

Abstract
Background
This study was to explore whether the efficacy of the EGFR tyrosine kinase inhibitor ZD1839 (Z, Iressa, gefitinib) plus chemotherapeutic agents docetaxel (D) and cisplatin (P) may benefit from sequencing of the combination.
Methods
Three head and neck cancer cell lines were used to study the effect of various combinations of and relative sequencing of D, P, and Z in cell growth inhibition. A population pharmacokinetic stimulation study was conducted on Z in silico, and used to together with the growth inhibition data to derive principles for future in vivo use of this drug combination.
Results
The inhibitory effects of Z on combinations of D and P were sequence dependent. Treatment simultaneously with DPZ or with DP followed by Z (DP→Z) showed synergistic effects in all three cell lines. However, sequencing with Z followed by DP (Z→DP), gave an antagonistic effect, suggesting that D and P should be administered when the effect of Z is low. The induction of apoptosis was also sequence dependent. The in silico pharmacokinetic study suggested the feasibility of deriving a 5 day on 2 day off regimen for Z, in which D and P administration commences when levels of Z are low, allowing levels of Z to accumulate sufficiently during the remainder of the cycle.
Conclusion
These data suggests that it is feasible to design clinical trials with these settings to maximize the efficacy of this combined drug regimen.
Keywords: EGFR, cytotoxic agent, docetaxel, cisplatin, apoptosis, head and neck cancer
INTRODUCTION
It was estimated that 45,660 patients would be diagnosed with head and neck cancer (SCCHN) and more than 11,210 patients would succumb to this disease in 20071. The expected 5 year overall survival rate of individuals diagnosed with locally advanced SCCHN is still poor. The standard of care includes radiation therapy with concurrent cisplatin, which may be combined with a taxane derivative such as docetaxel2. Cisplatin is believed to mediate cell death secondary to DNA cross-linking3, while docetaxel is believed to act by promoting tubulin assembly in microtubules and inhibiting depolymerization in proliferating cells, producing G2M arrest and p53-independent apoptosis.
Other approaches that suggest improved response rates and survival benefits include induction chemotherapy using docetaxel, cisplatin and 5-fluorouracil, followed by chemoradiation4, 5. Phase III trials are ongoing to assess induction chemotherapy followed by chemoradiation versus chemoradiation alone. Although these aggressive treatment regimens are efficacious and potentially curative, they are accompanied by toxicities that may severely impair the quality of life in a substantial proportion of patients6–8. One approach to minimize toxicity without compromising efficacy includes the incorporation of targeted agents to cytotoxic chemotherapy. Since over 80% of SCCHN cells express high levels of epidermal growth factor receptor (EGFR)9, it may be beneficial to include EGFR tyrosine kinase inhibitors (TKIs) in the treatment regimen, as was demonstrated in a clinical trial that combined the EGFR-directed monoclonal antibody cetuximab with radiation therapy10.
While modern TKIs have only modest activity as monotherapy agents11–13, they are well tolerated, making them suitable for patients with impaired performance status. An early clinical trial that combined ZD1839 (gefitinib) with 5-fluorouracil and hydroxyurea concomitantly with twice daily radiation has been shown to be feasible, effective and tolerable14. Furthermore, data from two phase II trials have shown promising results for the combination of a TKI with docetaxel and cisplatin in SCCHN15, 16.
Although phase II trials in non small cell lung cancer (NSCLC) demonstrated promising results, large phase III trials produced negative results, suggesting that the combination of TKIs with chemotherapy may be detrimental when used without a clearer mechanistic basis 17, 18. Therefore, this in vitro study was conducted using SCCHN cell lines (Tu177, Tu212, and SqCC/Y1), to assess the cell cycle effects of the TKI ZD1839 (gefitinib), and to address mechanistically whether the potentiation and/or inhibition of cancer cell death by a TKI during chemotherapy may be adversely related to the sequence and timing of drug application.
The effect of drug concentration on cell growth is best studied in vitro, while drug exposure (pharmacokinetics) can only be characterized in vivo. Although preclinical tumor growth models may be conducted in animals, it is often difficult to duplicate pharmacokinetic human pharamacokinetic profiles precisely in an animal model. Population pharmacokinetic modeling is used to derive mean parameter estimates and their variability across a treated population, and may be used to establish relationships between drug exposure and the relevant pharmacodynamic parameters. Simulation with these models may also be used to simulate ranges in drug concentration versus time as a function of dose regimen, in order facilitate the selection of dosing schedules19. In this study, parameters from a previous pharmacokinetic study on ZD18392, were used to simulate expected ranges of plasma concentrations versus time in humans, at the dose regimens of interest. Simulated plasma concentrations were then used together with the dose-response relationships developed in vitro, to help rationalize the observed successful and failing clinical trial regimens.
MATERIALS AND METHODS
Reagents
Docetaxel and ZD1839 were obtained from Aventis Pharmaceuticals Inc. (Bridgewater, NJ) and AstraZeneca Pharmaceuticals (Cheshire, England), respectively. Cisplatin was purchased from Sigma-Aldrich (St. Louis, MO). All three drugs were dissolved in sterile dimethyl sulfoxide (DMSO; Sigma-Aldrich) and stored at –20°C. Stock solutions were diluted in culture media to the final concentration immediately prior to use. In all cases, final concentrations of DMSO were less than 0.1% in the cell culture media.
Cell lines
The Tu177 cell line was established from a human laryngeal squamous cell carcinoma. Tu212 was established from a primary hypopharyngeal tumor. SqCC/Y1 was established from a patient’s SCC of the oral cavity. These cell lines were obtained from Dr. Gary L. Clayman (University of Texas M. D. Anderson Cancer Center, Houston, TX), Dr. Peter G. Sacks (New York University College of Dentistry, New York, NY), and Dr. Shi-Yong Sun (Emory University Winship Cancer Institute, Atlanta, GA), respectively. Cells were cultured in DMEM: Ham’s F-12 (1:1) supplemented with 10% heat-inactivated fetal bovine serum and antibiotics (streptomycin, penicillin G and amphotericin B) and maintained in a 37°, 5% CO2, humidified incubator.
Cell growth assay
SCCHN cell lines were seeded at a density of 3 × 103 cells/well into 96-well plates in triplicate and incubated at 37 °C and 5% humidity for 24 hours prior to drug treatment to allow attachment. Drugs were added to cell cultures as single agents at the following concentrations: docetaxel, 0.08 −20 nM; cisplatin, 0.2 – 50 µM; ZD1839, 0.1 – 30 µM, or in combination. The cells were then incubated for a further 72 hours. The concentration ratio of the drugs in the combination treatments was determined based on the IC50 of each drug (D:P:Z = 1:2500:1500). For sequential drug treatments, docetaxel and cisplatin were considered a group. In these cell lines one cell cycle is completed in approximately 24 hours of which about one third to one half is spent in G1 phase. To ensure that most of cells are accumulated in G1 arrest after addition of ZD1839, we chose 12 hours as the duration period for these experiments. To assess the effect of sequencing of ZD1839, this agent was added either concomitantly with, 12 hours prior or 12 hours after DP. Cell growth inhibition was measured by determining cell density using a sulforhodamine B assay. The percent of inhibition was calculated as the cell density ratio of drug-treated/untreated cells. All experiments were performed in triplicate. Multliple drug effect analysis was performed using the CalcuSyn software from Biosoft (Cambridge, UK), which calculates a combination index (C.I) using the approach of Chou and Talalay20. In this approach, synergy is defined as a C.I. < 1.0, antagonism as a C.I. > 1.0, and additivity as C.I. values not significantly different from 1.020.
Annexin V assay to assess apoptosis
The effects of single and combination drugs on apoptosis were analyzed in Tu177, Tu212 and SqCC/Y1 cells using flow cytometry. Exponentially growing cells (2.5 × 105) were seeded in 60 mm2 dishes. After seeding for 24 hours, they were treated with single, double, or triple drug combinations using the same concentrations as the growth inhibition assay (above). The second drug combination (DP or Z) was added 12 hours after the first treatment for sequential drug treatment. Control cultures were exposed to DMSO only (0.1%). Both adherent and floating cells were collected and analyzed with Annexin V assay after 72 hours of treatment, according to manufacturer’s protocol (BD PharMingen, Franklin Lakes, NJ). Briefly, pelleted cells were washed with PBS and resuspended in Annexin binding buffer (BD PharMingen). Cells were then incubated with Annexin V-phycoerythrin (Annexin V-PE; BD PharMingen) and 7-amino-actinomycin (7-AAD; BD PharMingen) for 15 minutes at room temperature. The stained cells were then analyzed using a fluorescence-activated cell sorting (FACS) Caliber bench-top flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Cells with no drug treatment (DMSO only) were used as a control. The experiments were repeated independently at least three times, and the average apoptosis rate was calculated and graphed using FlowJo software (Tree Star, Inc. Ashland, OR).
Cell cycle analysis
Cells were trypsinized, pelleted, washed twice with PBS and fixed in ice-cold 70% ethanol 24 hours after treatment with the various drug concentrations. Fixed cells were treated with 100 units/ml RNase and their DNA was fluorescently labeled with 0.05 mg/ml propidium iodide. Their morphological and cell cycle parameters were then measured by flow cytometry using the FACSCalibur System (BD Biosciences, Franklin Lakes, NJ). Data were analyzed using FlowJo 4.3.1 software (Tree Star, Ashland, OR). Analysis of cell cycle was performed by investigating the FL2-A profile of the cells identified to be in cycle. Experiments were repeated in triplicate and statistical differences were assessed based on a two-tailed t test.
Immunoblotting analysis
Immunoblotting was used to elucidate the underlying mechanism of enhanced growth inhibition and apoptosis in Tu212 cells. Cells were seeded at a density of 6 × 105 cells in 100 mm2 dishes for 24 hours before drug treatment. The cells were lysed with sample buffer after 72 hours of incubation with the single, double, or triple drug combinations. Protein samples (75 µg) from each experiment were separated on 8–15% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry fat milk in TBST and incubated with various primary antibodies including poly (ADP-ribose) polymerase (PARP), caspase-9, AKT, p-AKT, ERK, and p-ERK (anti-rabbit: Cell Signaling, Beverly, MA), caspase-8 (anti-mouse: Cell Signaling), EGFR and p-EGFR (anti-rabbit: Santa Crutz, Santa Cruz, CA), caspase-3 (anti-mouse: Imgenex, San Diego, CA) and with horseradish peroxidase (HRP)-conjugated secondary antibodies (Promega, Madison, WI). The binding signal was visualized using an enhanced chemiluminescense kit (ECL, Amersham, Buckinghamshire, UK).
Statistical analysis
Effects of the combined treatments on growth inhibition was analyzed statistically using a 1 sample two-tailed student t-test. Bonferroni correction to maintain the family wide error rate at 0.05 was carried since multiple hypotheses were carried for each experiment.
Pharmacokinetic simulation for ZD1839 (Z, Iressa, gefitinib)
The oral dose population pharmacokinetics of ZD1839 were previously described in a study conducted in 27 individuals with advanced cancer, using a model that included a rate limiting dissolution process with subsequent first order oral absorption into the central plasma compartment and two compartment disposition21. Parameters from the covariate free version of the model were used to simulate non-protein bound plasma concentrations of ZD1869 versus time profiles in 2,000 individuals taking oral daily doses of 250 or 500 mg of ZD1839, for 5 days followed by a 2 day washout period, using Trial Simulator (ver. 2.1.2, Pharsight Corp., Mountain View, CA, 2001). The 25th, median and 75th quantile (P25, median, P75) plasma concentrations versus time profiles were then compared with the IC50 versus the wild-type and mutant EGF receptor (0.1 µ M = 44.7 ng/mL and 0.015 µ M = 6.7 ng/mL, respectively), during and after the 7 day period (Figure 6). The unbound fraction of ZD1839 in plasma was assumed equal to 0.035221.
RESULTS
Treatment with docetaxel, cisplatin and ZD1839. The growth of the SCCHN cell lines Tu177, Tu212, and SqCC/Y1 were inhibited by docetaxel and ZD1839 in a dose-dependent manner with IC50s around 1 nM and 10 µM, respectively22. Their responses to cisplatin was also dose-dependent with an IC50 around 1.5 µM. Dual combinations comprising any pair of the agents docetaxel, cisplatin and ZD1839 inhibited the growth of the SCCHN cell lines, with the most substantial inhibition of cell growth resulting from the combination of docetaxel and ZD1839 (DZ) (52–58%), while docetaxel plus cisplatin (DP) and cisplatin plus ZD1839 (PZ) produced growth inhibition rates of 47–53% and 38–51% of control, respectively (Figure 1A–1C). The triple combination of docetaxel, cisplatin and ZD1839 (DPZ) induced 65–74% growth inhibition in each cell line and was significantly different from each single drug (Tu212 p <0.004-0.009; Tu177 p <0.0006-0.011; SqCC/Y1 p <0.032-0.068).
Figure 1. Growth inhibition assays for three SCCHN cell lines.
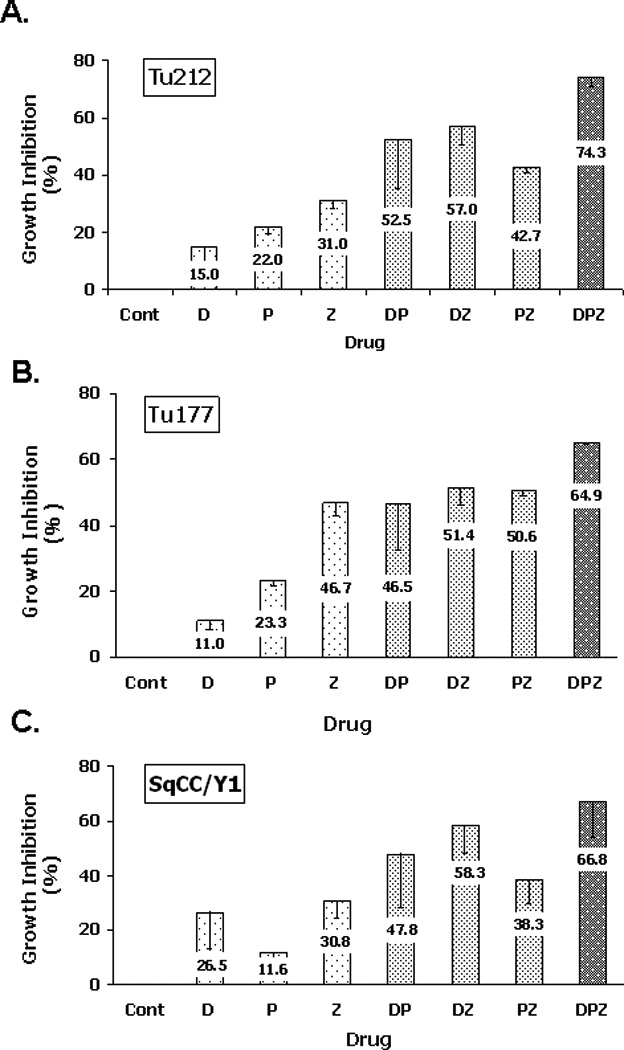
Cells of each cell line were seeded at a density of 3 X 103/well in a 96 well plate. After 24 hr, cells were treated with single or combination drugs (D, 0.7 nM; P, 1.6 µM; Z, 1 µM) for 72 hrs. Cells without treatment were used as a control.
Combination indices calculated according to the method of Chou and Talalay 19 demonstrated some synergy of DP in Tu212 and Tu177 cells. However, this combination had additive effects in SqCC/Y1 cells (Table 1). Dual combinations of ZD1839 with cisplatin or docetaxel resulted in additive or synergy effects in all three cell lines, and synergy in Tu212, Tu177 and SqCC/Y1 cells with average C.I. of 0.43, 0.63 and 0.48, respectively. Treatment with the triple drug combination DPZ resulted in even lower C.I. in Tu212, Tu177 and SqCC/Y1 cells: 0.37 (p=0.023), 0.52 (p=0.066) and 0.48 (p=0.117) at multiple effect levels (IC25~IC75) respectively, suggesting a greater degree of synergy (Table 1).
Table 1.
Combination Index of the SCCHN Cells Treated with Docetaxel, Cisplatin, and ZD1839
| DP | DZ | PZ | DPZ | DP→z | Z→DP | |
|---|---|---|---|---|---|---|
| Tu212 | 0.76 ± 0.20 | 0.41 ± 0.21 | 0.75 ± 0.08 | 0.37 ± 0.08 | 0.60 ± 0.27 | 0.80 ± 0.35 |
| Tu177 | 0.84 ± 0.28 | 0.60 ± 0.16 | 0.85 ± 0.14 | 0.52 ± 0.11 | 0.50 ± 0.22 | 1.10. ± 0.15 |
| SqCC/YI | 1.16 ± 0.46 | 0.47 ± 0.07 | 0.87 ± 0.20 | 0.48 ± 0.16 | 0.50 ± 0.08 | 1.16 ± 0.46 |
Note: DP, docetaxel plus cisplatin; DZ, docetaxel plus ZD1839; PZ, cisplatin plus ZD1839; DPZ, docetaxel, cisplatin, and ZD1839 simultaneously; DP→Z, docetaxel and cisplatin followed by ZD1839; Z→DP.ZD1839 followed by docetaxel and cisplatin.
Assessment of apoptosis in SCCHN cells
Annexin V flow cytometric analysis of Tu212, Tu177 and SqCC/Y1 cells was utilized to determine whether growth inhibition was related to increased apoptosis (Figure 2A–2C). The triple drug combination DPZ showed enhanced apoptotic rates relative to those of the single agents. Lysates of cells treated with the different drug combinations were analyzed by immunoblotting to determine whether apoptosis occurred via the extrinsic or the intrinsic apoptosis pathway23 (Figure 2D). The pathways of apoptotic cell death have been sub-divided into induction and execution phases24. The induction phase is initiated by mechanisms that involve either cell surface (extrinsic) signaling and subsequent signal transduction mechanisms, or activation of intracellular (intrinsic) signaling that results in the release of mitochondrial components. Caspase-8 and -9 are categorized as initiator caspases, which activate downstream caspases such as caspase-325. Some induction of PARP and caspase cleavage was observed when cells were treated with D and P as single agents. However, cells treated with ZD1839 alone did not demonstrate induction of either pathway. Cells treated with the dual combinations DP, DZ or DP all demonstrated activation of PARP and some caspase-3, -8 and -9 cleavage. DP had the least effect on caspase and PARP activation, corresponding to its lesser effects on growth inhibition. The triple combination DPZ showed the highest level of PARP, caspase-3 and -8 cleavage. However, caspase-9 cleavage was not increased relative to that seen with DP. These data suggest that apoptosis in cells treated with docetaxel and cisplatin with or without ZD1839 is mediated by both the death receptor and the intrinsic apoptotic pathways.
Figure 2. Effect of the treatments on apoptosis.
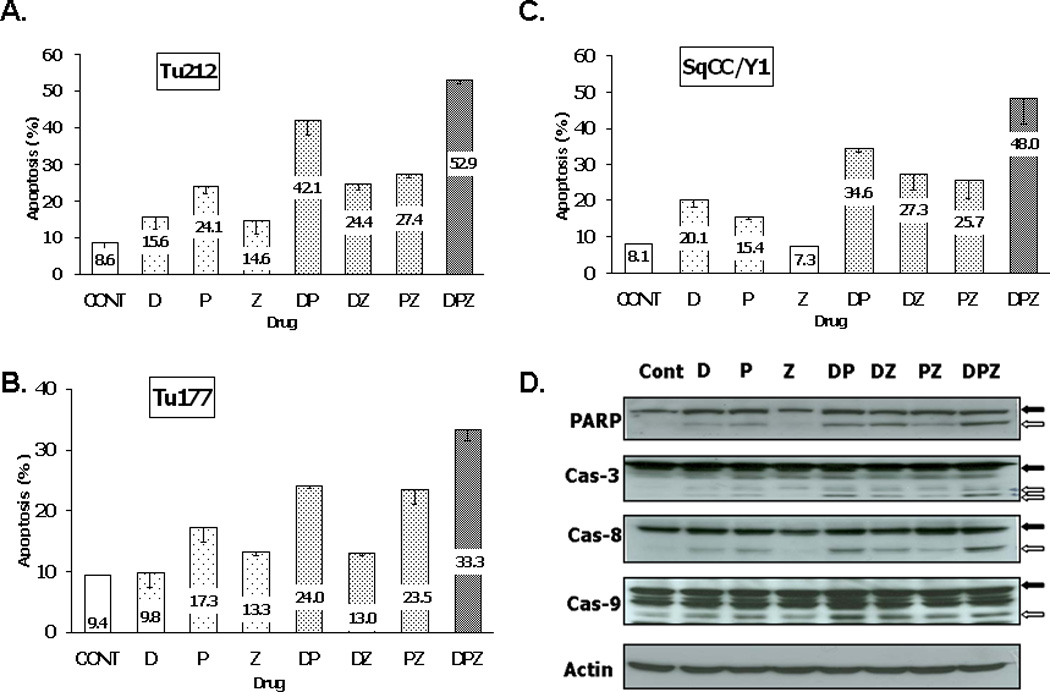
Cells of each cell line were seeded at a density of 2.5 X 105/well in a 6 cm dish. After 24 hr, cells were treated with single or combination drugs (D, 0.7 nM; P, 1.6 (M; Z, 1 (M) for 72 hrs. Untreated cells were used as a control. A-C. Average of apoptotic rate in each cell line (A. Tu177; B, Tu212: C, SqCC/Y1). D. Immunoblotting for apoptosis-related markers in the Tu212 cell line. The DPZ combination shows more cleavage of PARP and caspases than other single or double drug treatments (black arrows, pre-cleaved; white arrows, cleaved bands).
Effect of ZD1839 on EGFR-mediated signaling pathways
ZD1839 blocked activation of EGFR and phosphorylation of the downstream effector kinases ERK and AKT. Treatment of Tu212 cells with cisplatin or docetaxel did not alter the phosphorylation of either of these proteins. However, the addition of ZD1839 to either or both of these drugs resulted in decreased phosphorylation of EGFR, ERK and AKT in a similar manner to treatment with ZD1839 as a single agent (Figure 3).
Figure 3. Immunoblotting for EGFR-related signaling pathway proteins in the Tu212 cell line.

Z and any of its combinations efficiently inhibit the EGFR-mediated signaling pathways involving ERK and AKT.
Sequence-dependent growth inhibitory effects of ZD1839
The growth inhibitory effects of TKIs in combination with cytotoxic drugs may depend on the sequence of drug addition to cells. Therefore, we tested whether the synergistic interaction observed for the triple combination of ZD1839 with DP was sensitive to the sequence of addition of ZD1839 to the cells. Administration of DP followed 12 hours later by Z gave synergistic effects similar to that of simultaneous DPZ treatment in all three cell lines (Table 1). However, Z followed by the DP combination (Z→DP) gave an antagonistic effect in Tu177 and SqCC/Y1 cells (C.I. = 1.1 and 1.3, respectively), and a less synergistic effect (C.I= 0.8) in Tu212 cells. Concomitant treatment with all three drugs (DPZ) or treatment with the DP→Z sequence produced high apoptotic response rates in SCCHN cells, as measured using an annexin V flow cytometric assay (Figure 4A–C). However, when Z was administered prior to DP (Z→DP) a substantial decrease in apoptosis was observed. Furthermore, the antagonistic effect of the DP combination observed in SqCC/Y1 cells (40.7% apoptosis) was further reduced to 25.5% with the Z→DP regimen (Figure 4A–C). These results were supported by immunoblotting experiments performed on Tu212 cell lysates for apoptotic markers (Figure 4D), in which DPZ and DP→Z demonstrated the highest levels of activation of PARP, caspase-3, -8 and -9. These data lend support for the inhibition of both the extrinsic and intrinsic apoptotic pathways in cells treated with prior ZD1839.
Figure 4. Effect of sequential treatment on apoptosis and cell cycle.

A–C. Apoptotic rates in Tu212, Tu177, and SqCC/Y1 cells upon sequential treatment. Z→ DP consistently shows a lower apoptotic rate than the DP combination. D. Immunoblotting for apoptosis-related markers in sequentially treated Tu212 cell line. Z→DP shows less cleavage of PARP and caspases than DP, DPZ or DP→Z drug treatments (black arrows, pre-cleaved; white arrows, cleaved bands). E. Representative graph of SqCC/Y1 cells treated for 24 hours with DP, Z, DPZ, DP→ Z, or Z→DP. DP treatment induced S and G2/M arrest while Z induced G0/G1 arrest. The cell cycle distributions by DP, DPZ and DP→Z were similar. When Z was given first, there was an increase in G1 arrest (gray line: the control).
Sequential treatment causes different effects on cell cycle
The three agents used in this study could produce cell cycle arrest at different phases in the cycle. Therefore, cell cycle analysis was performed on SqCC/Y1 cells treated with different drug combinations for 24 hours (Figure 4E). G2M arrest was observed with docetaxel and cisplatin, even at concentrations of 0.7 nM for docetaxel and 1.6 µM for cisplatin. However, cells treated with ZD1839 alone underwent G1 arrest. Cells treated with either DPZ or DP→ Z demonstrated a decreased population of cells in the G1 phase and an increased population in the G2M phase. However, cells receiving Z prior to the other agents failed to accumulate in G2M phase, but were arrested in the G1 phase after 24 hours of incubation.
Pharmacokinetic simulation of ZD1839
Plasma concentrations (non-protein bound) of simulated individuals taking oral daily doses of 250 or 500 mg of ZD1839 (n = 2,000 per cohort) over a 28 day period, were in agreement with observed values in the study from which the population pharmacokinetic parameters were derived (data not shown) 20. Additional simulations were then performed at the 250 and 500 mg doses for 5 days followed by a 2 day washout period. The P25, median and P50 concentrations were then plotted versus time (Figure 5). Neither dose produced concentrations higher than the IC50 of wild-type EGFR (0.08 µM = 44.7 ng/mL ) for a clinically relevant period of time, while both produced concentrations greater than the IC50 of mutant EGFR, with maximal concentrations of at least 2-fold greater than the IC50 from day 2 to 5. After a 2 day washout period (day 7) predicted plasma concentrations in more than 90% of individuals in the 250 mg cohort and about 25% of individuals in the 500 mg group declined to concentrations below the IC50 of mutant EGFR (0.015 µ M = 6.7 ng/mL).
Figure 5.
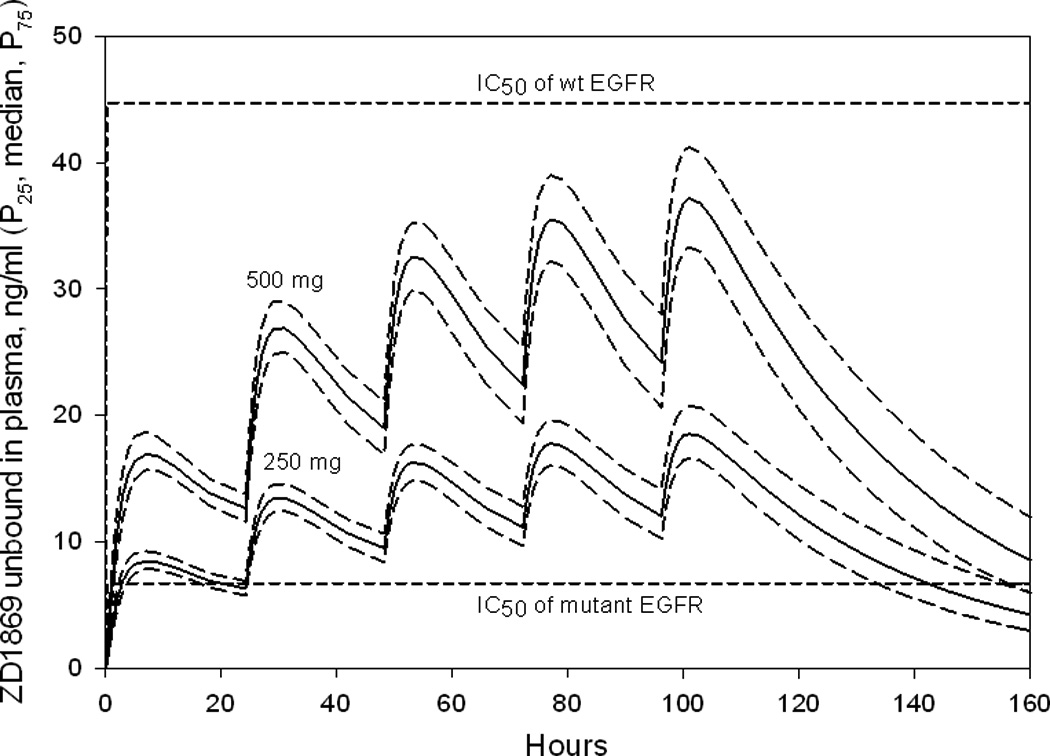
Simulated mean plasma concentrations of ZD1839 (P25, median and P75) in individuals (n = 2,000), receiving 250 or 500 mg daily for 5 days following 2 days off drug. Shown also is the IC50 concentration of ZD1839 versus wt and mutant EGFR (0.1 µM, corresponding to 44.7 ng/ml, versus 0.015µM, corresponding to 6.7 ng/ml, respectively).
DISCUSSION
Docetaxel and cisplatin are both efficacious in the treatment of SCCHN. However, disease-free survival continues to be rare in advanced SCCHN and new treatment strategies are needed. The in vitro data from this study suggest that the combination of the EGFR tyrosine kinase inhibitor ZD1839 with cisplatin and docetaxel produces synergistic growth inhibition, which is mediated by an increased rate of apoptosis. Furthermore, synergism was highly dependent upon the correct sequencing of the exposure of cells to ZD1839 relative to the other cytotoxic agents. Our studies suggest that when ZD1839 is administered first, it arrests carcinoma cells in G0/G1 phase, making them less sensitive to cell killing by drugs that exert their cytotoxic effects primarily during the G2M phase of the cell cycle. This study may provide a new paradigm of considering optimal drug sequencing when planning clinical trials that involve biologic agents and conventional cytotoxic chemotherapy.
Apoptosis was previously considered a mechanism of taxane-related cytotoxicity24. However, recent evidence suggests that the precise mechanisms of taxane-mediated cell death are dose- and cell line-specific, and that individual cells may die via a variety of apoptotic and mitotic cell death pathways24–29. Proteolytic cleavage of poly (ADP-ribose) polymerase (PARP) is an early biochemical event during apoptosis and a hallmark of caspase activation. All drug combinations in this study produced PARP cleavage, with DPZ demonstrating the highest levels of cleaved PARP. The intrinsic pathway may be a more prominent mechanism underlying our findings, since caspase-9 cleavage was observed in all cell samples treated with the various drug combinations. However, the extrinsic pathway was a major contributor to the enhanced apoptosis observed in cells treated with the triple drug combination DPZ, as evidenced by enhanced caspase-3 and -8 cleavage. The pathway of taxane-induced apoptosis is cell line-specific and may be mediated by either the intrinisic or extrinsic pathway with varying caspase cleavage patterns30, 31. Docetaxel induces caspase-3, -8 and -9 activation in a human oral squamous carcinoma cell line, which is consistent with our observations32. The single agents did not induce caspase activation noticeably at low doses. However, synergistic effects were observed with the triple combination DPZ, suggesting a role for both extrinsic and intrinsic apoptosis pathways.
Treatment of SCCHN with EGFR TKIs has gained wide acceptance in recent years33–35, in part, since their favorable toxicity profile makes them an attractive choice in patients who are elderly or have significant comorbidities. The two commercially available EGFR TKIs in the U.S., ZD1839 (gefitinib) and erlotinib, have demonstrated modest response rates in SCCHN when assessed with conventional RECIST criteria. However, substantial disease control rates including stable disease have been achieved in 34 to 53% of patients with metastatic or recurrent SCCHN11–13. Conventional cytotoxic chemotherapy of SCCHN with docetaxel and cisplatin results in response rates of about 30% to 40% in recurrent disease. There has been much debate concerning the possible activation of pro-survival pathways by cytotoxic drugs that could be responsible for chemoresistance and poor response rates. Upregulation of activated AKT has been shown to correlate with worse prognosis in different cancers36–38. While docetaxel and cisplatin as single agents lead to activation of EGFR-mediated pro-survival pathways, as evidenced by elevated levels of p-EGFR, p-ERK and p-AKT, the combination of both drugs has an even more profound effect. Activation of EGFR-mediated pro-survival pathways can be suppressed by the addition of ZD1839. In this study, ZD1839 downregulated AKT and ERK activation, even when combined with docetaxel and cisplatin which strongly induce these pathways. This suggests that survival in Tu212 cells treated with cisplatin and docetaxel is dependent on these two survival pathways, which are successfully abrogated by the addition of ZD1839.
While combinations of EGFR TKIs with cytotoxic chemotherapy have shown promising results in phase II trials in non small cell lung cancer, the results of phase III trials were disappointing17, 39, 40. The detailed mechanisms of these negative results need to be explored. In the preclinical setting, animals were given TKIs for only five days a week. However, patients took TKIs continuously for seven days a week11, 12, 41. Taxanes exert their antitumor effects by stabilizing microtubules during mitosis, which results in disruption of mitosis. However, when tumor cells are pretreated with cytostatic agents like TKIs, their entry into the mitotic phase of the cell cycle is delayed. Therefore, cytostatic agents like TKIs may protect tumor cells from taxane-mediated killing. Our data demonstrated that when ZD1839 is administered before cisplatin and docetaxel, the cell cycle was arrested in G0/G1 with a concomitant reduction in apoptosis42. We hypothesize that this G0/G1 arrest prevents cisplatin and docetaxel from exerting their apoptotic effects, since only cells in the G2M phase of the cell cycle are susceptible to these agents. Since SCCHN cells were compromised when cisplatin and docetaxel were administered following pre-treatment with ZD1839, future clinical trials may consider the administration of ZD1839 intermittently, while administering the cytotoxic agents (D and P) at intervals when plasma concentrations of ZD1839 are low. These observations are consistent with a study in lung cancer cell lines by Mahaffey et al. who also illustrated that the sequence of docetaxel followed by another EGFR-TKI erlotinib resulted in enhanced apoptosis compared with single agent docetaxel43. In addition, we performed an in silico dosing study of Z using mean and variance parameter estimates from a previously published population pharmacokinetic model to derive dosing principles for future in vivo use.
A feasibility dosing study of ZD1839 was performed in silico, to assess whether a regimen could be developed that maintains adequate plasma concentrations during periods when D and P are not administered, while yielding minimal effect levels of ZD1839 during the intervals when D and P are administered. Plasma concentration versus time curves were simulated for large cohorts taking ZD1839 at 250 or 500 mg daily (n = 2,000 per group) for 5 days, and no ZD1839 on days 6 and 7 (analogous to a Monday to Friday daily dose regimen, with no dose administered on Saturday and Sunday). Both regimens were predicted to produce plasma concentrations higher than the IC50 of mutant EGFR (IC50 = 0.08 µM) (Fig. 5)21, 44 for most of the 5 day regimen. Therefore, EGFR-TK should be adequately inhibited during that period. Based on the inter-individual variation in pharmacokinetics, relatively mild toxicity, and the poor vascular perfusion of certain tumors, Li, et al, suggested that the 500 mg daily regimen would be preferable21. However, these in vitro data suggest that certain chemotherapy agents such as D and P should be administered when concentrations of ZD1839 are low, to allow the targeted cancer cells to enter the G2M phase of the cell cycle, where maximal cell kill is expected. Therefore, the pharmacokinetic simulations suggest that it may be preferable to initiate chemotherapy with D and P on day one and then ZD1839 at a dose of 250 mg/day for 5 days followed by a 2 day absence of the drug, allowing cells to cycle to a more sensitive phase for killing by the D and P chemotherapy. This regimen could then be repeated for the remaining cycles of chemotherapy, to maintain sufficiently high plasma concentrations of ZD1839 while allowing the drug to decline to levels below the IC50 of mutant EGFR during the 2-day washout period in the vast majority of (> 90% versus 25% for the 500 mg dose regimen). However, the higher (500 mg) dose regimen of ZD1839 could be considered for maintenance once the D and P component of the therapy has been completed. However, the pharmacodynamic studies reported in this study were performed using dissociated cells grown in culture and lacks the heterogeneity in oxygen, pH and nutrients that occurs in solid tumors, due to varying distances of cells from the nearest blood vessels, and from the relatively disorganized vasculatures observed in solid tumors45. The in vitro study also did not consider the possibility of shielding of sub-populations of cells by the tumor interstitium46. However, the primary mechanism of action of gefitinib is to inhibit growth of cells that express the EGF receptor. Therefore, it was not surprising that the phase specific cytotoxic chemotherapy agents (docetaxel and cisplatin), examined in this study were most effective against EGFR positive cells when gefitinib levels were low. Thus, assuming that gefitinib and these cytotoxic agents reach adequate concentrations in the tumors, it would seem rational to sequence the administration of the cytotoxic agents when cell growth is not inhibited by high levels of gefitinib.
A phase II clinical trial combining docetaxel, cisplatin and ZD1839 (gefitinib) was presented at ASCO in 2005, which demonstrated a remarkable 66.7% disease control rate and 50% overall response rate 16. However, two thirds of the enrolled patients had newly diagnosed metastatic SCCHN, and only one third had recurrent SCCHN which carries significantly lower response rates. The primary tumor location in almost half of the patients was the oral cavity, which makes it difficult to compare these results with other trials in which most patients had primary oropharyngeal lesions. Our data suggest that holding the dose of TKI two days prior to cytotoxic chemotherapy could increase response rates by avoiding cell cycle arrest in the G0/G1 phase immediately prior to giving cisplatin and/or docetaxel. The combination of TKIs with conventional cytotoxic chemotherapy is promising. However, these combinations will only show maximum efficacy if clinical trials are designed that consider the importance of correct sequencing of the various modalities. A better understanding of the molecular mechanisms underlying the effects of chemotherapy should enable us to design improved clinical trials with targeted and conventional chemotherapy agents.
Acknowledgment
We thank Dr. Anthea Hammond for her critical reading and editing of the manuscript.
Grant support: This study was supported by NCI P50 CA128613, RO1 CA112643, and U01 101244 grant (D.M.S.), and Georgia Cancer Coalition Distinguished Scholar (D.M.S. and Z.C.).
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56(2):106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Schrijvers D, Vermorken JB. Taxanes in the treatment of head and neck cancer. Curr Opin Oncol. 2005;17(3):218–224. doi: 10.1097/01.cco.0000158735.91723.0e. [DOI] [PubMed] [Google Scholar]
- 3.Trimmer EE, Essigmann JM. Cisplatin. Essays Biochem. 1999;34:191–211. doi: 10.1042/bse0340191. [DOI] [PubMed] [Google Scholar]
- 4.Hitt R, Grau J, Lopez-Pousa A, et al. Randomized phase II/III clinical trial of induction chemotherapy (ICT) with either cisplatin/5-fluorouracil (PF) or docetaxel/cisplatin/5-fluorouracil (TPF) followed by chemoradiotherapy (CRT) vs. crt alone for patients (pts) with unresectable locally advanced head and neck cancer (LAHNC) Journal of Clinical Oncology, 2006 ASCO Annual Meeting Proceedings. 2006;24(18S):5515. [Google Scholar]
- 5.Posner MR, Hershock D, Le-Lann L, Devlin PM. Scientific Special Session: Docetaxel added to Induction Therapy in Head and Neck Cancer. SCO Annual Meeting. Atlanta, GA: 2006. Haddad RIftTSG. [Google Scholar]
- 6.Mehanna HM, Morton RP. Deterioration in quality-of-life of late (10-year) survivors of head and neck cancer. Clin Otolaryngol. 2006;31(3):204–211. doi: 10.1111/j.1749-4486.2006.01188.x. [DOI] [PubMed] [Google Scholar]
- 7.List MA, Siston A, Haraf D, et al. Quality of life and performance in advanced head and neck cancer patients on concomitant chemoradiotherapy: a prospective examination. J Clin Oncol. 1999;17(3):1020–1028. doi: 10.1200/JCO.1999.17.3.1020. [DOI] [PubMed] [Google Scholar]
- 8.Munker R, Purmale L, Aydemir U, et al. Advanced head and neck cancer: long-term results of chemo-radiotherapy, complications and induction of second malignancies. Onkologie. 2001;24(6):553–558. doi: 10.1159/000055143. [DOI] [PubMed] [Google Scholar]
- 9.Rubin Grandis J, Melhem MF, Barnes EL, Tweardy DJ. Quantitative immunohistochemical analysis of transforming growth factor-alpha and epidermal growth factor receptor in patients with squamous cell carcinoma of the head and neck. Cancer. 1996;78(6):1284–1292. doi: 10.1002/(SICI)1097-0142(19960915)78:6<1284::AID-CNCR17>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 10.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354(6):567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 11.Cohen EE, Rosen F, Stadler WM, et al. Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2003;21(10):1980–1987. doi: 10.1200/JCO.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 12.Cohen EE, Kane MA, List MA, et al. Phase II trial of gefitinib 250 mg daily in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. Clin Cancer Res. 2005;11(23):8418–8424. doi: 10.1158/1078-0432.CCR-05-1247. [DOI] [PubMed] [Google Scholar]
- 13.Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22(1):77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 14.Cohen EE, Haraf DJ, Stenson KM, et al. Integration of Gefitinib (G), into a Concurrent Chemoradiation (CRT) Regimen Followed by G Adjuvant Therapy in Patients with Locally Advanced Head and Neck Cancer (HNC) - a Phase II Trial. Journal of Clinical Oncology, 2005 ASCO Annual Meeting Proceedings. 2005;23(16S):5506. [Google Scholar]
- 15.Kim ES, Kies M, Sabichi A, et al. Phase II Study of Combination Cisplatin, Docetaxel and Erlotinib in Patients with Metastatic/Recurrent Head and Neck Squamous Cell Carcinoma (HNSCC) J Clin Oncol, 2005 Annual Meeting Proceedings. 2005;23(16S):5546. [Google Scholar]
- 16.Belón J, Irigoyen A, Rodríguez I, et al. Preliminary results of a Phase II study to evaluate gefitinib combined with docetaxel and cisplatin in patients with recurrent and/or metastatic squamous-cell carcinoma of the head and neck. Journal of Clinical Oncology, 2005 ASCO Annual Meeting Proceedings. 2005;23(16S):5563. [Google Scholar]
- 17.Herbst RS, Prager D, Hermann R, et al. TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol. 2005;23(25):5892–5899. doi: 10.1200/JCO.2005.02.840. [DOI] [PubMed] [Google Scholar]
- 18.Gazemeier U, Pluzanska A, Szczesna A, et al. Results of a Phase III trial of erlotinib (OSI-774) combined with cisplatin and gemcitabine (GC) chemotherapy in advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2004;22(14S):619. abstr 7010. [Google Scholar]
- 19.Zandvliet AS, Schellens JH, Beijnen JH, et al. Population pharmacokinetics and pharmacodynamics for treatment optimization in clinical oncology. Clin Pharmacokinetic. 47:857–513. doi: 10.2165/00003088-200847080-00001. [DOI] [PubMed] [Google Scholar]
- 20.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Karlsson MO, Brahmer J, et al. CYP3A phenotyping approach to predict systemic exposure to EGFR tyrosine kinase inhibitors. J Natl Cancer Inst. 2006;98(23):1714–1723. doi: 10.1093/jnci/djj466. [DOI] [PubMed] [Google Scholar]
- 22.Choe MS, Chen ZG, Klass CM, et al. Enhancement of docetaxel-induced cytotoxicity by blocking EGFR and COX-2 pathways in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13:3015–3023. doi: 10.1158/1078-0432.CCR-06-2959. [DOI] [PubMed] [Google Scholar]
- 23.Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55(3):178–194. doi: 10.3322/canjclin.55.3.178. [DOI] [PubMed] [Google Scholar]
- 24.Ricci MS, Zong WX. Chemotherapeutic approaches for targeting cell death pathways. Oncologist. 2006;11(4):342–357. doi: 10.1634/theoncologist.11-4-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 26.Wang LG, Liu XM, Kreis W, Budman DR. The effect of antimicrotubule agents on signal transduction pathways of apoptosis: a review. Cancer Chemother Pharmacol. 1999;44(5):355–361. doi: 10.1007/s002800050989. [DOI] [PubMed] [Google Scholar]
- 27.Blajeski AL, Kottke TJ, Kaufmann SH. A multistep model for paclitaxel-induced apoptosis in human breast cancer cell lines. Exp Cell Res. 2001;270(2):277–288. doi: 10.1006/excr.2001.5349. [DOI] [PubMed] [Google Scholar]
- 28.Torres K, Horwitz SB. Mechanisms of Taxol-induced cell death are concentration dependent. Cancer Res. 1998;58(16):3620–3626. [PubMed] [Google Scholar]
- 29.Morse DL, Gray H, Payne CM, Gillies RJ. Docetaxel induces cell death through mitotic catastrophe in human breast cancer cells. Mol Cancer Ther. 2005;4(10):1495–1504. doi: 10.1158/1535-7163.MCT-05-0130. [DOI] [PubMed] [Google Scholar]
- 30.Huisman C, Ferreira CG, Broker LE, et al. Paclitaxel triggers cell death primarily via caspase-independent routes in the non-small cell lung cancer cell line NCI-H460. Clin Cancer Res. 2002;8(2):596–606. [PubMed] [Google Scholar]
- 31.Perkins CL, Fang G, Kim CN, Bhalla KN. The role of Apaf-1, caspase-9, and bid proteins in etoposide- or paclitaxel-induced mitochondrial events during apoptosis. Cancer Res. 2000;60(6):1645–1653. [PubMed] [Google Scholar]
- 32.Taniguchi T, Takahashi M, Shinohara F, Sato T, Echigo S, Rikiishi H. Involvement of NF-kappaB and mitochondrial pathways in docetaxel-induced apoptosis of human oral squamous cell carcinoma. Int J Mol Med. 2005;15(4):667–673. [PubMed] [Google Scholar]
- 33.Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33(4):369–385. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 34.Cohen EE. Role of epidermal growth factor receptor pathway-targeted therapy in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2006;24(17):2659–2665. doi: 10.1200/JCO.2005.05.4577. [DOI] [PubMed] [Google Scholar]
- 35.Vokes EE, Chu E. Anti-EGFR therapies: clinical experience in colorectal, lung, and head and neck cancers. Oncology (Williston Park) 2006;20(5) Suppl 2:15–25. [PubMed] [Google Scholar]
- 36.Lim J, Kim JH, Paeng JY, et al. Prognostic value of activated Akt expression in oral squamous cell carcinoma. J Clin Pathol. 2005;58(11):1199–1205. doi: 10.1136/jcp.2004.024786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Massarelli E, Liu DD, Lee JJ, et al. Akt activation correlates with adverse outcome in tongue cancer. Cancer. 2005;104(11):2430–2436. doi: 10.1002/cncr.21476. [DOI] [PubMed] [Google Scholar]
- 38.Janmaat ML, Gallegos-Ruiz MI, Rodriguez JA, et al. Predictive factors for outcome in a phase II study of gefitinib in second-line treatment of advanced esophageal cancer patients. J Clin Oncol. 2006;24(10):1612–1619. doi: 10.1200/JCO.2005.03.4900. [DOI] [PubMed] [Google Scholar]
- 39.Herbst RS, Giaccone G, Schiller JH, et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 2. J Clin Oncol. 2004;22(5):785–794. doi: 10.1200/JCO.2004.07.215. [DOI] [PubMed] [Google Scholar]
- 40.Giaccone G, Herbst RS, Manegold C, et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 1. J Clin Oncol. 2004;22(5):777–784. doi: 10.1200/JCO.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Sirotnak FM, Zakowski MF, Miller VA, Scher HI, Kris MG. Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin Cancer Res. 2000;6(12):4885–4892. [PubMed] [Google Scholar]
- 42.Solit DB, She Y, Lobo J, et al. Pulsatile administration of the epidermal growth factor receptor inhibitor gefitinib is significantly more effective than continuous dosing for sensitizing tumors to paclitaxel. Clin Cancer Res. 2005;11(5):1983–1989. doi: 10.1158/1078-0432.CCR-04-1347. [DOI] [PubMed] [Google Scholar]
- 43.Mahaffey CM, Davies AM, Lara PN, et al. Schedule-dependent apoptosis in K-ras mutant non-small-cell lung cancer cell lines treated with docetaxel and erlotinib: rationale for pharmacodynamic separation. Clin Lung Cancer. 2007;8(9):548–553. doi: 10.3816/clc.2007.n.041. [DOI] [PubMed] [Google Scholar]
- 44.Barker AJ, Gibson KH, Grundy W, et al. Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett. 2001;11(14):1911–1914. doi: 10.1016/s0960-894x(01)00344-4. [DOI] [PubMed] [Google Scholar]
- 45.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J. Natl Cancer Inst. 2007;99:1141–1154. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]