. Transcriptional regulation of the melanoma prognostic marker melastatin (TRPM1) by MITF in melanocytes and melanoma. Cancer Res. 2004;64(2):509–516. doi: 10.1158/0008-5472.can-03-2440. [DOI] [PubMed] [Google Scholar]
- 35.Du J, et al. MLANA/MART1 and SILV/PMEL17/GP100 are transcriptionally regulated by MITF in melanocytes and melanoma. Am J Pathol. 2003;163(1):333–343. doi: 10.1016/S0002-9440(10)63657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoek KS, et al. Novel MITF targets identified using a two-step DNA microarray strategy. Pigment Cell Melanoma Res. 2008;21(6):665–676. doi: 10.1111/j.1755-148X.2008.00505.x. [DOI] [PubMed] [Google Scholar]
- 37.McGill GG, et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell. 2002;109(6):707–718. doi: 10.1016/s0092-8674(02)00762-6. [DOI] [PubMed] [Google Scholar]
- 38.Hatakeyama S, et al. Multiple gene duplication and expression of mouse bcl-2-related genes, A1. Int Immunol. 1998;10(5):631–637. doi: 10.1093/intimm/10.5.631. [DOI] [PubMed] [Google Scholar]
- 39.Price ER, et al. alpha-Melanocyte-stimulating hormone signaling regulates expression of microphthalmia, a gene deficient in Waardenburg syndrome. J Biol Chem. 1998;273(49):33042–33047. doi: 10.1074/jbc.273.49.33042. [DOI] [PubMed] [Google Scholar]
- 40.Nazarian R, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johannessen CM, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Villanueva J, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faber AC, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci USA. 2009;106(46):19503–19508. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cragg MS, et al. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest. 2008;118(11):3651–3659. doi: 10.1172/JCI35437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitzen JM, Chi LG, Uprichard AC, Lucchesi BR. Effects of combined thromboxane synthetase inhibition/thromboxane receptor antagonism in two models of sudden cardiac death in the canine: Limited role for thromboxane. J Cardiovasc Pharmacol. 1990;16(1):68–80. doi: 10.1097/00005344-199007000-00010. [DOI] [PubMed] [Google Scholar]
- 46.Vogler M, et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood. 2009;113(18):4403–4413. doi: 10.1182/blood-2008-08-173310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.