

Abstract
Recently, the development of poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP) inhibitors has brought a major breakthrough in the treatment of germline breast cancer susceptibility gene (BRCA)-mutant cancers.1-4 These agents target a DNA repair pathway via a novel mechanism of action. A better understanding of DNA damage repair mechanisms can extend the therapeutic application of this novel drug class to a wide range of sporadic cancers. Early clinical success has accelerated the development of various PARP inhibitors that are being explored in many cancers with single agents or in combination with chemotherapy or radiotherapy. In this article, the authors review DNA repair mechanisms and the role of PARP as a therapeutic target and summarize available PARP inhibitors, their clinical trials, biomarkers of PARP inhibitor sensitivity, and resistance mechanisms.
Keywords: Poly(ADP-ribose) polymerase, Inhibitor, Synthetic lethality, BRCA
DNA DAMAGE AND REPAIR MECHANISMS
DNA damage is generated by a variety of factors, not only from external factors such as chemical agents, UV light, or ionizing radiation but also from internal factors such as reactive oxygen species or intrinsic DNA replication errors during cell division. DNA damages, if let unrepaired, can cause errors of DNA synthesis during replication, leading to cell death or irreversible mutations resulting in long-term oncogenesis. Thus, individuals with an inherited defect in the DNA repair system are often at an increased risk of cancer.5 Common errors include (1) base modifications by frequently reactive oxygen species or chemical agents, such as loss of an amino group (deam-ination) or alkylation; (2) mismatch of nucleotide pairs by replication errors; (3) single-strand break (SSB) or double-strand break (DSB) by ionizing radiation; (4) failures in normal DNA metabolism by topoisomerases and nuclease; or (5) cross-links, covalent linkages between bases on intrastrand or interstrand. DSBs are the most critical form of DNA damage and can result in problems for transcription, replication, and chromosome segregation, eventually leading to apoptosis or carcinogenesis.5
To maintain genetic stability against constantly occurring DNA lesions, several strategies of DNA damage detection and repair have evolved. The principal and partly overlapping DNA repair pathways in humans can be largely divided into 2 groups: one is the repair pathway for DNA SSB and the other is for DNA DSB. The former can be subdivided into 3 different repair processes: (1) repair of base damage and SSBs by base excision repair (BER), (2) repair of bulky DNA adducts by nucleotide excision repair (NER), and (3) repair of mismatches and insertion/deletion loops by DNA mismatch repair (MMR). The DSB is repaired through (1) homologous recombination (HR) and (2) nonhomologous end joining (NHEJ).
Base Excision Repair
The BER pathway repairs damage to a single or a few bases caused by reactive oxygen species, alkylating agents, or ionizing radiation, such as oxidation, alkylation, hydrolysis, or deamination, that could cause mutations by incorrect base pairing or lead to breaks in DNA during replication if uncorrected (Fig. 1A).5,6 PARP1 and PARP2 are recruited to the site of the SSB in this pathway.
Fig. 1.
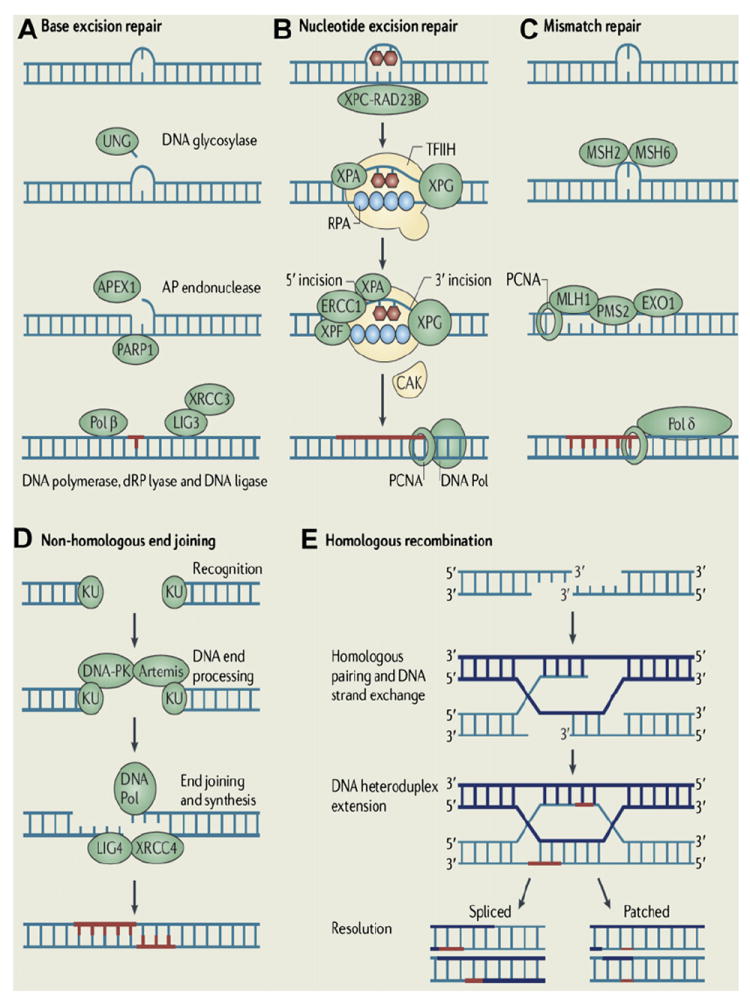
Major DNA damage repair mechanisms. (A) BER: a specific DNA glycosylase removes a damaged base, for example, uracil DNA glycosylase (UNG) for uracil. Subsequently, an apyrimidinic/apurinic (AP) site incision by an AP endonuclease 1 (APEX1) and the removal of 5’-deoxyribose-phosphate (dRP) residue by a dRP lyase occur followed by nucleotide gap filling by DNA polymerase β. (B) NER: after a DNA-distorting lesion is recognized with or without the xeroderma pigmentosum complementation group C (XPC)/Rad23 homolog B (RAD23B) protein complex, dual incisions on both sides of the lesion and excision of the damaged site occur by the general transcription factor TFIIH, XPG, and excision repair cross-complementation group 1 (ERCC1)/XPF complex. DNA polymerase fills the resulting nucleotide gap. (C) MMR: the mismatched bases are recognized by a heterodimer of MSH2/MSH6 and excised by exonuclease 1 (EXO1), which is recruited by a heterodimer of MLH1/PMS2. The resulting gap is filled by DNA polymerase δ along with proliferating cell nuclear antigen (PCNA) and replication protein A (RPA). (D) NHEJ: the 2 broken ends are processed and ligated directly by Ku70/Ku80 complex and the DNA-dependent protein kinase (DNA-PK) followed by DNA ligase IV (LIG4)/XRCC4. (E) HR: repair is initiated by resection of a DSB resulting in 3’ single-stranded DNA overhangs, which invade into a homologous sequence followed by DNA synthesis at the invading end. Subsequently, the second 3’ end is captured to form a Holliday junction. After gap-filling DNA synthesis and ligation, the structure is resolved at the Holliday junction in a crossover or noncrossover mode. (Adapted from Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer 2011;11:96–110; with permission. Copyright © 2011 Macmillan Publishers Ltd.)
Nucleotide Excision Repair
NER is used for the removal of large patches around DNA lesions that are bulky and/or alter the helical structure of the DNA molecule, including those caused by UV radiation, carcinogenic compounds, and cross-linking chemotherapeutic agents (see Fig. 1B).7
Mismatch Repair
MMR removes base mismatches by DNA polymerases and small insertion-deletion loops introduced during DNA replication and recombination (see Fig. 1C).5 A variety of base pair abnormalities resulting from DNA damage are also subject to processing by MMR, which include base pairs containing O6-methylguanine, carcinogen adducts, UV photo products, and cisplatin adducts.8 Defects in the MMR system dramatically increase mutation rates resulting in hereditary nonpolyposis colorectal cancer and some types of sporadic tumor.9
Nonhomologous End Joining
NHEJ can occur throughout the cell cycle, predominantly in the G1 phases. DNA break ends are directly ligated without the use of a homologous template, so NHEJ is prone to introducing errors ranging from small insertions and deletions at the break site to the joining of previously unlinked DNA ends (see Fig. 1D).10
Homologous Recombination
HR repairs DSB by using the undamaged sister chromatid or homologous chromosome, resulting in error-free repair, and occurs primarily during the S and G2 phases of the cell cycle. BRCA proteins are involved in this pathway (see Fig. 1E).
DNA REPAIR SYNTHETIC LETHALITY AS A THERAPEUTIC TARGET
PARP in DNA Repair
PARP is an abundant nuclear enzyme involved in several cellular processes involving mainly DNA repair and programmed cell death. PARP1 and PARP2 are the only members of the PARP family that are known to be involved in the DNA SSB repair via the BER pathway.11,12 Once PARP detects an SSB, it binds to the sites of DNA damage through its DNA-binding domain and begins the synthesis of a poly(ADP-ribose) (PAR) chain from the substrate nicotinamide adenine dinucleotide (NAD+) at sites of breakage. PARP induces poly(ADP-ribosyl)ation in PARP itself at the automodification domain and other proteins such as histone (H1 and H2B), which leads to chromatin decondensation to accommodate various DNA repair enzymes.13 The auto-poly(ADP-ribosyl)ation of PARP1 mediates the recruitment of BER proteins such as DNA polymerase β, DNA ligase III, and scaffolding proteins such as x-ray repair cross-complementing 1 (XRCC1) heterodimer.14 PARP1 then dissociates from the DNA because of its negative charge resulting from poly(ADP-ribosyl)ation, and the PAR chains are degraded by PAR glycohydrolase and possibly the ADP-ribose hydrolase ARH3 after repair of the DNA break.15 Besides its firmly established role in BER and DNA SSB repair, PARP1 is involved in multiple types of other DNA damage repair processes, including the repair of DNA DSBs, DNA cross-links, and stalled replication forks. PARP1 promotes restart of stalled replication forks and HR by recruiting HR factors including ATM, MRE11, and NBS1.16 Furthermore, PARP1 is recruited for DSB repair in the absence of essential components of the classical pathway of NHEJ such as Ku70.17 Although PARP1 is the predominant enzyme that synthesizes PAR in response to DNA damage, PARP2 has been shown to interact with PARP1 and is also implicated in BER.12 PARP2 is, however, unable to fully compensate for the loss of PARP1, accounting for approximately 10% of the total PARP activity of human cells.11,18
BRCA1 and BRCA2 in DNA Repair
BRCA1 plays a key role in the regulation of the cell cycle checkpoints and the HR-mediated DNA repair pathway. On DNA damage, BRCA1 is rapidly phosphorylated and redistributes to sites of DNA breaks, where it interacts with RAD51 and other proteins involved in DNA DSB repair, such as MRN and CtIP.19 Although BRCA1 appears to function upstream of RAD51 filament polymerization, BRCA2 directly binds to and translocates RAD51 to areas of DNA damage and stabilizes RAD51 filaments on DSB.20 Absence of either factor causes DNA repair defect, primarily in HR, which contributes to tumorigenesis. Heterozygous germline mutations in BRCA1 or BRCA2 predispose to various type of cancers, especially breast and ovarian cancers. This HR deficiency also has a critical impact on chemosensitivity. BRCA1-mutated breast cancer cells are highly sensitive to DNA interstrand cross-linking–inducing agents such as cisplatin and mitomycin C because they disrupt replication forks during S phase, which requires HR-mediated DSB repair for S phase progression and cell survival.21,22 Because of their heightened cytotoxicity in HR-defective cells, platinum agents have been extensively applied as chemotherapeutic drugs in BRCA1- or BRCA2-associated cancers with outcomes better than those of non-BRCA-associated cancers.23,24
Synthetic Lethality
The term synthetic lethality was coined in 1946. It describes a phenomenon in which 2 nonlethal mutations have no effect on cell viability in the presence of either mutation alone but lead to cell death in combination.25 This concept is now being exploited in cancer treatment. Because many cancer cells have cancer-relevant genetic lesions that are not present in normal cells, mimicking the effect of a second genetic mutation with a targeted agent should selectively kill only cancer cells with a large therapeutic window. To date, the most successful synthetic lethality relationship in DNA repair pathways for cancer treatment comes from PARP inhibition in BRCA-null tumors. Although PARP1 plays a critical role in DNA SSB repair, loss of PARP1 activity is not lethal in normal cells. When unrepaired DNA SSBs caused by the absence of PARP1 activity encounter DNA replication forks, they result in stalled replication forks that are subsequently converted to DNA DSBs. Although these DNA DSBs are effectively repaired by the HR pathway in normal cells,5,26 cells that are defective in HR, such as BRCA1- or BRCA2-deficient cells, cannot repair them, resulting in cell death. Therefore, PARP inhibitors could be selectively lethal to cells lacking functional BRCA1 or BRCA2 with minimal toxicity to normal cells.
Synthetic lethality in BRCA1- and BRCA2-deficient cells when exposed to PARP inhibitors was confirmed in in vitro and in vivo mouse models.27,28 Farmer and colleagues27 showed that PARP1 depletion with RNA interference decreased clonogenic survival of BRCA1- and BRCA2-deficient embryonic stem cells compared with wild-type cells. Furthermore, clonogenic cell survival assays showed that BRCA1- or BRCA2-deficient cells were much more sensitive to the potent PARP inhibitors, KU0058684 and KU0058948, than heterozygous mutant or wild-type cells.27 KU0058684 also blocked tumor growth in vivo in BRCA2-deficient cells. Similarly, Bryant and colleagues28 demonstrated that BRCA2-deficient V-C8 cells were profoundly sensitive to the PARP inhibitors NU1025 and AG014361, as compared with the wild-type V79 cells. Human breast cancer cell lines MCF-7 and MDA-MB-231 also displayed a similar sensitivity to NU1025 on depletion of BRCA2 with RNA interference.28
BRCAness
The primary challenge to the promise of PARP inhibitors in the treatment of BRCA-mutant cancers is the low frequency of germline mutations in BRCA1 or BRCA2, only 10% to 15% of unselected ovarian cancers29-31 and 5% to 10% of breast cancers.32-34 In addition, BRCA1 and BRCA2 genes are infrequently mutated somatically in sporadic cancers, with 4% to 9% of unselected or sporadic ovarian cancers.35-37 However, some sporadic cancers phenotypically behave like BRCA1/2-mutant cancers even though they do not have known BRCA mutations. These tumors may carry abnormalities in the expression or function of BRCA1 or BRCA2 or in other critical components of the HR DNA repair pathway. This phenomenon is called BRCAness and is characterized by defective HR.38 One of the mechanisms of BRCAness is the silencing of BRCA1 or BRCA2 through the aberrant methylation of the promoter and has been reported in 11% to 14% of sporadic breast cancers39-41 and 5% to 18% of ovarian cancers.36,39,40,42 Preclinical studies have shown that tumor cell lines with decreased BRCA expression by mutation or epigenetic silencing responded equally well to the PARP inhibitor AG014699 that additionally inhibited the growth of epigenetically silenced BRCA1 xenograft tumors.43 These suggest that PARP inhibitors may have a potential role in sporadic cancers as well as hereditary cancers. The amplification of the EMSY gene that encodes for EMSY protein that can interact with and silence the activation domain of BRCA2 has also been described as a potential mechanism of BRCA inactivation. The amplification of the EMSY gene was reported in 13% of sporadic breast cancers and 17% of high-grade serous ovarian cancers (HGSOCs).44 Because Fanconi anemia proteins are involved in the HR pathway, the epigenetic silencing of Fanconi anemia complementation group F (FANCF) gene through promoter methylation is another potential mechanism of BRCAness.45 The inhibition of the FANCF gene in ovarian cancer cell lines through promoter methylation was associated with enhanced sensitivity to DNA-damaging agents such as cisplatin; in reverse, demethylation of the FANCFpromoter resulted in cisplatin resistance.45 In addition to potential transcriptional and posttranslational abnormalities occurring in BRCA1 and BRCA2 pathways, deficiency of proteins involved in the HR pathway, such as RAD51, RAD54, DSS1, RPA1, NBS1, ATR, ATM, CHK1, or CHK2, can also induce sensitivity to PARP inhibitors.46,47
PARP INHIBITORS
The profound sensitivity of BRCA-mutant cells to PARP inhibition prompted the development of PARP inhibitors for cancer therapy (Table 1). At present, most PARP inhibitors in preclinical and clinical studies are third-generation PARP inhibitors and compete with the substrate NAD+ for the catalytic domain of the PARP enzyme, leading to reversible inhibition of the enzyme. AZD2281/KU-0059436 (olaparib) and ABT-888 (veliparib) are the most common inhibitors assessed to date in clinical trials. Both drugs are administered orally and are potent inhibitors of PARP1 and PARP2, with a half maximal inhibitory concentration (IC50 or Ki) in the nanomolar range for 2 enzymes.48,49 Preclinical activity of AG014361 has also been translated into the clinic as its clinical analogue CO338 (rucaparib, AG014699, PF-01367338) administered intravenously. It was initially tested in combination with temozolomide in melanoma.50 Other PARP inhibitors under active clinical investigation include MK4827, BMN 673, CEP-9722, and E7016/GPI 21016 (see Table 1). Of note, BSI-201 (iniparib) was initially described as having PARP inhibitory activity, which proceeded into a phase 3 trial in triple-negative breast cancer (TNBC) in combination with carboplatin and paclitaxel,51-53 but recent evidence suggests that iniparib does not seem to inhibit PARP1 and PARP2 at the clinical dose.54,55
Table 1.
PARP inhibitors in active clinical investigation
| Name | Route | Disease | Single/Combination Therapy | Phase of Clinical Developmenta | Company |
|---|---|---|---|---|---|
| Rucaparib (CO338, AGO 14699, PF-0367338) | IV | BRCA1 + ovarian/breast cancer Melanoma | Single | 2 | Clovis Oncology (Boulder, CO, USA) |
| Temozolomide | 2 | ||||
|
| |||||
| Olaparib (AZD2281) | Oral | BRCA+ ovarian cancer | Single agent | 2 | AstraZeneca (London, UK) |
| BRCA+ breast cancer | |||||
| High-grade SOC/TNBC | |||||
| Various solid tumors | Carboplatin, topotecan, paclitaxel, dacarbazine, or gemcitabine/cisplatin | 1 | |||
|
| |||||
| Veliparib (ABT-888) | Oral | Melanoma | Temozolomide | 2 | Abbott (Abbott Park, IL, USA) |
| Breast cancer | |||||
| Colorectal cancer | |||||
| Various solid tumors | Topotecan, irinotecan, cyclophosphamide, or doxorubicin/cyclophosphamide | 1 | |||
|
| |||||
| MK 4827 | Oral | HGSOC enriched with BRCA+ | Single | 1 | Merck (Whitehouse Station, NJ, USA) |
|
| |||||
| BMN 673 | Oral | Various solid tumors | Single agent | 1/2 | Biomarin (Novato, CA, USA) |
| Hematologic malignancies | |||||
|
| |||||
| CEP-9722 | Oral | Advanced solid tumor (phase 1)/cancer with defective DNA repair pathway (phase 2) | Single | 1/2 | Cephalon (Frazer, PA, USA) |
| Solid tumor | Temozolomide Gemcitabine/Cisplatin | 1 | |||
|
| |||||
| E7016 (GPI 21016) | Oral | Solid tumor | Temozolomide | 1 | MGI Pharma/Eisai (Bloomington, MN, USA) |
Abbreviations: BRCA, breast cancer susceptible gene; IV, intravenous; SOC, serous ovarian cancer; TNBC, triple-negative breast cancer.
Indicates the most advanced phase in studies that have been published or presented. In CEP-9722 and E7016, ongoing trials are shown because there are no published studies.
Data from ClinicalTrials.gov, National Library of Medicine, Bethesda, MD.
MONOTHERAPY WITH PARP INHIBITORS IN BRCA-MUTANT CANCERS
On the basis of preclinical data showing hypersensitivity to PARP inhibitors in tumor cells lacking BRCA1 or BRCA2, PARP inhibitor was used as a single agent in patients with BRCA mutation. The first landmark trial of PARP inhibitor as monotherapy was performed with olaparib enriched with BRCA1/2 mutation carriers and limited to patients with BRCA mutation ovarian cancer in the expansion phase.1 Olaparib was safe and well tolerated with no obvious differences in toxicities observed between BRCA1 or BRCA2 mutation carriers and noncarriers. The maximum tolerated dose (MTD) was 400 mg twice a day, and the dose-limiting toxicities (DLTs) included grade 3 mood alteration, fatigue, somnolence, and grade 4 thrombocytopenia. As expected, there was significant antitumor activity only in patients with BRCA1- or BRCA2-mutated cancers, including ovarian, breast, and prostate cancers, among whom 12 of 17 (63%) patients had clinical benefit and 9 of 19 (47%) patients achieved responses according to the Response Evaluation Criteria in Solid Tumors (RECIST). In the expansion cohort of 50 BRCA mutation carriers with ovarian, primary peritoneal, and fallopian tube cancers, 20 patients (40%) had a RECIST response, cancer antigen 125 responses, or both; 14 patients (28%) had RECIST responses.2 Pharmacodynamic assay demonstrated that a dose of 60 mg or more twice daily reduced PARP activity in peripheral blood mononuclear cells (PBMCs) by more than 90% as compared with the value at baseline. Plucked eyebrow hair follicles were collected before treatment and again 6 hours after treatment, showing induction of γ-H2AX, which did not seem to increase further at doses greater than 100 mg twice daily, the lowest dose of this analysis. Based on these results, a dose of 100 mg twice daily may be adequate for PARP inhibitory effect; this dose was further studied in phase 2 studies along with a twice-daily dose of 400 mg, the MTD.
Subsequently, 2 proof-of-concept phase 2 studies with olaparib in patients with advanced or recurrent breast (International Collaborative Expertise for BRCA Education and Research through Genetics [ICEBERG]1) or ovarian cancer (ICEBERG2) who had BRCA1 or BRCA2 mutations have provided further support for synthetic lethal approach to cancer therapy.3,4 Both studies recruited sequentially 2 dose cohorts treated with twice-daily doses of olaparib 400 mg (the established MTD) and then 100 mg (the lowest PARP inhibitory dose with clinical activity).1,2 In ICEBERG1 trial, the objective response rate in breast cancer seemed to be higher in the 400-mg cohort (41%, 11/27) than in the 100-mg cohort (22%, 6/27), and median progression-free survival (PFS) also seemed to be longer in the 400-mg cohort (5.7 vs 3.8 months).3 Similarly, ICEBERG2 trial in ovarian cancer showed that the high-dose cohort seemed to have better response rate (33% vs 13%) and median PFS (5.8 vs 1.9 months) than the low-dose cohort.4 Overall, both trials confirmed the tolerability of olaparib in BRCA 1/2 mutation carriers, with mainly grade 1 or 2 adverse events.
A subsequent randomized phase 2 study comparing the safety and efficacy of 2 doses of olaparib (200 or 400 mg twice daily continuously in 28-day cycles) with that of pegylated liposomal doxorubicin (PLD) (50 mg/m2 every 4 weeks) was done in patients with BRCA mutation–associated ovarian cancer who had failed previous platinum-based chemotherapy.56 A total of 97 patients were randomized into 3 groups. Although the 400-mg group had a numerically longer median PFS (8.8 vs 6.5 vs 7.1 months, respectively) and a higher response rate (25% vs 18% vs 31%, respectively) than the 200-mg or PLD groups, neither outcome reached statistical significance. These negative outcomes might be partially attributed to better PFS in the PLD group compared with historical cohorts. Regarding safety, both doses of olaparib were well tolerated with less toxicity compared with PLD. The PLD group had more grade 3 or 4 toxicities. Although the primary end point was not fulfilled, this study demonstrated the consistent efficacy with favorable safety of olaparib as monotherapy in BRCA-mutant ovarian cancer.
Another promising PARP inhibitor that has been studied in BRCA1/2 mutation carriers is MK4827, a potent oral PARP1/2 inhibitor with an IC50 of 3.8 and 2.1 nM for PARP1 and PARP2, respectively.57 A phase 1 study of MK4827 enriched with BRCA1/2 mutation carriers had established MTD at a dose of 300 mg once daily.58 DLTs included grade 3 fatigue and pneumonitis and grade 4 thrombocytopenia. Inhibition of PARP activity in PBMCs was demonstrated at doses of 80 mg or greater. The response rate was 37% (7/19) in patients with BRCA-mutated ovarian cancer and 20% (3/15) in patients with sporadic ovarian cancer. MK4827 was well tolerated with the most common toxicities of fatigue (52.5%), nausea (52.5%), vomiting (38.8%), diarrhea (21.3%), thrombo-cytopenia (33.8%), and neutropenia (21.3%), which were mostly graded 1 to 2 except for thrombocytopenia (grade 3/4, 15%).
Rucaparib was evaluated in a phase 2 study for BRCA1/2-mutated advanced ovarian and/or breast cancer in which 18 mg/m2 of rucaparib was given on days 1 to 5 of a 21-day cycle.59 PARP activity assessed in PBMC demonstrated a mean inhibition of 84% over baseline levels at 24 hours after a single dose of 18 mg/m2 of AG014699. Although the overall response rate was 5% (2/38), 26% (10/38) achieved stable disease for 4 or more months. Rucaparib had an acceptable safety profile with drug-related toxicity, mainly grade 1 and 2; the most common toxicities were fatigue (39%), nausea (26.8%), diarrhea (19.5%), and dizziness (17.1%).
Overall, these data clearly provide clinical validation for single-agent PARP inhibitor activity in patients with BRCA1/2 mutations.
MONOTHERAPY WITH PARP INHIBITORS IN SPORADIC CANCERS
The evidence of BRCAness has been shown particularly in a substantial proportion of sporadic ovarian cancer and TNBC or basal-like breast cancer. Overall, BRCA1/2 deficiency as a result of germline or somatic BRCA1/2 mutations, epigenetic loss of BRCA1, or expression loss by other mechanisms has been reported in 30% to 53% of ovarian cancers, which was associated with high-grade serous/undifferentiated tumor histology and improved PFS with chemotherapy.36,37 TNBC overlaps substantially with basal-like breast cancer. BRCA1-associated cancers generally cluster with the basal-like subtype in gene expression profiling studies.60 Most BRCA1-associated breast cancers share many phenotypic features with TNBC and basal-like breast cancer.61 This notion of shared BRCAness is the rationale for testing PARP inhibition in sporadic TNBC or HGSOC because even without germline BRCA1/2 mutations, these tumors may harbor other lesions that impair HR.
To address whether sporadic cancers with BRCAness would also be responsive to PARP inhibitors as are BRCA-mutant cancers, a phase 2 study of single-agent olaparib (400 mg twice daily continuously) in patients with HGSOC or undifferentiated ovarian cancer or TNBC was conducted in which patients were stratified according to whether they had a germline BRCA1/2 mutation or not (or unknown BRCA mutation status).62 Ninety patients (64 with ovarian cancer and 26 with breast cancer) received treatment with a median of 3 prior chemotherapy regimens. Although the breast cancer cohorts did not have any confirmed objective response, the ovarian cancer cohorts showed a response rate of 41% in patients with BRCA mutations and 24% in patients without mutations. The median PFSs were 221 days and 192 days, respectively. The most common adverse events were grade 1 or 2 fatigue, nausea, and vomiting. These results demonstrate the activity of a PARP inhibitor in patients with HGSOC without germline BRCA1/2 mutations.
In addition, promising results of a randomized, double-blind, placebo-controlled phase 2 study of maintenance olaparib in patients with platinum-sensitive relapsed HGSOC independent of germline BRCA status were also recently reported.63 Patients who had maintained an objective response following the last platinum-containing regimen were randomized to a twice-daily dose of 400 mg of olaparib (n = 136) or placebo (n = 129). The olaparib group had a significantly longer PFS, median 8.4 versus 4.8 months. Approximately 20% of patients in both groups had BRCA1/2 mutations, and a preplanned subgroup analysis showed that the PFS benefit from olaparib was not restricted to patients with BRCA mutation–associated ovarian cancer. No unexpected toxicities were seen.
Overall, these data provide clinical evidence for single-agent PARP inhibitor activity in cancers with BRCAness phenotype even without BRCA mutations.
COMBINATION OF PARP INHIBITORS WITH CHEMOTHERAPY
Radiotherapy and chemotherapeutic agents such as platinum compounds confer their antitumor effects by inducing DNA damage, which, if not repaired, triggers cell death. As PARP inhibitors could impair DNA repair mechanisms, the potential of PARP inhibitors as a chemosensitizer or radiosensitizer has been raised. Consequently, preclinical studies have shown activity of a PARP inhibitor in combination with DNA-damaging agents such as alkylating agents (such as cyclophosphamide and temozolomide), type I topoisomerase inhibitors (such as irinotecan and topotecan), platinum agents (such as cisplatin and carboplatin), and anthracyclines. These combinations are being tested clinically with various PARP inhibitors as discussed later.
Rucaparib (CO338, AG014699, PF-01367338)
Rucaparib was the first PARP inhibitor to undergo a phase 1 trial as an enhancing agent for chemotherapy. A phase 1 study of the combination of AG014699 with temozolomide in unselected advanced solid tumors determined that the recommended phase 2 dose was intravenous rucaparib, 12 mg/m2/d, and oral temozolomide, 200 mg/m2/d, for 5 days every 28 days. At this dose, PARP was inhibited by more than 70% in PBMC. DLTs included myelosuppression at the dose level of 18 mg/m2/d and temozolomide 200 mg/m2/d.50 A phase 2 study was conducted with rucaparib and temozolomide in untreated metastatic melanoma.64 However, significantly more myelosuppression was observed than was predicted from the phase 1 study. Of 46 patients treated, grade 4 thrombocytopenia and neutropenia were seen in 30% and 41% of patients in cycle 1, respectively, resulting in 39% dose reduction and/or dose delay. Given that severe myelosuppression is uncommon with temozolomide monotherapy, it has been suggested that PARP inhibitors may inhibit the repair of DNA damage from temozolomide in bone marrow stem cells. Partial response was seen in 18% of the 40 evaluable patients.
Olaparib (AZD2281, KU-0059436)
Several phase 1 trials of olaparib combined with chemotherapy have been reported in BRCA-mutant cancer or sporadic cancer. A phase 1 study of olaparib combined with carboplatin in BRCA1/2-mutated ovarian or breast cancer initially evaluated the continuous schedule of olaparib, 100 or 200 mg twice daily, on days 1 to 21 plus carboplatin AUC 3 on days 1 or 2 every 3 weeks.65 However, the schedule was changed to intermittent administration of olaparib (olaparib, 200 or 400 mg twice daily, on days 1 to 7 plus carboplatin AUC 3, 4, or 5 on days 1 or 2 every 3 weeks) because of thrombocytopenia and delayed recovery of neutropenia. The recommended dose was olaparib, 400 mg twice daily, on days 1 to 7 with carboplatin AUC 5 on days 1 or 2 every 3 weeks. Main toxicity was grade 3/4 marrow suppression. Of 27 evaluable patients, the response rate was 39% in ovarian cancer and 74% in breast cancer, and the clinical benefit rate was 83% and 100%, respectively. A phase 1 study of olaparib with topotecan in advanced solid tumors reported prolonged thrombocytopenia and neutropenia. Although the MTD was established as topotecan, 1.0 mg/m2/d, on days 1 to 3 plus olaparib, 100 mg twice daily, on days 1 to 21 every 3 weeks, no responses were seen. The investigators did not recommend further development of this combination. When olaparib was combined with weekly paclitaxel in patients with metastatic TNBC, significant myelosuppression required granulocyte colony–stimulating factor as secondary prophylaxis. The confirmed partial response rate was 37%. A phase 1 trial of the combination of olaparib and dacarbazine also showed higher incidence of neutropenia and delayed recovery than observed with single-agent dacarbazine.66 The optimal tolerated dose was defined as olaparib, 100 mg twice daily, on days 1 to 7 and dacarbazine, 600 mg/m2, on day 1 every 3 weeks. Although 2 patients with melanoma had partial responses in the initial dose escalation phase, there was no response in the dose confirmation cohort enrolling chemonaive patients with melanoma (overall response rate, 5%). In a phase 1 study of olaparib combined with gemcitabine/cisplatin, the MTD was olaparib, 100 mg once a day, on days 1 to 4; cisplatin, 50 mg/m2, on day 3; and gemcitabine, 400 mg/m2, on days 3 and 10 every 3 weeks or olaparib 100 mg twice daily on day 1, cisplatin 60 mg/m2 on day 1, and gemcitabine 500 mg/m2 on days 1 and 8 every 3 weeks.67 However, these MTDs were still associated with the high incidence of grade 3/4 neutropenia (50%) and thrombocytopenia (17%–50%). Of 21 evaluable patients, 2 (9.5%) had partial responses. Ongoing clinical trials of olaparib in combination with chemotherapy are summarized in Table 2.
Table 2.
Ongoing clinical trials of olaparib
| Phase | Regimen | Disease | Study Designation |
|---|---|---|---|
| 2 | Olaparib | Advanced solid tumor with BRCA1/2 mutation | NCT01078662 |
|
| |||
| 1 | Olaparib + cisplatin | Advanced solid tumor | NCT00782574 |
|
| |||
| 1 | Olaparib + gemcitabine | Advanced pancreatic cancer | NCT00515866 |
|
| |||
| 1 | Olaparib + liposomal doxorubicin | Advanced solid tumor | NCT00819221 |
|
| |||
| 1 | Olaparib + irinotecan | Advanced colorectal cancer | NCT00535353 |
|
| |||
| 1 | Olaparib + paclitaxel vs olaparib + carboplatin vs olaparib + paclitaxel/carboplatin | Advanced TNBC or ovarian cancer | NCT00516724 |
|
| |||
| 1 | Olaparib + temozolomide | Advanced glioblastoma | NCT01390571 |
|
| |||
| 1 | Group 1: olaparib on days 1–7 + carboplatin on day 1 every 3 wk | Refractory gynecologic cancers such as breast, ovarian, fallopian, primary peritoneal, uterine, cervical cancer, or malignant mixed müllerian tumors | NCT01237067 |
| Group 2A: olaparib on days 1–7 + carboplatin on day 8 in cycle 1 followed by carboplatin on day 1 + olaparib on days 2–8 in cycle 2 | |||
| Group 2B: reverse sequence | |||
|
| |||
| 1/2 | Phase 1: irinotecan/cisplatin/mitomycin (ICM) | Advanced pancreatic cancer | NCT01296763 |
| Phase 2: IC/ICM ± olaparib | |||
|
| |||
| 2 | Olaparib + paclitaxel vs placebo + paclitaxel | Advanced gastric cancer | NCT01063517 |
|
| |||
| 2 | Paclitaxel/carboplatin ± olaparib | Advanced ovarian cancer | NCT01081951 |
Abbreviations: BRCA, breast cancer susceptible gene; TNBC, triple-negative breast cancer.
Data from ClinicalTrials.gov, National Library of Medicine, Bethesda, MD.
Veliparib (ABT-888)
Veliparib was initially evaluated in the first nontherapeutic phase 0 clinical trial in oncology that guided the design of subsequent phase 1 trials of veliparib in combination with chemotherapeutic agents.68 This study suggested that an appropriate phase 1 starting dose of veliparib in combination with DNA-damaging agents is 10 mg twice daily, based on the measured levels of PARP inhibition between paired blood and tumor specimens. In a subsequent phase 1 study of veliparib combined with topotecan for refractory solid tumors and lymphomas, there were 3 DLTs in the 6 patients on dose level 1 (veliparib, 10 mg twice daily, on days 1–7; topotecan, 1.2 mg/m2/d, on days 1–5 in a 3-week cycle).69 Further dose reduction of topotecan to 0.6 mg/m2/d showed no DLT in 6 patients that was defined as the MTD combined with veliparib, 10 mg twice daily, on days 1 to 5. There was no tumor response. A phase 1 study of the combination of veliparib and weekly irinotecan determined that the MTD was veliparib, 40 mg twice daily, on days 1 to 14 and irinotecan, 100 mg/m2, on days 1 and 8 every 3 weeks with DLTs including grade 3/4 fatigue, leukopenia/neutropenia, and diarrhea.70 Of 30 evaluable patients, 6 (20%) had partial responses and the clinical benefit rate was 63%. A phase 1 study of veliparib in combination with a fixed dosing of doxorubicin (60 mg/m2) and cyclophosphamide (600 mg/m2) for breast cancer and other solid tumors showed that the MTD was veliparib, 100 mg, twice daily on days 1 to 4 in a 3-week cycle.71 The most common toxicities were fatigue and myelosuppression. There were 3 partial responses in 5 BRCA mutation carriers (60%), and 79% of the patients with breast cancer had clinical benefit.
In contrast, when veliparib was combined with metronomic oral cyclophosphamide in a phase 1 study for refractory solid tumor and lymphoma, myelosuppression was not prominent.72 The MTD was veliparib, 60 mg, and cyclophosphamide, 50 mg, both given once a day. Confirmed partial responses were observed in 3 among 18 patients (17%); 2 BRCA-mutant ovarian cancers and 1 TNBC. At present, a phase 2 study of this regimen is ongoing in patients with BRCA-mutant ovarian cancer, HGSOC/TNBC, or low-grade non-Hodgkin lymphoma. A phase 1 study of veliparib combined with intravenous cyclophosphamide for advanced solid tumor also showed a favorable toxicity profile.73 DLTs included myelosuppression. The highest dose evaluated so far is 200 mg twice daily on days 1 to 4 with cyclophosphamide, 750 mg/m2, on day 3 in a 3-week cycle. The most common toxicities were fatigue, anemia, and neutropenia. There was one partial response among 30 patients.
Two phase 2 trials of veliparib with temozolomide have been reported in metastatic breast cancer and colorectal cancer, respectively.74,75 In both trials, treatment consisted of veliparib, 40 mg, twice daily on days 1 to 7 and temozolomide 150 mg/m2/d on days 1 to 5 every 4 weeks.74 The objective response rate was 4.1% and the disease control rate was 22% in these heavily treated patients with a median number of prior chemotherapeutic regimen of 3.5. The median duration of disease control was 22 weeks (range, 15–40 weeks). In the breast cancer study, frequent grade 4 thrombocytopenia led to general decrease of veliparib to 30 mg twice daily with better tolerability.75 The objective response rate and clinical benefit rate was 37.5% and 62.5%, respectively, in BRCA mutation carriers (n = 8). The median PFS was 5.5 months (BRCA carriers) and 1.8 months (noncarriers) (P = .0042). Recently, a randomized phase 2 placebo-controlled study compared temozolomide (150 mg/m2/d for 5 days) plus veliparib (20 or 40 mg twice daily on days 1-7) every 4 weeks versus temozolomide plus placebo in patients with advanced melanoma.76 The primary end point of PFS showed 113 days in the 20-mg group versus 110 days in the 40-mg group versus 60 days in the placebo group; these trends were not significant. Grade 3/4 adverse events, mainly of hematological toxicities, were seen in 38% (placebo), 54% (veliparib 20 mg), and 57% (veliparib 40 mg) of patients. Ongoing clinical trials of veliparib in combination with chemotherapy or chemoradiotherapy are summarized in Tables 3 and 4.
Table 3.
Ongoing clinical trials of veliparib
| Phase | Regimen | Disease | Study Designation |
|---|---|---|---|
| 1 | Veliparib | BRCA1/2-mutated cancer; platinum-refractory ovarian, fallopian tube, or primary peritoneal cancer; or basal-like breast cancer | NCT00892736 |
|
| |||
| 1 | Veliparib ± mitomycin | Advanced solid tumor with a defective FA pathway | NCT01017640 |
|
| |||
| 1 | Veliparib + PLD | Recurrent ovarian cancer, fallopian tube cancer, or primary peritoneal cancer or metastatic breast cancer | NCT01145430 |
|
| |||
| 1 | Veliparib + gemcitabine | Advanced solid tumor | NCT01154426 |
|
| |||
| 1 | Veliparib + gemcitabine + cisplatin | Advanced biliary, pancreatic, urothelial, or non–small cell lung cancer | NCT01282333 |
|
| |||
| 1 | Veliparib + carboplatin | Advanced TNBC or estrogen receptor and/or progesterone receptor (+), HER2 (−) breast cancer with a defective FA pathway | NCT01251874 |
|
| |||
| 1 | Veliparib + gemcitabine + carboplatin | Advanced solid tumor | NCT01063816 |
|
| |||
| 1 | Veliparib + weekly paclitaxel + weekly carboplatin | Advanced solid tumors | NCT01281150 |
|
| |||
| 1 | Veliparib + triweekly paclitaxel + triweekly carboplatin | Patients with hepatic or renal dysfunction and advanced solid tumors | NCT01366144 |
|
| |||
| 1 | Veliparib + triweekly paclitaxel + triweekly carboplatin | Advanced solid cancer (stratum I), advanced solid cancer with a germline BRCA1/2 mutation (stratum II) | NCT00535119 |
|
| |||
| 1 | Veliparib + paclitaxel + carboplatin + bevacizumab | Newly diagnosed stage II–IV ovarian epithelial, fallopian tube, or primary peritoneal carcinoma | NCT00989651 |
|
| |||
| 1 | Veliparib + cisplatin + vinorelbine | Advanced TNBC and/or BRCA1/2-mutant breast cancer | NCT01104259 |
|
| |||
| 1 | Veliparib + temozolomide | Metastatic castration-resistant prostate cancer | NCT01085422 |
|
| |||
| 1 | Veliparib + FOLFIRI (irinotecan 150 mg/m2 or 180 mg/m2) | Escalation phase: advanced solid tumor | NCT01123876 |
| Expansion phase: advanced colorectal cancer | |||
|
| |||
| 1/2 | Veliparib + triweekly paclitaxel + triweekly cisplatin | Stage IVB, recurrent, or persistent cervix cancer | NCT01281852 |
|
| |||
| 1/2 | Veliparib + topotecan | Advanced solid tumors (phase 1), relapsed or refractory ovarian cancer or primary peritoneal cancer (phase 2) | NCT01012817 |
|
| |||
| 1/2 | Veliparib + bendamustine + rituximab | Advanced lymphoma, multiple myeloma, or solid tumors | NCT01326702 |
|
| |||
| 2 | Veliparib ± carboplatin | BRCA1/2-mutant advanced breast cancer | NCT01149083 |
|
| |||
| 2 | Cyclophosphamide ± veliparib | Estrogen receptor and/or progesterone receptor (+) and HER2 (−) metastatic breast cancer | NCT01351909 |
|
| |||
| 2 | Cyclophosphamide ± veliparib | BRCA1/2-mutant ovarian cancer, primary peritoneal or ovarian high-grade carcinoma, or fallopian tube cancer; TNBC (not responsive to hormone-related therapy); or low-grade non-Hodgkin lymphoma | NCT01306032 |
|
| |||
| 2 | Veliparib + temozolomide | Advanced solid tumor | NCT01193140 |
|
| |||
| 2 | Veliparib + temozolomide | Advanced hepatocellular carcinoma progressing following sorafenib or intolerant to sorafenib | NCT01205828 |
|
| |||
| 2 | Veliparib + temozolomide vs PLD alone | Recurrent high-grade serous ovarian, fallopian tube, or primary peritoneal cancer | NCT01113957 |
|
| |||
| 2 | Neratinib vs veliparib + carboplatin vs paclitaxel + trastuzumab followed by doxorubicin + cyclophosphamide vs AMG 386 | Stage II or III or T4, any N, M0 breast cancer | NCT01042379 |
|
| |||
| 1 | Veliparib + WBRT | Metastatic brain tumor | NCT00649207 |
|
| |||
| 1 | Veliparib + LDFWAR | Advanced solid tumor with peritoneal carcinomatosis | NCT01264432 |
|
| |||
| 1/2 | Veliparib + temozolomide + radiotherapy | Newly diagnosed glioblastoma multiforme | NCT00770471 |
|
| |||
| 1/2 | Veliparib + chemoradiotherapy with paclitaxel + carboplatin | Unresectable stage III non–small cell lung cancer | NCT01386385 |
Abbreviations: BRCA, breast cancer susceptible gene; FA, Fanconi anemia; FOLFIRI, 5-fluorouracil/leucovorin/irinotecan; HER2, human epidermal growth factor receptor 2; LDFWAR, low-dose fractionated whole abdominal radiation therapy; PLD, pegylated liposomal doxorubicin; TNBC, triple-negative breast cancer; WBRT, whole brain radiation therapy.
Data from ClinicalTrials.gov, National Library of Medicine, Bethesda, MD.
Table 4.
Ongoing clinical trials of MK4827, BMN 673, CEP-9722, or E7016
| Phase | Regimen | Disease | Study Designation |
|---|---|---|---|
| 1 | MK4827 | Advanced solid tumor | NCT01226901 |
| 1 | MK4827 | Dose escalation phase: advanced solid tumor (part A). Expansion phase: prostate, recurrent platinum-resistant HGSOC (part B); T-cell prolymphocytic leukemia or chronic lymphocytic leukemia (part C); advanced colorectal, endometrial, triple- negative or estrogen receptor–positive breast cancer, or partially platinum-sensitive epithelial ovarian cancer (part D) | NCT00749502 |
| 1 | MK4827 + PLD | Advanced solid tumor (part A) and platinum-resistant/refractory HGSOC (part B) | NCT01227941 |
| 1 | MK4827 + temozolomide | Advanced solid tumor (part A); advanced glioblastoma multiforme or melanoma (part B) | NCT01294735 |
| 1 | BMN 673 | Hematologic malignancies | NCT 01399840 |
| 1 | BMN 673 | Advanced solid tumor | NCT 01286987 |
| 1/2 | CEP-9722 | Advanced solid tumor | NCT01311713 |
| 1 | CEP-9722 ± temozolomide | Advanced solid tumor | NCT00920595 |
| 1 | CEP-9722 + gemcitabine + cisplatin | Advanced solid tumor | NCT01345357 |
| 1 | E7016 + temozolomide | Advanced solid tumor (escalation phase) and glioma (expansion phase) | NCT01127178 |
Abbreviations: HGSOC, high-grade serous ovarian cancer; PLD, pegylated liposomal doxorubicin.
Data from ClinicalTrials.gov, National Library of Medicine, Bethesda, MD.
MK4827
MK4827 had been combined with carboplatin with/without paclitaxel or PLD and temozolomide (Table 4).
BMN 673
Two single agent phase I trial in solid tumor and hematologic malignancies are ongoing at this time (see Table 4).
CEP-9722
Two phase 1 studies of CEP-9722 in combination with temozolomide and gemcitabine/cisplatin, respectively, in patients with advanced solid tumors are ongoing (see Table 4).
E7016/GPI 21016
A phase 1 study of E7016 combined with temozolomide in patients with advanced solid tumors is ongoing (see Table 4).
BIOMARKERS FOR EFFICACY OF PARP INHIBITORS
Biomarkers for PARP Inhibitor Sensitivity
Potential biomarkers of PARP inhibitor sensitivity in sporadic cancer include BRCA1/2 somatic mutation, BRCA1 promoter methylation, BRCA1 suppression in the absence of methylation, phosphatase and tensin homolog (PTEN) deficiency, ATM mutation, MRE11-dominant negative mutations in MMR–deficient cancers, and FANCF promoter methylation. PTEN is a tumor suppressor gene that inactivates the PI3K/AKT survival pathway, and its loss of function is a frequent event in a variety of human cancers, occurring through mutations, deletions, or promoter hypermethylation.77,78 Preclinical data have demonstrated that PTEN has a role in maintaining chromosomal stability and controlling DNA DSB repair by regulating the transcription of RAD51, and PTEN-deficient tumor cells are defective in HR repair and sensitive to PARP inhibitors such as olaparib and veliparib.79-83 Of note, it was reported that a patient with PTEN-deficient endometrial adenocarcinoma without BRCA1 or BRCA2 mutations had a clear response to olaparib.84 Future clinical trials are needed to confirm this promising finding.
In addition to these specific molecular defects, there has been an approach to identify unifying biomarkers of HR deficiency that all these diverse mechanisms share. One example is the potential signature of BRCAness that has been reported based on the differences in gene expression between hereditary BRCA1/2-related cancers and sporadic cancers.85,86 These gene expression signatures were associated with responsiveness to DNA-damaging chemotherapy that provides interesting proofs of principle. The other example is to assess the HR pathway competency using surrogate markers, such as RAD51 foci formation on DNA damage.87 Although these approaches are promising, further assessment and validation is needed for use in the clinical setting.
Biomarkers for PAPR Inhibitor Resistance
Recently, genetic reversion of BRCA1 or BRCA2 mutations, which causes restoration of normal BRCA1 or BRCA2 protein function, was recognized as a mechanism of acquired resistance to both platinum and PARP inhibitors in BRCA1- or BRCA2-mutated tumor cells.88-90 A recent study in 110 patients with ovarian cancer with a BRCA1/2 mutation has shown that a secondary somatic BRCA1/2 mutation was found more frequently in recurrent cancer than in primary cancer (28.3% vs 3.1%, P = .0003) and in platinum-resistant recurrences than in platinum-sensitive recurrences (46.2% vs 5.3%, P = .003).91 In addition, 12 (92%) of 13 recurrent cancers with secondary mutations were platinum resistant, and 2 of 3 patients with platinum-resistant recurrent cancer with secondary mutations were also refractory to olaparib, although 1 patient responded to veliparib combined with carboplatin/gemcitabine. By contrast, all 3 patients with platinum-resistant recurrent cancer without secondary mutations achieved responses (2 complete responses, 1 partial response) to olaparib ± carboplatin. Larger studies are needed to reveal to what extent this mechanism will drive clinical resistance to PARP inhibitors in patients with BRCA1/2-related cancers. In addition to genetic reversion, recent evidence also suggested a role for P-glycoprotein efflux pump in the development of resistance to olaparib.92 Other potential resistance mechanisms include the p53 binding protein (53BP1) loss that partially rescues the HR defect in BRCA1-deficient cells and reverts their hypersensitivity to DNA-damaging agents.93,94 Furthermore, 53BP1-deficient BRCA1Δ11/Δ11 cells were no longer sensitive to PARP inhibition.93 The expression of 53BP1 was lost or reduced in subsets of BRCA1/2-associated and sporadic TNBC/basal-like breast cancers.94
SUMMARY
The use of PARP inhibitors to create synthetic lethality or modulate cytotoxicity may be one of the most exciting developments in cancer research in this decade. This new class of drugs shows promise as a treatment of a wide range of cancers, not just those associated with BRCA1/2 genetic mutations. The evaluation of PARP inhibitors is being expanded to combined therapy with chemotherapy, radiotherapy, or other molecularly targeted agents and to a variety of treatment settings, including neoadjuvant, adjuvant, or prophylaxis setting. At the same time, many unanswered questions remain about the optimal use of PARP inhibitors. Could a single-agent PARP inhibitor be a possible alternative front-line treatment of BRCA-associated cancer or other HR-defective cancer? If PARP inhibitors are to be combined with chemotherapy or radiotherapy, what is the best administration schedule, dose, or chemotherapeutic partner to maximize efficacy while minimizing toxicity? What are robust predictive biomarkers of sensitivity and resistance to PARP inhibitors alone or combined with chemotherapy or radiotherapy? To what extent the PARP enzyme needs to be inhibited for the best efficacy? What creates the differences between the individual agents in the PAPR inhibitor class resulting in distinct toxicity and efficacy profiles? What are the consequences of long-term administration of PARP inhibitor? This question may be especially important because this class moves into the adjuvant setting or for long-term administration in prevention studies in BRCA mutation carriers. Carefully designed clinical trials incorporating translational end points are required to get answers about these important challenges.
KEY POINTS.
Many mechanisms are present in a cell to repair DNA damage, which when effective allow for cell survival. Understanding these mechanisms are important for both therapeutic treatment of cancer and overcoming resistance.
The concept of synthetic lethality has be tested in treating patients with BRCA mutation with a PARP inhibitor. Initial results with olaparib was encouraging and supportive of synthetic lethality principle.
The role of PARP inhibitor outside of BRCA mutation tumors await confirmation. Multiple studies are ongoing investigating PARP inhibitor with chemotherapy to see if inhibiting DNA repair will lead to tumor reduction and ultimately benefit to the patients.
Footnotes
Disclosure: The authors declared no conflicts of interest.
References
- 1.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 2.Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–9. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 3.Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–44. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 4.Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–51. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 5.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 6.Chan KK, Zhang QM, Dianov GL. Base excision repair fidelity in normal and cancer cells. Mutagenesis. 2006;21(3):173–8. doi: 10.1093/mutage/gel020. [DOI] [PubMed] [Google Scholar]
- 7.Muniandy PA, Liu J, Majumdar A, et al. DNA interstrand crosslink repair in mammalian cells: step by step. Crit Rev Biochem Mol Biol. 2010;45(1):23–49. doi: 10.3109/10409230903501819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iyer RR, Pluciennik A, Burdett V, et al. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106(2):302–23. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 9.Peltomaki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;21(6):1174–9. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- 10.Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412(6847):607–14. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 11.Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26(8):882–93. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 12.Schreiber V, Ame JC, Dolle P, et al. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem. 2002;277(25):23028–36. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- 13.de Murcia G, Huletsky A, Lamarre D, et al. Modulation of chromatin superstructure induced by poly(ADP-ribose) synthesis and degradation. J Biol Chem. 1986;261(15):7011–7. [PubMed] [Google Scholar]
- 14.El-Khamisy SF, Masutani M, Suzuki H, et al. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31(19):5526–33. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oka S, Kato J, Moss J. Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J Biol Chem. 2006;281(2):705–13. doi: 10.1074/jbc.M510290200. [DOI] [PubMed] [Google Scholar]
- 16.Haince JF, McDonald D, Rodrigue A, et al. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem. 2008;283(2):1197–208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- 17.Wang M, Wu W, Rosidi B, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34(21):6170–82. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ame JC, Rolli V, Schreiber V, et al. PARP-2, a novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem. 1999;274(25):17860–8. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- 19.Scully R, Chen J, Ochs RL, et al. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90(3):425–35. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 20.Esashi F, Galkin VE, Yu X, et al. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat Struct Mol Biol. 2007;14(6):468–74. doi: 10.1038/nsmb1245. [DOI] [PubMed] [Google Scholar]
- 21.Moynahan ME, Cui TY, Jasin M. Homology-directed DNA repair, mitomycin-C resistance, and chromosome stability is restored with correction of a BRCA1 mutation. Cancer Res. 2001;61(12):4842–50. [PubMed] [Google Scholar]
- 22.Tassone P, Di Martino MT, Ventura M, et al. Loss of BRCA1 function increases the antitumor activity of cisplatin against human breast cancer xenografts in vivo. Cancer Biol Ther. 2009;8(7):648–53. doi: 10.4161/cbt.8.7.7968. [DOI] [PubMed] [Google Scholar]
- 23.Narod SA. BRCA mutations in the management of breast cancer: the state of the art. Nat Rev Clin Oncol. 2010;7(12):702–7. doi: 10.1038/nrclinonc.2010.166. [DOI] [PubMed] [Google Scholar]
- 24.Tan DS, Rothermundt C, Thomas K, et al. “BRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol. 2008;26(34):5530–6. doi: 10.1200/JCO.2008.16.1703. [DOI] [PubMed] [Google Scholar]
- 25.Dobzhansky T. Genetics of natural populations. Xiii. Recombination and variability in populations of Drosophila pseudoobscura. Genetics. 1946;31(3):269–90. doi: 10.1093/genetics/31.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001;307(5):1235–45. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- 27.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 28.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 29.Risch HA, McLaughlin JR, Cole DE, et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet. 2001;68(3):700–10. doi: 10.1086/318787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pal T, Permuth-Wey J, Betts JA, et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005;104(12):2807–16. doi: 10.1002/cncr.21536. [DOI] [PubMed] [Google Scholar]
- 31.Risch HA, McLaughlin JR, Cole DE, et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J Natl Cancer Inst. 2006;98(23):1694–706. doi: 10.1093/jnci/djj465. [DOI] [PubMed] [Google Scholar]
- 32.Eeles RA. Screening for hereditary cancer and genetic testing, epitomized by breast cancer. Eur J Cancer. 1999;35(14):1954–62. doi: 10.1016/s0959-8049(99)00246-4. [DOI] [PubMed] [Google Scholar]
- 33.Langston AA, Malone KE, Thompson JD, et al. BRCA1 mutations in a population-based sample of young women with breast cancer. N Engl J Med. 1996;334(3):137–42. doi: 10.1056/NEJM199601183340301. [DOI] [PubMed] [Google Scholar]
- 34.Malone KE, Daling JR, Neal C, et al. Frequency of BRCA1/BRCA2 mutations in a population-based sample of young breast carcinoma cases. Cancer. 2000;88(6):1393–402. doi: 10.1002/(sici)1097-0142(20000315)88:6<1393::aid-cncr17>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 35.Merajver SD, Pham TM, Caduff RF, et al. Somatic mutations in the BRCA1 gene in sporadic ovarian tumours. Nat Genet. 1995;9(4):439–43. doi: 10.1038/ng0495-439. [DOI] [PubMed] [Google Scholar]
- 36.Press JZ, De Luca A, Boyd N, et al. Ovarian carcinomas with genetic and epigenetic BRCA1 loss have distinct molecular abnormalities. BMC Cancer. 2008;8:17. doi: 10.1186/1471-2407-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hennessy BT, Timms KM, Carey MS, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28(22):3570–6. doi: 10.1200/JCO.2009.27.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 39.Catteau A, Harris WH, Xu CF, et al. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene. 1999;18(11):1957–65. doi: 10.1038/sj.onc.1202509. [DOI] [PubMed] [Google Scholar]
- 40.Esteller M, Silva JM, Dominguez G, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92(7):564–9. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- 41.Rice JC, Ozcelik H, Maxeiner P, et al. Methylation of the BRCA1 promoter is associated with decreased BRCA1 mRNA levels in clinical breast cancer specimens. Carcinogenesis. 2000;21(9):1761–5. doi: 10.1093/carcin/21.9.1761. [DOI] [PubMed] [Google Scholar]
- 42.Baldwin RL, Nemeth E, Tran H, et al. BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res. 2000;60(19):5329–33. [PubMed] [Google Scholar]
- 43.Drew Y, Mulligan EA, Vong WT, et al. Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst. 2011;103(4):334–46. doi: 10.1093/jnci/djq509. [DOI] [PubMed] [Google Scholar]
- 44.Hughes-Davies L, Huntsman D, Ruas M, et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell. 2003;115(5):523–35. doi: 10.1016/s0092-8674(03)00930-9. [DOI] [PubMed] [Google Scholar]
- 45.Taniguchi T, Tischkowitz M, Ameziane N, et al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003;9(5):568–74. doi: 10.1038/nm852. [DOI] [PubMed] [Google Scholar]
- 46.McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 47.Bryant HE, Helleday T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006;34(6):1685–91. doi: 10.1093/nar/gkl108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menear KA, Adcock C, Boulter R, et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem. 2008;51(20):6581–91. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 49.Penning TD, Zhu GD, Gandhi VB, et al. Discovery of the Poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-car-boxamide (ABT-888) for the treatment of cancer. J Med Chem. 2009;52(2):514–23. doi: 10.1021/jm801171j. [DOI] [PubMed] [Google Scholar]
- 50.Plummer R, Jones C, Middleton M, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14(23):7917–23. doi: 10.1158/1078-0432.CCR-08-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mendeleyev J, Kirsten E, Hakam A, et al. Potential chemotherapeutic activity of 4-iodo-3-nitrobenzamide. Metabolic reduction to the 3-nitroso derivative and induction of cell death in tumor cells in culture. Biochem Pharmacol. 1995;50(5):705–14. doi: 10.1016/0006-2952(95)00189-7. [DOI] [PubMed] [Google Scholar]
- 52.Bauer PI, Mendeleyeva J, Kirsten E, et al. Anti-cancer action of 4-iodo-3-nitrobenza-mide in combination with buthionine sulfoximine: inactivation of poly(ADP-ribose) polymerase and tumor glycolysis and the appearance of a poly(ADP-ribose) poly-merase protease. Biochem Pharmacol. 2002;63(3):455–62. doi: 10.1016/s0006-2952(01)00872-3. [DOI] [PubMed] [Google Scholar]
- 53.O’Shaughnessy J, Schwartzberg LS, Danso MA, et al. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC) J Clin Oncol. 2011;29(Suppl 15) abstract: 1007. [Google Scholar]
- 54.Ji J, Lee MP, Kadota M, et al. Pharmacodynamic and pathway analysis of three presumed inhibitors of poly (ADP-ribose) polymerase: ABT-888, AZD2281, and BSI201. Programs and abstracts of the 102nd Annual Meeting of American Association for Cancer Research; Orlando. April 2–6; 2011. abstract 4527. [Google Scholar]
- 55.Maegley KA, Bingham P, Tatlock JH, et al. All PARP inhibitors are not equal: an in vitro mechanistic comparison of PF-01367338 to iniparib. J Clin Oncol. 2011;29(Suppl 15) abstract: e13576. [Google Scholar]
- 56.Kaye S, Kaufman B, Lubinski J, et al. Phase II study of the oral PARP inhibitor olaparib (AZD2281) versus liposomal doxorubicin in ovarian cancer patients with BRCA1 and/or BRCA2 mutations. Ann Oncol. 2010;21(Suppl 8):viii304. abstract: 9710. [Google Scholar]
- 57.Jones P, Altamura S, Boueres J, et al. Discovery of 2-{4-[(3S)-piperidin-3-yl] phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose) polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J Med Chem. 2009;52(22):7170–85. doi: 10.1021/jm901188v. [DOI] [PubMed] [Google Scholar]
- 58.Schelman WR, Sandhu SK, Moreno Garcia V, et al. First-in-human trial of a poly (ADP)-ribose polymerase (PARP) inhibitor MK-4827 in advanced cancer patients with antitumor activity in BRCA-deficient tumors and sporadic ovarian cancers. J Clin Oncol. 2011;29(Suppl 15) abstract: 3102. [Google Scholar]
- 59.Drew Y, Ledermann JA, Jones A, et al. Phase II trial of the poly(ADP-ribose) polymerase (PARP) inhibitor AG-014699 in BRCA 1 and 2-mutated, advanced ovarian and/or locally advanced or metastatic breast cancer. J Clin Oncol. 2011;29(Suppl 15) abstract: 3104. [Google Scholar]
- 60.Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100(14):8418–23. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–48. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 62.Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–61. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 63.Ledermann JA, Harter P, Gourley C, et al. Phase II randomized placebo-controlled study of olaparib (AZD2281) in patients with platinum-sensitive relapsed serous ovarian cancer (PSR SOC) J Clin Oncol. 2011;29(Suppl 15) abstract: 5003. [Google Scholar]
- 64.Plummer R, Lorigan P, Evans J, et al. First and final report of a phase II study of the poly(ADP-ribose) polymerase (PARP) inhibitor, AG014699, in combination with temozolomide (TMZ) in patients with metastatic malignant melanoma (MM) J Clin Oncol. 2006;24(Suppl 18) abstract: 8013. [Google Scholar]
- 65.Lee J, Annunziata CM, Minasian LM, et al. Phase I study of the PARP inhibitor olaparib (O) in combination with carboplatin (C) in BRCA1/2 mutation carriers with breast (Br) or ovarian (Ov) cancer (Ca) J Clin Oncol. 2011;29(Suppl 15) abstract: 2520. [Google Scholar]
- 66.Khan OA, Gore M, Lorigan P, et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer. 2011;104(5):750–5. doi: 10.1038/bjc.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giaccone G, Rajan A, Kelly RJ, et al. A phase I combination study of olaparib (AZD2281; KU-0059436) and cisplatin (C) plus gemcitabine (G) in adults with solid tumors. J Clin Oncol. 2010;28(Suppl 15) abstract: 3027. [Google Scholar]
- 68.Kummar S, Kinders R, Gutierrez ME, et al. Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol. 2009;27(16):2705–11. doi: 10.1200/JCO.2008.19.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kummar S, Chen A, Ji J, et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011;71(17):5626–34. doi: 10.1158/0008-5472.CAN-11-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.LoRusso P, Ji JJ, Li J, et al. Phase I study of the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of the poly(ADP-ribose) polymerase (PARP) inhibitor veliparib (ABT-888; V) in combination with irinotecan (CPT-11; Ir) in patients (pts) with advanced solid tumors. J Clin Oncol. 2011;29(Suppl 15) doi: 10.1158/1078-0432.CCR-15-0652. abstract: 3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan AR, Toppmeyer D, Stein MN, et al. Phase I trial of veliparib, (ABT-888), a poly(ADP-ribose) polymerase (PARP) inhibitor, in combination with doxorubicin and cyclophosphamide in breast cancer and other solid tumors. J Clin Oncol. 2011;29(Suppl 15) abstract: 3041. [Google Scholar]
- 72.Kummar S, Chen AP, Ji JJ, et al. A phase I study of ABT-888 (A) in combination with metronomic cyclophosphamide (C) in adults with refractory solid tumors and lymphomas. J Clin Oncol. 2010;28(Suppl 15) abstract: 2605. [Google Scholar]
- 73.Tan AR, Gibbon D, Stein MN, et al. Preliminary results of a phase I trial of ABT-888, a poly(ADP-ribose) polymerase (PARP) inhibitor, in combination with cyclo-phosphamide. J Clin Oncol. 2010;28(Suppl 15) abstract: 3000. [Google Scholar]
- 74.Pishvaian MJ, Slack R, Witkiewicz A, et al. A phase II study of the PARP inhibitor ABT-888 plus temozolomide in patients with heavily pretreated, metastatic colorectal cancer. J Clin Oncol. 2011;29(Suppl 15) abstract: 3502. [Google Scholar]
- 75.Isakoff SJ, Overmoyer B, Tung NM, et al. A phase II trial of the PARP inhibitor veliparib (ABT888) and temozolomide for metastatic breast cancer. J Clin Oncol. 2010;28(Suppl 15) abstract: 1019. [Google Scholar]
- 76.Middleton M, Friedlander P, Hamid O, et al. Efficacy of veliparib (ABT-888) plus temozolomide versus temozolomide alone: a randomized, double-blind, placebo-controlled trial in patients with metastatic melanoma. Programs and abstracts of the 36th European Society for Medical Oncology Congress; Stockholm. Sep 23–27, 2011. abstract: 13LBA. [Google Scholar]
- 77.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 78.Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27(41):5477–85. doi: 10.1038/onc.2008.248. [DOI] [PubMed] [Google Scholar]
- 79.Shen WH, Balajee AS, Wang J, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 80.Gupta A, Yang Q, Pandita RK, et al. Cell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repair. Cell Cycle. 2009;8(14):2198–210. doi: 10.4161/cc.8.14.8947. [DOI] [PubMed] [Google Scholar]
- 81.Mendes-Pereira AM, Martin SA, Brough R, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1(6–7):315–22. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dedes KJ, Wetterskog D, Mendes-Pereira AM, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2(53):53ra75. doi: 10.1126/scitranslmed.3001538. [DOI] [PubMed] [Google Scholar]
- 83.McEllin B, Camacho CV, Mukherjee B, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010;70(13):5457–64. doi: 10.1158/0008-5472.CAN-09-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Forster MD, Dedes KJ, Sandhu S, et al. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat Rev Clin Oncol. 2011;8(5):302–6. doi: 10.1038/nrclinonc.2011.42. [DOI] [PubMed] [Google Scholar]
- 85.Konstantinopoulos PA, Spentzos D, Karlan BY, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–61. doi: 10.1200/JCO.2009.27.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rodriguez AA, Makris A, Wu MF, et al. DNA repair signature is associated with anthracycline response in triple negative breast cancer patients. Breast Cancer Res Treat. 2010;123(1):189–96. doi: 10.1007/s10549-010-0983-z. [DOI] [PubMed] [Google Scholar]
- 87.Mukhopadhyay A, Elattar A, Cerbinskaite A, et al. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin Cancer Res. 2010;16(8):2344–51. doi: 10.1158/1078-0432.CCR-09-2758. [DOI] [PubMed] [Google Scholar]
- 88.Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 89.Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–20. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Swisher EM, Sakai W, Karlan BY, et al. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68(8):2581–6. doi: 10.1158/0008-5472.CAN-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29(22):3008–15. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–84. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bunting SF, Callen E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–95. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]