

Abstract
Chronic inflammation contributes to numerous diseases, and regulation of inflammation is crucial for disease control and resolution. Sex hormones have potent immunoregulatory abilities. Specifically, estrogen influences immune cells and inflammation, which contributes to the sexual dimorphism of autoimmunity and protection against disease seen during pregnancy in multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE). Although long thought to act primarily on T cells, recent evidence demonstrated that myeloid cells, such as dendritic cells (DCs), are essential in mediating estrogen's protective effects. Estriol (E3), a pregnancy-specific estrogen, has therapeutic efficacy in MS and EAE, and we evaluated whether E3 could act exclusively through DCs to protect against the inflammatory autoimmune disease EAE. Levels of activation markers (CD80 and CD86) and inhibitory costimulatory markers (PD-L1, PD-L2, B7–H3, and B7–H4) were increased in E3 DCs. E3 DCs had decreased proinflammatory IL-12, IL-23, and IL-6 mRNA expression, increased immunoregulatory IL-10 and TGF-β mRNA expression, and a decreased ratio of IL-12/IL-10 protein production. Importantly, transfer of E3 DCs to mice prior to active induction of EAE protected them from developing EAE through immune deviation to a Th2 response. This protection was apparent, even in the face of in vitro and in vivo inflammatory challenge. In summary, our results showed that E3 generates tolerogenic DCs, which protect against the inflammatory autoimmune disease EAE. Targeted generation of tolerogenic DCs with immunomodulatory therapeutics, such as E3, has potential applications in the treatment of numerous autoimmune and chronic inflammatory diseases.
Chronic inflammation significantly contributes to numerous diseases, including autoimmunity, cardiovascular disease, allergic disease, and cancer, and the ability to regulate such inflammation is crucial for disease control and resolution. Sex hormones have long been known to have potent effects on the immune system and are capable of proinflammatory and immunoregulatory actions. Sexual dimorphism in autoimmune disease and dramatic immunological alterations that occur during pregnancy highlight the profound immunomodulatory capabilities of steroid sex hormones. Estrogens are a group of sex hormones with well-described effects on adaptive immunity, and they can influence pathogenesis and protection in autoimmune and inflammatory disease (1, 2).
Multiple sclerosis (MS) is an inflammatory chronic demyelinating disease of the CNS with no known cure. Although women have an increased incidence of autoimmune diseases, such as MS, pregnancy and pregnancy-related estrogens mediate some of the most potent naturally occurring protection from ongoing MS disease (3–5). Studies with MS and its animal model, experimental autoimmune encephalomyelitis (EAE), have been instrumental in uncovering some of the mechanisms by which estrogens, particularly 17-β estradiol (E2), influence immune cells. However, estrogens, such as E2, have limited therapeutic applications because of toxicity and untoward side effects, including uterine bleeding, hypertension, and increased breast and uterine cancer risk (6). Estrogen-related therapies that minimize such risks, while maintaining immunomodulatory effects, have broad applications in treating autoimmune and chronic inflammatory diseases. Estriol (E3) is a pregnancy-associated estrogen with similar protective effects as E2 but fewer side effects and increased safety (6, 7). Similar to E2, systemic E3 prevents induction of EAE (5, 8, 9); however, unlike E2, E3 has shown protection against established MS, EAE, and autoreactive T cells (3, 10–12). E3 is thought to have potential anti-inflammatory (e.g., limiting infiltration of autoreactive T cells) and neuroprotective actions (13). Interestingly, although E3 is currently in phase II clinical trials for treatment of MS, relatively little is known about E3's specific effects on various immune cells.
EAE is an animal model with clinical, immunological, and histopathologic similarities to MS, which has facilitated our understanding of how sex hormones affect adaptive-immune responses. Active EAE is induced by immunization with neuroantigen to promote disease-causing CD4+ Th1 or Th17 cells, whereas Th2 or regulatory T cells (Tregs) protect from disease (14, 15). It is generally accepted that estrogens impact the development of adaptive immunity, with low levels of estrogen promoting pathogenic Th1/Th17 responses and high (pregnancy) levels of estrogen promoting Th2/Treg responses (1–3, 16, 17). Much of our understanding of estrogen's immunomodulatory abilities has resulted from studies exploring E2's effects in EAE, and it was long thought that these actions were mediated primarily through direct actions on T cells. However, recent evidence suggested that myeloid cells, such as dendritic cells (DCs), are the primary cells responsible for mediating estrogen's protective abilities in EAE (18, 19).
DCs are innate immune cells of the myeloid lineage uniquely able to drive the differentiation of naive Th cells into Th1, Th2, Th17, and Tregs through a combination of MHC class II–TCR–peptide interactions, costimulatory molecules, and cytokine production (20, 21). Long known to promote inflammatory immune responses, DCs are increasingly being recognized for their potent regulatory capabilities in limiting inflammation, with possible therapeutic applications in the treatment of autoimmune and inflammatory diseases (22–24). Such tolerogenic DCs (Tol-DCs) are programmed by a variety of factors, including innate immune receptor signaling, cell–cell interactions, and microenvironmental cues (e.g., steroid hormones, cytokines, other soluble mediators) (20, 25–32). Steroid hormones, such as glucocorticoids and vitamin D, are potent generators of Tol-DCs. Studies showed that these Tol-DCs are arrested in an immature state, expressing low CD40, CD80, CD86, and MHC class II expression and decreased Ag-processing and -presentation capabilities (20, 25–29). Tol-DC populations are diverse and regulate immune responses through numerous potential mechanisms, including altered costimulatory molecule expression, inhibition of proinflammatory mediators (e.g., IL-12, TNF-α, NO, NF-κB), enhanced production of immunoregulatory factors (e.g., IL-10, TGF-β, IDO, arginase), or increased expansion and/or differentiation of Tregs (20, 21, 33–36).
Estrogen's effects on DCs are less well studied than glucocorticoids or vitamin D; however, DCs, like other myeloid cells, are fully able to express estrogen receptors and respond to estrogens (37, 38). Most studies use E2 and show an anti-inflammatory effect of E2 on mature DCs and a potent effect on resting and inflammatory DC differentiation (37, 39, 40). Studies in autoimmune models demonstrated that E2 exposure can diminish Ag presentation, enhance Th2 responses, and enhance production of immunoregulatory IDO or Tregs (18, 33, 41). Although E2 influences DC–T cell interactions, the effects of other estrogens, such as E3, have not been thoroughly investigated, although E3 was shown to decrease proinflammatory NO and TNF-α in mouse microglia and enhance IL-10 production in CD68+ monocytes/macrophages in E3-treated MS patients (11, 36). Given that E3 is protective in EAE and MS and that estrogens (i.e., E2) mediate protection through myeloid cells, such as DCs, this study evaluated whether E3 generates Tol-DCs that could protect against autoimmunity.
We found that E3 exposure generates Tol-DCs with stable regulatory abilities that are resistant to inflammatory challenge. In vivo E3 exposure increased the expression levels of activation markers CD80, CD86, and MHC class II and inhibitory costimulatory markers PD-L1, PD-L2, B7–H3, and B7–H4 in DCs compared to levels seen in placebo (Pb)-treated mice. E3 Tol-DCs also expressed decreased proinflammatory IL-12, IL-23, and IL-6 mRNA levels and increased immunoregulatory IL-10 and TGF-β mRNA levels compared with Pb DCs; had a decreased ratio of IL-12/IL-10 protein production; and diminished T cell proliferation. Importantly, E3 Tol-DCs protected mice from developing EAE through immune deviation to a Th2 response, and in vitro and in vivo inflammatory stimuli failed to abrogate the protective effect of E3 Tol-DCs. Taken together, these data suggest that immunomodulatory compounds, such as E3, generate Tol-DCs that may be useful in the treatment of autoimmune or chronic inflammatory diseases.
Materials and Methods
Mice
Female C57BL/6 (H-2b) mice (4–6 wk old) were purchased from Jackson Laboratories (Bar Harbor, ME), housed four or five/cage, and maintained on a 12-h light/dark cycle. Mice were allowed to rest for 7 d prior to pellet implantation; 5–7 d later they were given Flt3L to expand DCs, and age-matched mice were used for DC transfer and EAE studies. All animals received identical commercially available chow and were housed and cared for according to the institutional guidelines in the University Laboratory Animal Resources at The Ohio State University. Myelin oligodendrocyte glycoprotein (MOG) TCR transgenic (Tg) mice were maintained in-house and screened by flow cytometry for expression of the Vα3.2/Vβ11 MOG-specific TCR.
Induction of EAE and clinical scoring of EAE
Mice were immunized s.c. in four sites on the back (left and right shoulder, left and right flank) with MOG peptide 35–55 (MEVGWYRSPFSRVVHLYRNGK; Princeton Biomolecules Corporation, Langhorne, PA) emulsified in equal volumes of CFA (containing 200 μg heat-killed Mycobacterium tuberculosis Jamaica strain). Mice received 200 ng pertussis toxin in 0.2 ml PBS (List Biological Laboratories, Campbell, CA, USA) i.p. at the time of immunization and 48 h later. Mice were evaluated daily for clinical signs of EAE for ≥35 d and scored as follows: 0, no clinical signs; +1, limp tail or waddling gait with tail tonicity; +2, ataxia or waddling gait with tail limpness; +3, partial hind limb paralysis; +4, total hind limb paralysis; and +5 moribund/death.
Hormone pellet administration and in vivo DC expansion
Sixty day-release 5-mg estriol (E3) pellets or Pb pellets containing carrier without hormone (Innovative Research of America, Sarasota, FL) were implanted s.c. between the shoulder blades; mid- to late-pregnancy levels were achieved by day 5 postimplantation, as previously reported (42). Mice were allowed to recover for ≥4 d prior to Flt3 ligand (Flt3L) administration. Human recombinant Flt3L (hFL; kindly provided by Amgen Thousand Oaks, CA) was used to expand DCs in vivo through daily administration of hFL (200 ng/ml hFL in 200 μl 0.1% mouse serum albumin) s.c. in the nape of the neck for 9 d. Control mice were given vehicle alone (200 μl 0.1% mouse serum albumin). Alternatively, DCs were expanded in vivo by administering 4 million B16 melanoma cells (expressing murine Flt3L) s.c., and mice were sacrificed 3–4 wk after administration. All comparisons of DC populations were from identically treated mice (i.e., E3 and Pb DCs from nonexpanded mice were compared or E3 or Pb DCs from Flt3L-expanded mice were compared).
CD11c+ and CD4+ cell preparation and culture
Splenic DCs from C57BL/6 female mice were positively selected and purified using CD11c magnetic bead separation (Miltenyi Biotec, Auburn, CA) and pulsed with MOG35–55 for 2 h in supplemented RPMI 1640 containing 10% FBS, 25 mM HEPES, 2 mM l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, and 5 × 10−5 M 2-ME. Purified CD11c+ DCs were washed and administered i.v. to naive recipients (1–2 × 106 to 8–10 × 106 DCs) 1 d prior to EAE induction or used for companion in vitro cocultures. CD11c+ cells were phenotypically evaluated for cell surface marker expression (flow cytometry) or cytokine production (ELISPOT) and in functional CD11c+/CD4+ cocultures (proliferation assays, ELISA, ELISPOT, and flow cytometry). Spleen and lymph node CD4+ T cells were purified using CD4+ magnetic bead separation (Miltenyi Biotec, Auburn, CA) for cocultures, and DCs were plated at a ratio of 1:5 or 1:10 with 2 × 105 purified CD4+ T cells and 4 × 104 or 2 × 104 DCs, respectively. Purity of the negative and positive fractions was assessed by flow cytometry analysis, and cell populations with a purity >95% were used for experimentation. Cells were cultured in supplemented RPMI 1640 with or without the following stimuli: with medium, 10 μg/ml MOG35–55, 5 μg/ml LPS, 3 μg/ml ConA, or 3 μg/ml anti-CD3 for 24–96 h. Cells were cultured in round-bottom 96-well plates (2 × 105 T cells/well) or in transwell 96-well plates (separating purified DCs from responder [2 × 105] splenocytes) for 24–96 h, with responder cell numbers remaining constant. Cell viability was assessed by 7-aminoactinomycin D staining, with subsequent evaluation by flow cytometry at 24, 48, 72, and 96 h of culture.
Proliferation assays
Cellular proliferation was evaluated by culture of cells for 48–72 h, with incorporation of [3H]thymidine for the last 18 h of culture. Cells were harvested and counted using Perkin Elmer Top Count NXT, with Packard's Top Count NXT software. Results are expressed as the total cpm (mean cpm of cultures with Ag/mean cpm of cultures with medium alone) ± SEM for all animals in the group.
Evaluation of cytokines: ELISA and ELISPOT
For cytokine protein determination, supernatants were collected from in vitro cultures of DCs 24 h after purification or from DC–T cell cocultures. OPT-EIA Sandwich ELISA kits (Pharmingen, San Diego, CA) were used to determine the levels of IL-12p40, IFN-γ, TNF-α, IL-2, IL-4, and IL-10, and colorimetric changes were read with a SpectraMax Plus384 spectrophotometer and analyzed with SoftMax Pro software (Molecular Devices, Sunnyvale, CA). The frequency of cytokine-secreting cells was determined for IL-12, IFN-γ, IL-2, IL-4, and TNF-α using ELISPOT development modules (R&D Systems, Minneapolis, MN), and samples were run in triplicate with medium, MOG35–55, LPS, ConA, or anti-CD3. Computer-assisted image analysis, using KS ELISPOT software and microscope control processor MCP4 (Carl Zeiss Vision GmbH, Thornwood, NY), were used to analyze developed plates. Data are expressed as the number of cytokine-producing cells/million ± SEM for all animals in a group.
Flow cytometry
Single-cell suspensions of spleen and lymph nodes cells were evaluated for CD4, CD25, and intracellular Foxp3 expression. For intracellular staining, cells were stained first for surface markers and then fixed and permeabilized prior to intracellular Foxp3 staining. DC populations from pelleted mice were evaluated for overall CD11c expression and gated on CD11c+ (from nonpurified splenocytes) or CD8α+ (from purified CD11c+ populations) to determine levels of CD80/B7-1, CD86/B7-2, PD-1, CD40 (BD Biosciences, San Jose, CA), PD-L1/B7-H1, PD-L2/B7-DC, B7-H3, and B7-H4 (eBiosciences, San Diego, CA). Three-color flow cytometry was performed on DC and T cell populations using FITC-, PE-, or allophycocyanin-conjugated mAbs, and the corresponding isotype controls (BD Biosciences, San Jose, CA) and cells were processed on a BD FACSCalibur flow cytometer and analyzed using Cell Quest analysis software (BD Biosciences, San Jose, CA).
RT-PCR and real-time RT-PCR
Purified CD11c+ DCs from mice implanted with E3 or Pb pellets were frozen at −80°C in TRIzol reagent and then used for RNA extraction. The following primers were used in RT-PCR: 5′-AGGTGCGTTCCTCGTAGAGA-3′ and 3′-AAAGCCACCAAGCAGAAGA-5′ for IL-12p40; 5′-ACCCACAAGGACTCAAGGACAACA-3′ and 3′-AGGCCAAGGGCTCGAGACTTTATT-5′ for IL-23p19; 5′-ACAGATGACATGGTGAAGACG-3′ and 3′-TCGTTCTTGTGTAGTTCCAGTG-5′ for IL-12p35; 5′-CCGGAGAGGAGACTTCACAG-3′ and 3′-CAAGAGACCCTTTAGCACCT-5′ for IL-6; 5′-CTCTTCAAGGGACAAGGCTG-3′ and 3′-GAATCTGAAACGCCTCAGGC-5′ for TNF-α; 5′-AGCCCGAAGCGGACTACTAT-3′ and 3′-TCCACATGTTGCTCCACACT for TGF-β; 5′-CTATGCAGTTGATGAAGATGTCAAA-3′ and 3′-ACCTGGTAGAAGTGATGCCCCAGGCA-5′ for IL-10; 5′-CATCCTGGAGGAAGTGGGCCGA-3′ and 3′-ACGGGCCCGGTACTCATTCTG-5′ for inducible NO synthase; 5′-AGCTCCGAGAAGAAGTCGAGAA-3′ and 3′-TGTAACCTGTGTCCCCTCAGTTC-5′ for IDO; 5′-TTGGGTGGATGCTCACACTGACA-3′ and 3′-CCCCAGGGTCTACGTCTCGCAA-5′ for Arg2 (arginase II); and 5′G-CCCATCCTCTTCCTCCCTGGAGAA-3′ and 3′-GGAGGGGCCGGACTCATCGTACTC-5′ for β-actin. Adjusted average Δcycle threshold (CT) values were calculated (20 – AvgΔCT) per cytokine. The RT-PCR data were normalized to the housekeeping gene β-actin. The fold difference number is (20 – AvgΔCT), where 20 is the expected maximum cycle threshold of the housekeeping gene. Individual p values were determined based on triplicates of individual experiments and are reported only when a consistent difference (trend or p < 0.05) was seen in at least three separate experiments.
Statistical analysis
A two-tailed Student t test was used to determine statistical differences when comparing two groups with parametric data, as in the ELISA, ELISPOT, adjusted average ΔCT values, and proliferation assays. A one-way ANOVA was used for the percentage-expression assays. χ2 Analysis was used for determining differences in disease incidence.
Results
In vivo E3 exposure increases DCs expressing activation and inhibitory cell surface markers
To determine whether pregnancy levels of estrogen could distinctly affect DCs, splenic DCs from mice implanted with E3 (pregnancy levels) or Pb pellets were phenotypically evaluated. E3 exposure increased the levels of activation markers CD80, CD86, and MHC class II in DCs compared with mice implanted with Pb pellets (Fig. 1A). CD40 levels did not differ significantly, although trends of increased CD40 levels in E3 DCs were seen in some experiments (data not shown). Mean fluorescence data suggested that although E3 increased the percentage of these markers, only CD80 mean fluorescence levels seemed to increase in DCs exposed to E3 (Fig. 1C). CD86, CD40, and MHC class II mean fluorescence levels did not increase, and MHC class II levels may have decreased slightly in E3-exposed DCs. These results suggested that E3 increases the overall level of activation in the splenic DC compartment but may not upregulate the expression of activation markers on individual DCs. We next evaluated whether E3 affected the expression of inhibitory costimulatory molecules. Similar to what was seen in activation markers, levels of the inhibitory markers PD-L1, PDL2, B7-H3, and B7-H4 were increased after E3 exposure (Fig. 1B), whereas mean fluorescence levels did not differ significantly for most markers, with the exception of slight increases in B7-H3 (Fig. 1D). E3 did not seem to influence ICOS ligand (ICOSL) or CD40, suggesting that it increases select stimulatory and inhibitory markers in DC populations, which may contribute to the tolerogenic ability of E3 Tol-DCs.
FIGURE 1.

E3 increases levels of activation and inhibitory markers in CD11c+ DCs. Flow cytometric analysis comparing splenocytes expanded under the influence of E3 (E3 DCs) or placebo (Pb DC) pellets. Splenocytes were gated on CD11c expression and the relative percentage expression of maturation/activation markers CD80, CD86, MHC class II, and CD40 (A) and inhibitory markers PD-L1, PD-L2, B7-H3, and B7-H4 (B). C and D, Graph overlays of mean fluorescence of activation and inhibitory markers from panels A and B, respectively, representing gated CD11c+ DCs from E3 (black)- and Pb (gray)-treated mice. Positive fractions are values >101 (based on isotype controls) for each marker of interest. Purified CD11c+ E3 or Pb DC populations from seven separate experiments were used to determine the statistical significance in the percentage of positive cells. E3 and Pb DCs were obtained from splenocytes of mice implanted with E3 or Pb pellets from seven separate experiments and ≥20 mice per E3 or Pb treatment. Bar graphs show mean percent positive ± SEM. *p < 0.05.
Given that specific CD8α+ and CD8α− DCs can directly impact Th1/Th2 responses, we next determined whether E3 differentially alters the levels of CD8α+ DCs and the expression of stimulatory or inhibitory markers in CD8α+ and CD8α− DC subpopulations (43). E3 consistently increased CD8α+ DCs ~10–15% over Pb treatment (Fig. 2A), suggesting that increased CD8α+ levels may contribute to the tolerogenic abilities of transferred E3 DCs. Similar to the overall CD11c+ population, mean fluorescence levels did not differ between E3 and Pb DCs for most markers. Only in the CD8α−CD11c+ subset were mild increases in CD80 and PD-L1 and decreases in ICOSL and MHC class II seen (Fig. 2C), suggesting that altered levels of these molecules in the CD8α+ DC subset may significantly contribute to the tolerogenic ability of E3 Tol-DCs.
FIGURE 2.

Relative percentage and phenotypic profile of CD8a+ and CD8a− DC subsets within CD11c+ DC population. A, Percentage of CD8α+ DCs of purified CD11c+ DC populations from mice implanted with E3 or Pb pellets. Graphs show mean (± SEM) CD8a levels from five separate experiments (≥10 mice per E3 or Pb treatment). *p < 0.05. B, Phenotypic-expression profiles of maturation/activation markers CD80, CD86, and CD40 and inhibitory markers PD-L1, PD-L2, B7-H3, and B7-H4) of CD8α+CD11c+ (B) and CD8α-CD11c+ (C) DCs of E3 (black)- or Pb (gray)-treated mice. Positive fractions are values >101 (based on isotype controls) for each marker of interest. E3 and Pb DCs were obtained from pooled splenocytes of mice implanted with E3 or Pb pellets.
Expression of proinflammatory mediator mRNA is decreased, whereas anti-inflammatory mediator mRNA is increased in E3 Tol-DCs
Expression of proinflammatory and immunoregulatory factor mRNA from E3 and Pb DCs was investigated using real time RT-PCR. E3 DCs demonstrated decreased expression of proinflammatory IL-12p40, IL-23p19, IL-6, and inducible NO synthase (NOS2) mRNA and increased expression of immunoregulatory TGF-β and IL-10 mRNA (Fig. 3). Decreased IL-12 mRNA levels are consistent with DC–T cell coculture results in Fig. 2B and, together with decreased IL-12p40, IL-23p19, and IL-6 mRNA levels, suggested that E3 may decrease the proinflammatory capabilities of DCs, which may affect the generation of Th1 and Th17 cells. Similarly, increased expression of immunoregulatory cytokines, such as TGF-β and IL-10 mRNA, may also contribute to the immunoregulatory role of E3 DC-mediated protection. No consistent differences were seen in the expression of TNF-α and immunoregulatory mediators arginase (Arg2) and IDO mRNA between E3 and Pb DCs, suggesting that these factors are not involved in E3 DC action (Fig. 3). Interestingly, slight increases in IL-12p35 expression were noted in E3 DCs (Fig. 3).
FIGURE 3.
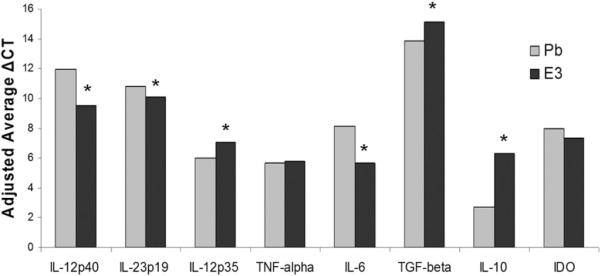
E3 DCs express decreased proinflammatory and increased immunoregulatory mRNA levels. E3 and Pb DCs were purified with CD11c magnetic beads and evaluated by real-time RT-PCR to determine the expression of proinflammatory and immunoregulatory mediators. Adjusted average ΔCT values are shown for proinflammatory IL-12p40, IL-23p19, IL-12p35, IL-6, TNF-α, and immunoregulatory TGF-β and IL-10. E3 and Pb DCs were obtained from pooled splenocytes of mice implanted with E3 or Pb pellets. *p < 0.05 of triplicates. Individual p values were determined based on triplicates of individual experiments and reported when a consistent difference (trend or p < 0.05) was seen in at least three separate experiments (at least six mice per E3 or Pb treatment).
E3 DCs have an increased IL-10/IL-12 ratio due to decreased IL-12 production and not through increased IL-10 production
Given the described roles of IL-12 and IL-10 in modulating T cell responses and influencing EAE, E3 and Pb DCs were cultured in vitro with or without LPS, and IL-12 and IL-10 protein expression was measured (44). E3 DCs produced less IL-12p40 after 96 h in culture in the media control group and after stimulation with LPS (Fig. 4A). To determine whether IL-12 production by E3 DCs could be enhanced or altered by Ag exposure, the neuroantigen MOG was added to DCs and cocultures of DCs with naive Ag-specific CD4+ T cells specific to MOG. Fig. 4B demonstrates that IL-12 production on a per-cell basis was lower in E3 DC cultures compared with Pb DCs in the media control group (98 ± 14 and 174 ± 3, respectively), as well as following MOG stimulation (124 ± 4 and 192 ± 11, respectively). These results demonstrated that the decreased IL-12 production by E3 DCs remains, even when DCs are stimulated directly with LPS or indirectly by Ag-stimulated T cells. Given the importance of IL-10 in regulating immune function in select Tol-DCs, we evaluated IL-10 production following in vitro stimulation of E3 and Pb DCs. IL-10 production did not differ between E3 and Pb DCs in the media control or following LPS stimulation (Fig. 3C), but the ratio of IL-10/IL-12 was increased for E3 DCs in the media-treated mice (ratio of 1) and LPS-stimulated mice (ratio of 1.59) compared with Pb DCs (ratios of 0.5 and 0.53 for media and LPS, respectively). This increased IL-10/IL-12 ratio occurred as a result of decreased IL-12 rather than because of elevated IL-10.
FIGURE 4.

E3 DCs produce less IL-12 and similar IL-10 compared with Pb DCs. A, Purified CD11c+ E3 or Pb DCs were cocultured with purified CD4+ naive MOG-specific T cells for 96 h, and IL-12p40 and IL-10 protein were evaluated in culture supernatant by ELISA. B, Cell-specific IL-12 production (ELISPOT) was lower in E3 DC cultures compared with Pb DC cultures in the media control group and following MOG stimulation. C, IL-10 cytokine production between E3 and Pb DCs show decreased IL-12 production in E3 DC groups. Data are representative of at least three separate experiments. Bar graphs show mean ± SEM and are representative of three separate experiments (at least six mice per E3 or Pb treatment). *p < 0.05.
T cells proliferate less when cocultured with E3 Tol-DCs
To assess whether the described E3 Tol-DC phenotype had functional consequences on T cell differentiation, E3 DCs and Pb DCs were cultured with purified naive CD4+ MOG-specific T (Th0) cells from MOG TCR Tg mice. MOG Th0 cells cultured with E3 DCs proliferated less than did those cultured with Pb DCs in the presence of media, the neuroantigen MOG, and APC-stimulating LPS, but they did not differ when T cells were specifically stimulated with anti-CD3 (Fig. 5A). These results suggested that E3 DCs have a baseline ability to inhibit T cell proliferation (i.e., media group) that is resistant to DC-activating stimuli (i.e., LPS group). Importantly, E3 DCs are also able to inhibit T cell proliferation in response to Ag (i.e., MOG group) but not in response to anti-CD3 (Fig. 5A). Additionally, soluble factors produced by DCs separated from splenocytes in a transwell system (Fig. 5B) and splenocytes cultured with E3 Tol-DC–conditioned media (data not shown) suggested that cell–cell interaction is necessary for the full spectrum of regulatory effects of E3 DCs on T cells.
FIGURE 5.
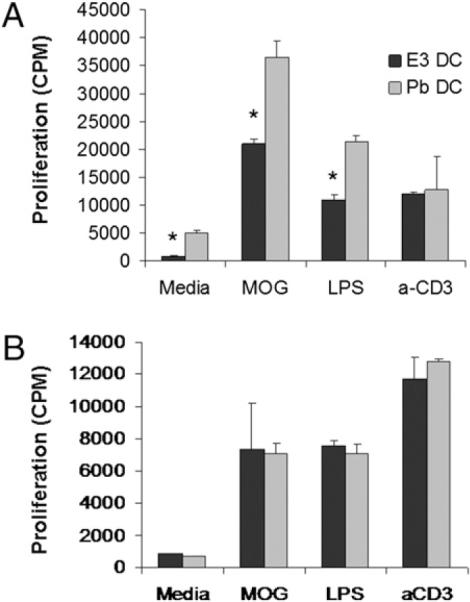
T cells proliferate less in the presence of E3 DCs, and cell–cell contact is required for this effect. Purified CD11c+ E3 DCs or Pb DCs were cultured directly with CD4+ TCR transgenic T cells specific for MOG (A) or separated from MOG Tg T cells by transwell culture inserts for 96 h in the presence of MOG, anti-CD3, and LPS (B). Cellular proliferation was determined by the amount of incorporated [3H]thymidine and read as cpm. Data are representative of five or three separate experiments, respectively. Bar graphs show mean ± SEM. *p < 0.05 for standard proliferation assays from at least three separate experiments (12 mice per E3 or Pb treatment) and two separate experiments (5 mice per E3 or Pb treatment) for transwell experiments.
E3 Tol-DC recipient mice develop less severe EAE
To assess the biological relevance of the E3 DC regulatory capabilities, we evaluated the protective ability of E3 DCs in the inflammatory autoimmune disease EAE. Regardless of the mechanism, simple hypoproduction of proinflammatory mediators (e.g., IL-12) by E3 Tol-DCs is likely insufficient to explain the prevention of EAE. Specifically, mice receiving E3 Tol-DCs have a full complement of resident DCs presumably capable of generating IL-12 and IL-23 necessary to promote Th1 and/or Th17 responses responsible for EAE clinical disease. Mice receiving E3 DCs developed less severe disease than did Pb DC recipients, had a decreased cumulative disease score (CDS) and decreased clinical severity, and failed to relapse or develop significant chronic EAE compared with Pb DC recipients (Fig. 6, Table I). The protective effect of E3 DCs was not influenced by exposure to LPS in vitro or to CFA and pertussis toxin adjuvants in vivo (Fig. 7, Table II). These results suggested that E3 DCs are resistant to in vitro and in vivo inflammatory signals and that the regulatory abilities of these E3 DCs are maintained in vivo.
FIGURE 6.
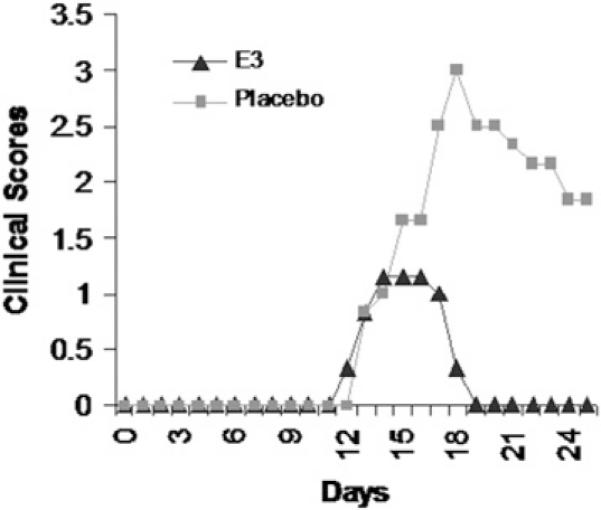
E3 DC recipients have reduced autoimmunity. Clinical scores of mice actively immunized 1 d after receiving 8–10 million E3 or Pb DCs (i.v.) pulsed in vitro with MOG35–55. Data are representative of seven separate experiments of at least five mice per group.
Table I.
Incidence, day of disease onset, peak day of clinical disease, and CDS of E3 and Pb DC recipients
| DC Treatment | Incidence (%) | Onset (d) | Peak (d) | CDS |
|---|---|---|---|---|
| E3 DC | 66 | 13.0 ± 9.2 | 1.2 ± 0.7* | 1.3 ± 0.8* |
| Pb DC | 100 | 14.3 ± 8.3 | 3.2 ± 1.8 | 26.3 ± 15.2 |
DC recipients have reduced autoimmunity; they had decreased peak severity, clinical severity, and resolution of clinical signs. Data are representative of seven separate experiments of at least five mice per group.
p < 0.05.
FIGURE 7.
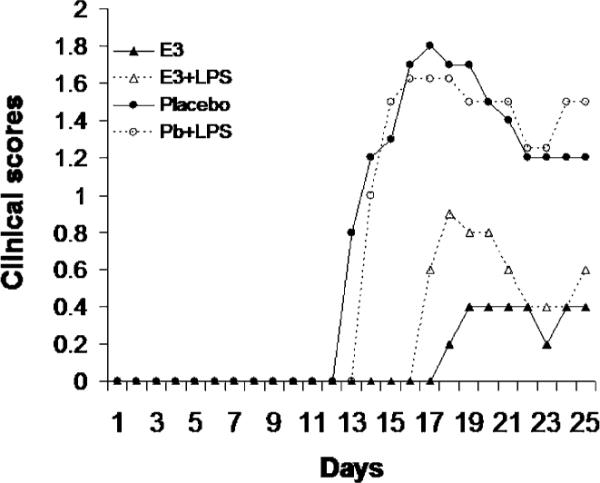
E3 DC regulatory function is not affected by inflammatory challenge, and protection from autoimmunity is maintained. Actively immunized mice received 1–2 million purified E3 or Pb DCs exposed to MOG35–55, with or without LPS, in vitro 24 h prior to EAE induction. Delayed onset and protection from clinical EAE was evident in E3 (± LPS) DC recipients. Data are representative of two independent experiments with five mice per group.
Table II.
Peak day of clinical disease and CDS of mice receiving E3 DCs and Pb DCs with or without LPS exposure
| DC Treatment | CDS | Peak (d) |
|---|---|---|
| E3 | 5.2 ± 5.2* | 0.4 ± 0.4* |
| E3 + LPS | 7.0 ± 4.3** | 0.9 ± 0.6 |
| Pb | 23.6 ± 7.7 | 1.9 ± 0.6 |
| Pb + LPS | 21.9 ± 6.4 | 1.8 ± 0.5 |
Protection from autoimmunity was maintained, even with inflammatory challenge. Recipients had decreased peak severity, clinical severity, and resolution of clinical signs. Data are representative of three separate experiments, with a total of 10 mice per group. All groups were compared with Pb DC recipients.
p < 0.01
p < 0.05.
E3 DC recipients promote immune deviation without generating Tregs
Active induction of EAE generates Th1 and/or Th17 cells that drive development of disease, whereas Th2 and Treg responses are protective. To assess whether E3 DCs protected mice through altering the Th1/Th2/Th17/Treg balance, peripheral immune organs were evaluated at day 10 postimmunization. Lymph node cells from immunized mice showed decreased Ag-specific proliferation to the immunizing neuroantigen MOG in E3 DC recipients (Fig. 8A). Proliferation did not differ when lymph node cells were stimulated with anti-CD3 or LPS (Fig. 8A). Because Tregs are potent inhibitors of T cell proliferation, the ability of E3 DCs to expand Tregs was evaluated. Surprisingly, there were no differences in the percentage of CD4+CD25+ T cells (Fig. 8B) or CD4+CD25+Foxp3+ cells within the lymph nodes (Fig. 8C) or spleen (data not shown) between E3 Tol-DC and Pb DC recipients. Because Tregs did not seem to be involved in the E3 DC-mediated protection against EAE, cytokine profiles were evaluated in E3 and Pb DC recipients. Lymph node cells from immunized mice receiving E3 DCs prior to immunization produced less IFN-γ and increased IL-4 following MOG stimulation compared with Pb DC recipients (Fig. 9A). Although IL-17 levels were slightly higher in the Pb DC recipients, the levels did not reach statistical significance (Fig. 9B). Taken together, these data suggested that E3 Tol-DCs alone were able to protect mice from developing EAE and do so through Ag-specific immune deviation to a Th2 response. Additionally, this protection is resistant to inflammatory challenge in vitro (i.e., LPS) or in vivo (i.e., Th1/Th17-promoting adjuvants).
FIGURE 8.
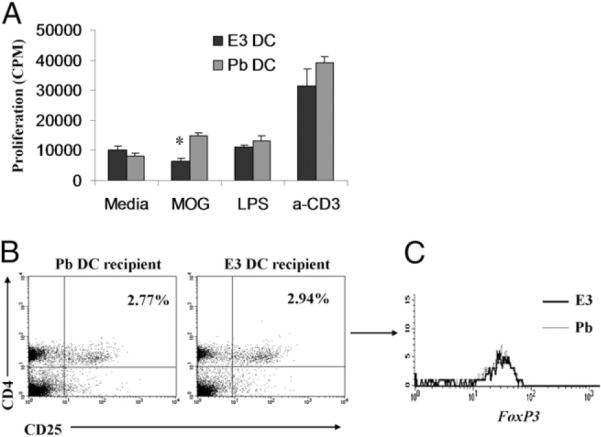
E3 DC recipients proliferate less to immunizing Ag, and Tregs do not mediate the protection. The proliferative ability of lymph node cells in E3 DC recipient mice was evaluated. A, Lymph node cells from E3 DC recipient mice proliferated less to MOG than did Pb DC recipients. The decreased proliferative ability was not through increased numbers of Tregs. E3 DC recipients are not protected by increased numbers of CD4+CD25+Foxp3+ Tregs. Lymph node cells were evaluated by flow cytometry for the presence of phenotypic markers CD4 and CD25 and the intracellular transcription factor foxp3 at day 10 postimmunization. No difference was seen in the number of CD4+CD25+ T cells within the lymph nodes at day 10 post-immunization (B) or in the intracellular Foxp3 expression in the CD4+CD25+ T cell population (C). Data are representative of three independent experiments with five mice per group. Bar graphs show mean ± SEM. *p < 0.05.
FIGURE 9.
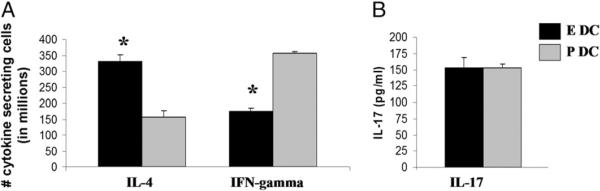
E3 DC recipients are protected from EAE by immune deviation to a Th2 response. Lymph node cells obtained 10 d after EAE induction were evaluated for production of IL-4, IFN-γ, and IL-17. A, Cell-specific cytokine secretion of lymph node cells by ELISPOT demonstrated increased IL-4 and decreased IFN-γ production in E3 DC recipients. B, Overall IL-17 production from culture supernatants detected by ELISA showed no differences between E3 and Pb DC recipients. Data are representative of three independent experiments with five mice per group. Bar graphs show mean ± SEM. *p < 0.05.
Discussion
Chronic inflammation is recognized as a significant contributor to a wide array of human diseases, including autoimmunity, cardiovascular disease, asthma, allergies, and cancer. The ability to regulate such inflammation is crucial for control and resolution of autoimmune and chronic inflammatory diseases. Although Tregs are known to have potent regulatory abilities, increasing evidence demonstrates that myeloid cells can dramatically regulate innate and adaptive immune responses and can be used therapeutically to regulate inflammation. Tol-DCs are one such regulatory myeloid cell population, and there is much interest in immunotherapy with these cells (20, 21). A primary difficulty in Tol-DC immunotherapy is the inadequate understanding of these cells and the factors that program them to become tolerogenic (32). Given the importance of myeloid cells in estrogen-mediated protection against autoimmunity, we asked whether E3 could generate Tol-DCs capable of protecting against the inflammatory autoimmune disease EAE. We found that E3 exposure altered the subset composition, levels of activation, and inhibitory markers in DCs and generated functional Tol-DCs. Importantly, transferred E3 Tol-DCs alone could protect mice from EAE, and the protective abilities of E3 Tol-DCs seem to be resistant to in vitro and in vivo inflammatory challenge. Other estrogens, such as E2, were shown to mediate protection through actions on myeloid cells, such as DCs, but safety concerns limit the therapeutic application of E2 (3, 6, 9, 12, 13, 45). This study demonstrated that E3, an estrogen with efficacy in MS patients and an increased safety profile compared with E2, programmed DCs to become tolerogenic and that E3 Tol-DCs protected mice against inflammatory autoimmune disease (3, 6, 9, 11, 12).
Tol-DCs are diverse and promote Th2 and other regulatory responses through a variety of mechanisms, including actions through specific DC subsets, soluble factors, and cell–cell contact. Our results showed that E3 increased the levels of activation and inhibitory molecules in the overall splenic DC compartment and specifically increased the percentage of CD8α+ DCs (Figs. 1, 2). E3 did not affect the levels of plasmacytoid DCs, which were consistently 3–5% of our purified population (data not shown). Numerous studies suggested that DC subsets impact tolerogenic versus regulatory immune responses, with CD8α+ potently producing IL-12 and promoting Th1 responses (46–50). Thus, it was surprising that E3 increased the CD8α+ subset, yet decreased IL-12 production was seen. Although increased CD8α+ levels may result from Flt3L's described propensity to increase CD8α+ subsets, the reason for overall decreased IL-12 production, even in the face of increased CD8α+ DCs, is not known. It is possible that E3 decreases the functional ability of CD8α+ DCs to produce IL-12 or promote Th2 responses, allowing the effects of CD8α− DCs to be more pronounced in the E3 DCs. Indeed, it was reported that CD8α− DCs are more tolerogenic (43, 46). However, Pb DCs have increased CD8α− levels, so increased CD8α− DC levels alone do not promote protective responses. Altered expression of surface markers in CD8α− DCs (e.g., increased CD80 and PD-L2) or alterations in IL-10 and IL-12 expression in either subset may contribute to E3 Tol-DC action. Given IL-12's important role in determining the Th1/Th2 balance in vivo, decreased IL-12 production by DCs, particularly CD8α+ DCs, may contribute to the Th2 response seen in E3 Tol-DC recipients (44, 47). The relative contribution of CD8α+ and CD8α− subsets and their cytokine profiles to mediated E3 DC protection are not known but are the subject of ongoing studies.
It is well known that IL-12 and IL-23 potently drive pathogenic T cell differentiation in EAE (i.e., Th1 and Th17, respectively), whereas IL-10 and TGF-β can dramatically regulate inflammation and EAE (51). Although E3 DCs produce less IL-12 protein, our results also showed decreased mRNA levels of proinflammatory IL-12, IL-23, IL-6, and NO, suggesting that E3 decreases the expression of numerous proinflammatory mediators. Impaired NF-κB signaling may be responsible for decreased proinflammatory mediators in E3-exposed DCs. E2 is known to influence genomic and nongenomic actions of NF-κB and other signaling pathways, and studies showed impaired or altered NF-κB signaling in E2-exposed CNS macrophages or splenic CD11c+ cells (52–54). Although E3 was shown to inhibit NF-κB in T cells, its actions on DCs are not known and were beyond the scope of this study; however, it is likely that E3 alters NF-κB, similar to E2 (55). Such alterations may contribute to the hypoproduction of proinflammatory mediators, such as IL-12 seen in our studies and reported by other investigators in human monocyte-derived and mouse splenic DCs following estrogen exposure (18, 41, 55). Alternatively, TGF-β is known to reduce the stability of IL-12p40 mRNA and may contribute to the resultant decrease in IL-12 protein (56, 57). Regardless of the mechanism, simple hypo-production of proinflammatory mediators (e.g., IL-12) by E3 Tol-DCs is likely insufficient to explain the prevention of EAE. Specifically, mice receiving E3 Tol-DCs have a full complement of resident DCs presumably capable of generating IL-12 and IL-23 necessary to promote Th1 and/or Th17 responses responsible for EAE clinical disease. Thus, active immunoregulatory mechanisms likely contribute to E3 Tol-DC–mediated protection.
TGF-β and IL-10 are potent immunoregulatory factors used by Tol-DC populations. IL-10, in particular, was shown to be used by other Tol-DCs (20, 58) and is upregulated in DCs exposed to E2 and in splenic immune cells exposed to E3 (18, 23, 41, 59). Our data showed that E3 Tol-DCs had increased expression of IL-10 and TGF-β mRNA (Fig. 3), suggesting that both of these immunoregulatory factors may contribute to E3 Tol-DC actions. Surprisingly, IL-10 protein levels increased after in vitro culture, but this is most likely attributed to suboptimal IL-10 production with LPS stimulation. Interestingly, a recent study suggested that CD40 signaling is important in inducing IL-10 Tol-DCs, so the absence of alterations in CD40 may explain why IL-10 protein is not increased in E3 Tol-DCs (60). Alternatively, similar overall IL-10 levels in E3 and Pb DCs suggested that CD8α− DCs in the E3 Tol-DCs may produce proportionately more IL-10 than similar CD8α− DCs in the Pb DC group, given that a lower percentage of E3 DCs are CD8α− than are Pb DCs. It is also possible that distinct increases in IL-10 production are not essential for regulation by E3 Tol-DCs but that an increased IL-10/IL-12 ratio is more important in determining the resultant immune response (47, 61).
Other immunoregulatory cytokines, such as TGF-β, may contribute to E3 Tol-DCs' regulatory action. TGF-β is a complex cytokine that influences the Treg/Th17 balance, is an important contributor to oral tolerance and the generation of Tregs and Th3 cells, and is known to control autoimmunity through actions on DCs (62–64). The increased mRNA levels of TGF-β seen in E3 Tol-DCs suggest that TGF-β contributes to regulatory actions of these cells. Given the complexity of TGF-β regulation (i.e., post-transcriptional and post-translational modifications and latent forms), assessment of biological activity by ELISA is reportedly imprecise and were beyond the scope of this study, but our data are intriguing, and further studies are needed to evaluate the contribution of TGF-β in E3 Tol-DC regulatory actions. Although other soluble mediators, such as IDO and arginase, were reported to play important roles for other Tol-DCs or regulatory myeloid cells, we found that they did not seem to be acting in E3 Tol-DCs based on similar mRNA levels seen between E3 or Pb DCs (33, 34, 65, 66). Rather, Fig. 5B suggests that cell–cell contact mechanisms are a more important mechanism by which E3 Tol-DCs regulate T cells.
Activation and inhibitory markers are important cell contact-mediated mechanisms by which DCs regulate adaptive immune responses. Decreased expression of activation markers, such as CD80, CD86, and MHC class II, are classically associated with dexamethasone or vitamin D Tol-DCs and mediate tolerance through their arrest in an immature state. Our data suggested that E3 acts in a distinct manner to generate Tol-DCs and that E3 Tol-DCs are not immature, based on increased expression of CD80, CD86, and MHC class II. In fact, E3 actually seems to increase the overall activation state of the splenic DC compartment, as evidenced by increased levels in CD80, CD86, and MHC class II expression in E3 versus Pb DCs. Although levels of activation markers are increased, so too are numerous inhibitory markers, such as PD-L1, PD-L2, B7-H3, and B7-H4. The inhibitory abilities of PD-L1, PD-L2, B7-H3, B7-H4, and ICOSL are well known, and the interactions of these molecules with their ligands on T cells may directly contribute to E3 Tol-DC regulatory actions (67–72). Data from transwell studies (Fig. 5) and E3 DC-conditioned supernatant (data not shown) strongly suggest that cell–cell interactions in E3 Tol-DCs directly contribute to suppression of proliferation and may be a primary mechanism by which E3 Tol-DCs protect against development of EAE. Studies showing enhanced PD-1 expression on APCs after E2 exposure suggest that the interactions between PD-1 and its ligands are one potential means by which estrogens influence DC function (16). Although these studies did not evaluate PD-L1 or PD-L2 on E2-exposed APCs, we found upregulation of these ligands on E3 Tol-DCs, highlighting the potential role of PD-1/PD-L interactions in E3 Tol-DC regulation. Less is known about the B7-H3 and B7-H4 mechanistic actions, but B7-H3 and B7-H4 can dramatically influence EAE through a variety of potential mechanisms, including inhibition of anti-CD3–induced T cell proliferation, altering cytokine production, influencing cell cycle arrest, and activating NFAT, NF-κB, and AP-1 (67, 69–77). The fact that PD-L2 and B7-H3 expression on E3 DCs or DC subsets suggest that these particular inhibitory molecules (potentially in combination with increased CD80) may play a more prominent role than others. Thus, E3 Tol-DCs may be actively regulating immune responses through alternative activation, expression of multiple inhibitory molecules, or a combination of these. At present, the relative balance of costimulatory molecules is thought to dramatically influence the resultant immune responses, but a thorough understanding of the complex interplay and hierarchical signaling of stimulatory versus inhibitory molecules is lacking (78). Interestingly, the effects of E3 do not universally affect all costimulatory molecules because CD40 and ICOSL levels did not differ between E3 and Pb DCs, suggesting a specific effect of E3 on Tol-DC phenotype.
Our findings show that exposure of DCs to E3 programs DCs to become Tol-DCs and that E3 Tol-DCs protect through immune deviation away from pathogenic Th1/Th17 cells to a protective Th2 response. These findings mirror the Th2 response (i.e., increased IL-4 and IL-5 and decreased IFN-γ) reported in MS patients treated with E3 (10, 11). Although the mechanisms by which E3 Tol-DCs promote Th2 responses are unknown, it is surprising that neither IL-17 nor Tregs were altered, particularly given the differences seen in IL-23, IL-6, and TGF-β levels between E3 and Pb DCs. These cytokines play important roles in regulating the generation and balance of Th17 and Tregs, and more mechanistic studies are required to understand the relative contributions of these cytokines and other factors (e.g., retinoic acid) in affecting the generation of Th17 and CD4+CD25+Foxp3+ Tregs following E3 Tol-DC exposure (16, 79–81). The absence of CD4+CD25+Foxp3+ Tregs in our study was particularly surprising given that other investigators found increased Tregs upon E2 exposure (3). It is possible that, although Treg numbers do not differ, Treg functionality may differ between E3 and Pb DC recipients or that Tregs were generated at times not evaluated in this study. Alternatively, E3 may preferentially enhance other Foxp3− regulatory cells that mediate protection, such as CD45dimVLA-4+ regulatory cells (82, 83). Whether our results represent distinct actions of E3 versus E2 or whether Tregs or other regulatory cells were simply not detected at the time points evaluated remains to be determined. Importantly, differences in estrogen, as well as model systems, may dramatically impact results. Specifically, our study evaluated in vivo effects of E3 on Flt3L DCs, whereas the majority of other studies evaluated E2 effects (typically in vitro) on GM-CSF DCs (16, 18, 84). It is known that GM-CSF generates inflammatory DCs while Flt3L generates steady-state (homeostatic) DCs (85). The additional impact of estrogen on both DC populations and regulatory responses (e.g., Th2, Tregs, and other regulatory cells) may likely differ depending on the relative inflammatory or resting state (40, 86). Estrogens (E2 and E3) have known effects on bone marrow DC differentiation, and our data showed that the composition of DC subsets can be altered by estrogen exposure. Thus, the very nature of DC populations may differ following estrogen exposure, which would further contribute to DC-mediated regulation of inflammation (40, 87).
In summary, we showed that the protective effects of E3 can be mediated through DCs alone and that E3 generates Tol-DCs that protect mice from developing EAE through immune deviation to a Th2 response. E3 Tol-DCs are resistant to in vitro and in vivo inflammatory stimuli, and cell–cell contact seems to be important in E3 Tol-DC actions. The precise mechanisms by which E3 alters DC actions are not known, but they may be similar to some E2-described actions, such as NF-κB inhibition, decreased production of inflammatory cytokines, inhibition through cell–cell interactions, and altered cellular migration, viability, and/or turnover (55, 56, 88, 89). E3 programs DCs to become tolerogenic, and E3 Tol-DCs protect against autoimmunity, even in the face of inflammatory challenge. The ability to program DCs to induce Th2 or tolerogenic responses has enormous therapeutic applications, and targeted generation of stable Tol-DCs to regulate inflammation is a promising therapy for the treatment of autoimmune and numerous chronic inflammatory diseases.
Acknowledgments
We thank Dr. Prosper Boyaka for helpful discussions during preparation of this manuscript and Amgen, Inc. for providing hFL.
Abbreviations used in this article
- CDS
cumulative disease score
- CT
cycle threshold
- DC
dendritic cell
- E2
17-β estradiol
- E3
estriol
- EAE
experimental autoimmune encephalomyelitis
- Flt3L
Flt3 ligand
- hFL
human recombinant Flt3L
- ICOSL
ICOS ligand
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- Pb
placebo
- Tg
transgenic
- Tol-DC
tolerogenic DC
- Treg
regulatory T cell.
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Whitacre CC. Sex differences in autoimmune disease. Nat. Immunol. 2001;2:777–780. doi: 10.1038/ni0901-777. [DOI] [PubMed] [Google Scholar]
- 2.Whitacre CC, Reingold SC, O'Looney PA. A gender gap in autoimmunity. Science. 1999;283:1277–1278. doi: 10.1126/science.283.5406.1277. [DOI] [PubMed] [Google Scholar]
- 3.Offner H, Polanczyk M. A potential role for estrogen in experimental autoimmune encephalomyelitis and multiple sclerosis. Ann. N. Y. Acad. Sci. 2006;1089:343–372. doi: 10.1196/annals.1386.021. [DOI] [PubMed] [Google Scholar]
- 4.McClain MA, Gatson NN, Powell ND, Papenfuss TL, Gienapp IE, Song F, Shawler TM, Kithcart A, Whitacre CC. Pregnancy suppresses experimental autoimmune encephalomyelitis through immunoregulatory cytokine production. J. Immunol. 2007;179:8146–8152. doi: 10.4049/jimmunol.179.12.8146. [DOI] [PubMed] [Google Scholar]
- 5.Voskuhl RR, Palaszynski K. Sex hormones in experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Neuroscientist. 2001;7:258–270. doi: 10.1177/107385840100700310. [DOI] [PubMed] [Google Scholar]
- 6.Head KA. Estriol: safety and efficacy. Altern. Med. Rev. 1998;3:101–113. [PubMed] [Google Scholar]
- 7.Taylor M. “Bioidentical” estrogens: hope or hype? Sexuality, Reproduction & Menopause. 2005;3:68–71. [Google Scholar]
- 8.Palaszynski KM, Liu H, Loo KK, Voskuhl RR. Estriol treatment ameliorates disease in males with experimental autoimmune encephalomyelitis: implications for multiple sclerosis. J. Neuroimmunol. 2004;149:84–89. doi: 10.1016/j.jneuroim.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 9.Kim S, Liva SM, Dalal MA, Verity MA, Voskuhl RR. Estriol ameliorates autoimmune demyelinating disease: implications for multiple sclerosis. Neurology. 1999;52:1230–1238. doi: 10.1212/wnl.52.6.1230. [DOI] [PubMed] [Google Scholar]
- 10.Sicotte NL, Liva SM, Klutch R, Pfeiffer P, Bouvier S, Odesa S, Wu TC, Voskuhl RR. Treatment of multiple sclerosis with the pregnancy hormone estriol. Ann. Neurol. 2002;52:421–428. doi: 10.1002/ana.10301. [DOI] [PubMed] [Google Scholar]
- 11.Soldan SS, Alvarez Retuerto AI, Sicotte NL, Voskuhl RR. Immune modulation in multiple sclerosis patients treated with the pregnancy hormone estriol. J. Immunol. 2003;171:6267–6274. doi: 10.4049/jimmunol.171.11.6267. [DOI] [PubMed] [Google Scholar]
- 12.Subramanian S, Matejuk A, Zamora A, Vandenbark AA, Offner H. Oral feeding with ethinyl estradiol suppresses and treats experimental autoimmune encephalomyelitis in SJL mice and inhibits the recruitment of inflammatory cells into the central nervous system. J. Immunol. 2003;170:1548–1555. doi: 10.4049/jimmunol.170.3.1548. [DOI] [PubMed] [Google Scholar]
- 13.Gold SM, Voskuhl RR. Estrogen treatment in multiple sclerosis. J. Neurol. Sci. 2009;286:99–103. doi: 10.1016/j.jns.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falcone M, Bloom BR. A T helper cell 2 (Th2) immune response against non-self antigens modifies the cytokine profile of autoimmune T cells and protects against experimental allergic encephalomyelitis. J. Exp. Med. 1997;185:901–907. doi: 10.1084/jem.185.5.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shevach EM. Regulatory T cells in autoimmmunity*. Annu. Rev. Immunol. 2000;18:423–449. doi: 10.1146/annurev.immunol.18.1.423. [DOI] [PubMed] [Google Scholar]
- 16.Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J. Neurosci. Res. 2006;84:370–378. doi: 10.1002/jnr.20881. [DOI] [PubMed] [Google Scholar]
- 17.Polanczyk MJ, Hopke C, Huan J, Vandenbark AA, Offner H. Enhanced FoxP3 expression and Treg cell function in pregnant and estrogen-treated mice. J. Neuroimmunol. 2005;170:85–92. doi: 10.1016/j.jneuroim.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 18.Liu HY, Buenafe AC, Matejuk A, Ito A, Zamora A, Dwyer J, Vandenbark AA, Offner H. Estrogen inhibition of EAE involves effects on dendritic cell function. J. Neurosci. Res. 2002;70:238–248. doi: 10.1002/jnr.10409. [DOI] [PubMed] [Google Scholar]
- 19.Polanczyk MJ, Jones RE, Subramanian S, Afentoulis M, Rich C, Zakroczymski M, Cooke P, Vandenbark AA, Offner H. T lymphocytes do not directly mediate the protective effect of estrogen on experimental autoimmune encephalomyelitis. Am. J. Pathol. 2004;165:2069–2077. doi: 10.1016/S0002-9440(10)63257-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 21.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 22.Griffiths KL, O'Neill HC. Dendritic cells as immune regulators: the mouse model. J. Cell. Mol. Med. 2008;12:1909–1914. doi: 10.1111/j.1582-4934.2008.00378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006;108:1435–1440. doi: 10.1182/blood-2006-03-006403. [DOI] [PubMed] [Google Scholar]
- 24.Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nat. Med. 2004;10:475–480. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 25.Szatmari I, Nagy L. Nuclear receptor signalling in dendritic cells connects lipids, the genome and immune function. EMBO J. 2008;27:2353–2362. doi: 10.1038/emboj.2008.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moser M, De Smedt T, Sornasse T, Tielemans F, Chentoufi AA, Muraille E, Van Mechelen M, Urbain J, Leo O. Glucocorticoids down-regulate dendritic cell function in vitro and in vivo. Eur. J. Immunol. 1995;25:2818–2824. doi: 10.1002/eji.1830251016. [DOI] [PubMed] [Google Scholar]
- 27.Adorini L, Penna G. Dendritic cell tolerogenicity: a key mechanism in immunomodulation by vitamin D receptor agonists. Hum. Immunol. 2009;70:345–352. doi: 10.1016/j.humimm.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Penna G, Adorini L. 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J. Immunol. 2000;164:2405–2411. doi: 10.4049/jimmunol.164.5.2405. [DOI] [PubMed] [Google Scholar]
- 29.Piemonti L, Monti P, Allavena P, Sironi M, Soldini L, Leone BE, Socci C, Di Carlo V. Glucocorticoids affect human dendritic cell differentiation and maturation. J. Immunol. 1999;162:6473–6481. [PubMed] [Google Scholar]
- 30.Kaliński P, Schuitemaker JH, Hilkens CM, Kapsenberg ML. Prostaglandin E2 induces the final maturation of IL-12-deficient CD1a+CD83+ dendritic cells: the levels of IL-12 are determined during the final dendritic cell maturation and are resistant to further modulation. J. Immunol. 1998;161:2804–2809. [PubMed] [Google Scholar]
- 31.Wada Y, Hisamatsu T, Kamada N, Okamoto S, Hibi T. Retinoic acid contributes to the induction of IL-12-hypoproducing dendritic cells. Inflamm. Bowel Dis. 2009;15:1548–1556. doi: 10.1002/ibd.20934. [DOI] [PubMed] [Google Scholar]
- 32.Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat. Immunol. 2010;11:647–655. doi: 10.1038/ni.1894. [DOI] [PubMed] [Google Scholar]
- 33.Xiao BG, Liu X, Link H. Antigen-specific T cell functions are suppressed over the estrogen-dendritic cell-indoleamine 2,3-dioxygenase axis. Steroids. 2004;69:653–659. doi: 10.1016/j.steroids.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 34.Liu Q, Zhang C, Sun A, Zheng Y, Wang L, Cao X. Tumor-educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J. Immunol. 2009;182:6207–6216. doi: 10.4049/jimmunol.0803926. [DOI] [PubMed] [Google Scholar]
- 35.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 36.Drew PD, Chavis JA, Bhatt R. Sex steroid regulation of microglial cell activation: relevance to multiple sclerosis. Ann. N. Y. Acad. Sci. 2003;1007:329–334. doi: 10.1196/annals.1286.031. [DOI] [PubMed] [Google Scholar]
- 37.Papenfuss TL, Whitacre CC. Sex hormones, pregnancy, and immune function. In: Arnold AP, Etgen AM, Fahrbahc SE, Rubin RT, Pfaff DW, editors. Hormones Brain and Behavior. Elsevier; Oxford, U.K.: 2009. pp. 367–393. [Google Scholar]
- 38.Polazzi E, Monti B. Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog. Neurobiol. 2010;92:293–315. doi: 10.1016/j.pneurobio.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 39.Kyurkchiev D, Ivanova-Todorova E, Hayrabedyan S, Altankova I, Kyurkchiev S. Female sex steroid hormones modify some regulatory properties of monocyte-derived dendritic cells. Am. J. Reprod. Immunol. 2007;58:425–433. doi: 10.1111/j.1600-0897.2007.00526.x. [DOI] [PubMed] [Google Scholar]
- 40.Carreras E, Turner S, Paharkova-Vatchkova V, Mao A, Dascher C, Kovats S. Estradiol acts directly on bone marrow myeloid progenitors to differentially regulate GM-CSF or Flt3 ligand-mediated dendritic cell differentiation. J. Immunol. 2008;180:727–738. doi: 10.4049/jimmunol.180.2.727. [DOI] [PubMed] [Google Scholar]
- 41.Uemura Y, Liu TY, Narita Y, Suzuki M, Matsushita S. 17 Beta-estradiol (E2) plus tumor necrosis factor-alpha induces a distorted maturation of human monocyte-derived dendritic cells and promotes their capacity to initiate T-helper 2 responses. Hum. Immunol. 2008;69:149–157. doi: 10.1016/j.humimm.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 42.Bebo BF, Jr., Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J. Immunol. 2001;166:2080–2089. doi: 10.4049/jimmunol.166.3.2080. [DOI] [PubMed] [Google Scholar]
- 43.Coquerelle C, Moser M. DC subsets in positive and negative regulation of immunity. Immunol. Rev. 2010;234:317–334. doi: 10.1111/j.0105-2896.2009.00887.x. [DOI] [PubMed] [Google Scholar]
- 44.Maldonado-López R, Maliszewski C, Urbain J, Moser M. Cytokines regulate the capacity of CD8alpha(+) and CD8alpha(−) dendritic cells to prime Th1/Th2 cells in vivo. J. Immunol. 2001;167:4345–4350. doi: 10.4049/jimmunol.167.8.4345. [DOI] [PubMed] [Google Scholar]
- 45.Gold SM, Voskuhl RR. Estrogen and testosterone therapies in multiple sclerosis. Prog. Brain Res. 2009;175:239–251. doi: 10.1016/S0079-6123(09)17516-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhattacharya P, Gopisetty A, Ganesh BB, Sheng JR, Prabhakar BS. GM-CSF-induced, bone-marrow-derived dendritic cells can expand natural Tregs and induce adaptive Tregs by different mechanisms. J. Leukoc. Biol. 2011 doi: 10.1189/jlb.0310154. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maldonado-López R, De Smedt T, Michel P, Godfroid J, Pajak B, Heirman C, Thielemans K, Leo O, Urbain J, Moser M. CD8alpha+ and CD8alpha− subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 1999;189:587–592. doi: 10.1084/jem.189.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pulendran B, Smith JL, Caspary G, Brasel K, Pettit D, Maraskovsky E, Maliszewski CR. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc. Natl. Acad. Sci. USA. 1999;96:1036–1041. doi: 10.1073/pnas.96.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, Sher A. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J. Exp. Med. 1997;186:1819–1829. doi: 10.1084/jem.186.11.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Racke MK, Bonomo A, Scott DE, Cannella B, Levine A, Raine CS, Shevach EM, Röcken M. Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J. Exp. Med. 1994;180:1961–1966. doi: 10.1084/jem.180.5.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pozzi S, Benedusi V, Maggi A, Vegeto E. Estrogen action in neuroprotection and brain inflammation. Ann. N. Y. Acad. Sci. 2006;1089:302–323. doi: 10.1196/annals.1386.035. [DOI] [PubMed] [Google Scholar]
- 53.Stice JP, Knowlton AA. Estrogen, NFkappaB, and the heat shock response. Mol. Med. 2008;14:517–527. doi: 10.2119/2008-00026.Stice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang L, Hu Y, Hou Y. Effects of 17beta-estradiol on the maturation, nuclear factor kappa B p65 and functions of murine spleen CD11c-positive dendritic cells. Mol. Immunol. 2006;43:357–366. doi: 10.1016/j.molimm.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 55.Zang YC, Halder JB, Hong J, Rivera VM, Zhang JZ. Regulatory effects of estriol on T cell migration and cytokine profile: inhibition of transcription factor NF-kappa B. J. Neuroimmunol. 2002;124:106–114. doi: 10.1016/s0165-5728(02)00016-4. [DOI] [PubMed] [Google Scholar]
- 56.Du C, Sriram S. Mechanism of inhibition of LPS-induced IL-12p40 production by IL-10 and TGF-beta in ANA-1 cells. J. Leukoc. Biol. 1998;64:92–97. doi: 10.1002/jlb.64.1.92. [DOI] [PubMed] [Google Scholar]
- 57.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 58.Pulendran B, Banchereau J, Maraskovsky E, Maliszewski C. Modulating the immune response with dendritic cells and their growth factors. Trends Immunol. 2001;22:41–47. doi: 10.1016/s1471-4906(00)01794-4. [DOI] [PubMed] [Google Scholar]
- 59.Pettersson A, Ciumas C, Chirsky V, Link H, Huang YM, Xiao BG. Dendritic cells exposed to estrogen in vitro exhibit therapeutic effects in ongoing experimental allergic encephalomyelitis. J. Neuroimmunol. 2004;156:58–65. doi: 10.1016/j.jneuroim.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 60.Tuettenberg A, Fondel S, Steinbrink K, Enk AH, Jonuleit H. CD40 signalling induces IL-10-producing, tolerogenic dendritic cells. Exp. Dermatol. 2010;19:44–53. doi: 10.1111/j.1600-0625.2009.00975.x. [DOI] [PubMed] [Google Scholar]
- 61.Pulendran B, Kumar P, Cutler CW, Mohamadzadeh M, Van Dyke T, Banchereau J. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J. Immunol. 2001;167:5067–5076. doi: 10.4049/jimmunol.167.9.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, Peng H, Cravens PD, Racke MK, Lovett-Racke AE. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J. Exp. Med. 2009;206:1549–1564. doi: 10.1084/jem.20082584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laouar Y, Town T, Jeng D, Tran E, Wan Y, Kuchroo VK, Flavell RA. TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA. 2008;105:10865–10870. doi: 10.1073/pnas.0805058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weiner HL. Oral tolerance: immune mechanisms and the generation of Th3-type TGF-beta-secreting regulatory cells. Microbes Infect. 2001;3:947–954. doi: 10.1016/s1286-4579(01)01456-3. [DOI] [PubMed] [Google Scholar]
- 65.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 66.Mellor AL, Baban B, Chandler P, Marshall B, Jhaver K, Hansen A, Koni PA, Iwashima M, Munn DH. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J. Immunol. 2003;171:1652–1655. doi: 10.4049/jimmunol.171.4.1652. [DOI] [PubMed] [Google Scholar]
- 67.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu. Rev. Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 68.Hashiguchi M, Kobori H, Ritprajak P, Kamimura Y, Kozono H, Azuma M. Triggering receptor expressed on myeloid cell-like transcript 2 (TLT-2) is a counter-receptor for B7-H3 and enhances T cell responses. Proc. Natl. Acad. Sci. USA. 2008;105:10495–10500. doi: 10.1073/pnas.0802423105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, Tushima F, Azuma M, Yagita H, Sayegh MH, Khoury SJ. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med. 2003;198:71–78. doi: 10.1084/jem.20022119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prasad DV, Nguyen T, Li Z, Yang Y, Duong J, Wang Y, Dong C. Murine B7-H3 is a negative regulator of T cells. J. Immunol. 2004;173:2500–2506. doi: 10.4049/jimmunol.173.4.2500. [DOI] [PubMed] [Google Scholar]
- 72.Sica GL, Choi IH, Zhu G, Tamada K, Wang SD, Tamura H, Chapoval AI, Flies DB, Bajorath J, Chen L. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity. 2003;18:849–861. doi: 10.1016/s1074-7613(03)00152-3. [DOI] [PubMed] [Google Scholar]
- 73.Suh WK, Gajewska BU, Okada H, Gronski MA, Bertram EM, Dawicki W, Duncan GS, Bukczynski J, Plyte S, Elia A, et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat. Immunol. 2003;4:899–906. doi: 10.1038/ni967. [DOI] [PubMed] [Google Scholar]
- 74.Ling V, Wu PW, Spaulding V, Kieleczawa J, Luxenberg D, Carreno BM, Collins M. Duplication of primate and rodent B7-H3 immunoglobulin V- and C-like domains: divergent history of functional redundancy and exon loss. Genomics. 2003;82:365–377. doi: 10.1016/s0888-7543(03)00126-5. [DOI] [PubMed] [Google Scholar]
- 75.Fukushima A, Sumi T, Fukuda K, Kumagai N, Nishida T, Yamazaki T, Akiba H, Okumura K, Yagita H, Ueno H. B7-H3 regulates the development of experimental allergic conjunctivitis in mice. Immunol. Lett. 2007;113:52–57. doi: 10.1016/j.imlet.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 76.Prasad DV, Richards S, Mai XM, Dong C. B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity. 2003;18:863–873. doi: 10.1016/s1074-7613(03)00147-x. [DOI] [PubMed] [Google Scholar]
- 77.Zang X, Loke P, Kim J, Murphy K, Waitz R, Allison JP. B7x: a widely expressed B7 family member that inhibits T cell activation. Proc. Natl. Acad. Sci. USA. 2003;100:10388–10392. doi: 10.1073/pnas.1434299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coyle AJ, Gutierrez-Ramos JC. The expanding B7 superfamily: increasing complexity in costimulatory signals regulating T cell function. Nat. Immunol. 2001;2:203–209. doi: 10.1038/85251. [DOI] [PubMed] [Google Scholar]
- 79.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 80.Awasthi A, Kuchroo VK. Th17 cells: from precursors to players in inflammation and infection. Int. Immunol. 2009;21:489–498. doi: 10.1093/intimm/dxp021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur. J. Immunol. 2010;40:1830–1835. doi: 10.1002/eji.201040391. [DOI] [PubMed] [Google Scholar]
- 82.Matejuk A, Bakke AC, Hopke C, Dwyer J, Vandenbark AA, Offner H. Estrogen treatment induces a novel population of regulatory cells, which suppresses experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2004;77:119–126. doi: 10.1002/jnr.20145. [DOI] [PubMed] [Google Scholar]
- 83.Matejuk A, Afentoulis M. Association of CD45(dim)VLA-4 (+) cells with the NKT cell lineage and their selective expression of IL-13, IP-15, and CCR3 transcripts. Arch. Immunol. Ther. Exp. (Warsz.) 2006;54:183–191. doi: 10.1007/s00005-006-0021-3. [DOI] [PubMed] [Google Scholar]
- 84.Zhang QH, Hu YZ, Cao J, Zhong YQ, Zhao YF, Mei QB. Estrogen influences the differentiation, maturation and function of dendritic cells in rats with experimental autoimmune encephalomyelitis. Acta Pharmacol. Sin. 2004;25:508–513. [PubMed] [Google Scholar]
- 85.Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH. Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J. Immunol. 2007;179:7577–7584. doi: 10.4049/jimmunol.179.11.7577. [DOI] [PubMed] [Google Scholar]
- 86.Kovats S, Carreras E. Regulation of dendritic cell differentiation and function by estrogen receptor ligands. Cell. Immunol. 2008;252:81–90. doi: 10.1016/j.cellimm.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Paharkova-Vatchkova V, Maldonado R, Kovats S. Estrogen preferentially promotes the differentiation of CD11c+ CD11b(intermediate) dendritic cells from bone marrow precursors. J. Immunol. 2004;172:1426–1436. doi: 10.4049/jimmunol.172.3.1426. [DOI] [PubMed] [Google Scholar]
- 88.Härkönen PL, Väänänen HK. Monocyte-macrophage system as a target for estrogen and selective estrogen receptor modulators. Ann. N. Y. Acad. Sci. 2006;1089:218–227. doi: 10.1196/annals.1386.045. [DOI] [PubMed] [Google Scholar]
- 89.Steinman RM, Lustig DS, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. 3. Functional properties in vivo. J. Exp. Med. 1974;139:1431–1445. doi: 10.1084/jem.139.6.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]