

Abstract
Pediatric low grade astrocytomas are the commonest brain tumors in children. They sometimes have similar microscopic and clinical features, making accurate diagnosis difficult. For patients whose tumors are in locations that do not permit full resection, or those with an intrinsically aggressive biology, more effective therapies are required. Until recently, little was known about the molecular changes that drive the initiation and growth of pilocytic and other low grade astrocytomas beyond the association of a minority of cases, primarily in the optic nerve, with neurofibromatosis type 1. Over the last several years, a wide range of studies have implicated the BRAF oncogene and other members of this signaling cascade in the pathobiology of pediatric low grade astrocytoma. In this review, we attempt to summarize this rapidly developing field, and discuss the potential for translating our growing molecular knowledge into improved diagnostic and prognostic biomarkers and new targeted therapies.
Keywords: Pilocytic astrocytoma, BRAF, NF1, RAF1
CLINICAL AND DEMOGRAPHIC FEATURES
Pediatric low grade astrocytomas (PLGA) arise throughout the central nervous system (CNS), but are found most often in the cerebellum, followed by the cerebrum and deep midline structures, optic pathways, brainstem and spinal cord (1). According to 2012 CBTRUS data, pilocytic astrocytomas (PA) are the second most common brain tumor (after embryonal neoplasms) in the 0–4 year age group, the commonest tumors in children 5–14 years of age, and the second most common tumors (after pituitary neoplasms) in 15–19 year-olds (http://www.cbtrus.org). A graphical representation of primary pediatric brain tumor diagnoses based on current CBTRUS data is shown in Figure 1. Most non-infiltrative PLGA (i.e. pilocytic/pilomyxoid astrocytoma, pleomorphic xanthoastrocytoma, subependymal giant cell astrocytoma) show variable contrast enhancement on imaging studies, while infiltrating diffuse low grade astrocytomas are generally non-enhancing.
Figure 1.

Primary CNS tumor distribution in children ages 0–14 based on CBTRUS data.
Outcomes for this group of tumors are good overall, but vary depending on the extent of surgical resection and histopathological classification. In one large institutional series of children with low grade astrocytomas, 5 year overall survival (OS) was 96% for PA patients and 48% for diffuse astrocytoma patients (2). Gross total resection has consistently been strongly prognostic of long-term survival, but can often only be achieved when tumors are localized to the cerebellum or superficial cerebrum (3–5). It has also been suggested that PA behave in a more aggressive fashion when arising in adults(6).
HISTOLOGICAL CLASSIFICATION
Pilocytic Astrocytoma (Grade I)
PA are generally characterized by a biphasic growth pattern including both compacted bipolar cells (Figure 2A) and regions with microcysts and more loosely textured cells (Figure 2B). Characteristic elements include Rosenthal fibers and eosinophilic granular bodies (Figure 2A, B). Oligodendroglial-appearing cells are often encountered in PA, and in some cases can comprise a significant portion of the tumor (7), making diagnosis difficult in small samples. Most lesions are cytologically bland, although some atypia can be present. Mitotic figures are only present in the minority of cases, and the lack of infiltration, as well as the presence of more specific features such as eosinophilic granular bodies, allows most cases to be differentiated from high grade astrocytomas (8).
Figure 2.

Histopathological features of PLGA. (A) PA with compacted glial cells and Rosenthal fibers (arrow). (B) Microcysts (asterisks) and eosinophilic granular bodies (EGB, arrow) in a looser region of a PA. (C) Pilomyxoid astrocytoma comprised of monomorphous cells with a pronounced perivascular growth pattern. (D) Spindled, pleomorphic cells in a PXA. (E) Enlarged ganglion-cell like elements in a SEGA. (F) Diffuse astrocytoma are only modestly hypercellular with scattered atypical cells.
A number of potential histological and immunohistochemical prognostic markers have been investigated in PA. It has been suggested that tumors with an elevated Ki67 proliferation index are associated with worse progression-free survival (9, 10), although in other studies proliferation was not prognostic (11–13). Anaplastic features, including cytological atypia, hypercellularity, high mitotic activity, and necrosis, can also portend worse outcomes (14). Morphological or immunohistochemical evidence of oligodendroglial differentiation may also predict aggressive behavior in PA patients (7, 11).
Pilomyxoid Astrocytoma (Grade II)
Pilomyxoid astrocytomas are comprised of monomorphous bipolar cells, often arrayed around vessels, with a mucoid background matrix, and lack Rosenthal fibers and eosinophilic granular bodies (Figure 2C). Referred to in early reports as “infantile” pilocytic astrocytomas, their distinct microscopic features and more aggressive clinical behavior were highlighted in 1999 by Tihan and colleagues (15), leading to their recognition as a potentially distinct variant in the 2007 World Health Organization (WHO) classification (8). They are often found in hypothalamus, optic chiasm, and around the third ventricle, although other sites can also be involved. Recently, transitional tumors with mixed pilocytic and pilomyxoid features have been described, as well as cases in which a pilomyxoid astrocytoma “matured” into a classic pilocytic astrocytoma over time (16), suggesting that these lesions represent part of a spectrum rather than completely distinct entities.
Pleomorphic Xanthoastrocytoma (PXA, Grade II)
PXA generally show greater cellularity and atypia than other entities discussed in this review, and can sometimes be misdiagnosed as high grade glioma. They occur most frequently in teenagers and young adults, often involve superficial cortex and meninges, and can contain lipidized “xanthomatous” astrocytes (8). Eosinophilic granular bodies as well as spindled and pleomorphic glial elements are all common, and the tumors frequently attract a lymphocytic infiltrate and contain abundant extracellular reticulin (Figure 2D). CD34 expression has been proposed as a useful immunohistochemical feature (17).
Subependymal Giant Cell Astrocytoma (SEGA, Grade I)
These benign tumors are tightly associated with the autosomal dominant inherited condition tuberous sclerosis, and arise almost exclusively in the walls of the lateral ventricles (8). Spindled, gemistocytic and ganglion-cell like elements can be present, with the latter cells representing the most characteristic finding (Figure 2E). Immunohistochemical and ultrastructural studies have shown mixed glial and neuronal differentiation in tumor cells (18–20), and despite their designation as an “astrocytoma” these are probably best regarded as mixed glial-neuronal neoplasms.
Diffuse Astrocytoma (Grade II)
Infiltrating fibrillary astrocytomas in children are morphologically similar to their WHO grade II counterparts in adults. They are characterized by modest cellularity, diffuse infiltration of normal brain elements, and a lack of significant mitotic activity, vascular proliferation or necrosis (Figure 2F)(8). Because of their ability to spread diffusely, these tumors are difficult to completely resect and have worse outcomes than PA in children (2). As in adults, they can progress to high grade glioma, although this often does not occur. Emerging data suggest that diffuse astrocytomas in the pediatric population are molecularly distinct compared to their morphologically similar adult counterparts.
Diagnostic Difficulties in PLGA
Tumors which are difficult to classify are unfortunately all too often encountered among the spectrum of PLGA. In one institutional study of 278 consecutive PLGA resected between 1965 and 1996, 75 cases (27%) did not clearly fit into a WHO diagnostic category. A more recent review of 1,670 pediatric brain tumors of all types diagnosed at our institution between 2003 and 2008 identified 302 cases which could not be classified, and 7 of the 10 most common problems with diagnosis involved low grade gliomas (personal communication, Dr. Peter Burger). In some cases diagnostic dilemmas arise due to small biopsies, but in others they reflect inability of the current scheme to fit a heterogeneous spectrum of lesions. Molecular studies have the potential to help with these problematic issues, and as described below are beginning to shed light on some diagnostic difficulties.
MOLECULAR ADVANCES
Aside from PA arising in neurofibromatosis type 1 (NF1) patients, and SEGA in children and young adults with tuberous sclerosis, for many years little was known about the molecular underpinnings of PLGA. Sporadic PA do not inactivate NF1, and generally lack changes to the oncogenes and tumor suppressors altered in adult diffuse astrocytomas (8, 21). Early cytogenetic studies of PA were notable for a lack of detectable chromosomal alterations, with largely normal karyotypes in the more than 100 cases initially studied (8). Mutations in IDH1 have been identified in the majority of low grade gliomas in adults, but interestingly are almost never detected in pediatric low or high grade glioma, and when present have mostly been reported in children at least 14 years old (22–24). This suggests that adolescents with IDH1-mutant tumors may represent the youngest patients with “adult” gliomas. Over the last few years, however, it has become clear that most non-syndromic PLGA harbor genomic alterations which affect the function of BRAF, and that mitogen activated protein kinase (MAPK) represents the dominant genetically altered pathway in these tumors (Figure 3).
Figure 3.

Signaling pathways commonly activated in PLGA.
BRAF Fusions and MAPK Activation in PLGA
In 2008, five different groups identified gains at 7q34 approximately 2 megabases in size in most PLGA using array-based comparative genomic hybridization (25–29) (Figure 4A). Fluorescent in situ hybridization (FISH) and other molecular analyses showed that these represented segmental duplications in the region (Figure 4B)(25, 28). In these initial studies, between 53% and 77% of the PLGA examined (mostly PA) contained the duplication at 7q34, with relatively few other chromosomal alterations detected, suggesting this represented the dominant change in PA.
Figure 4.

Molecular alterations involving BRAF. (A) Segmental gains at 7q34 approximately 2 megabases in size are commonly identified in PA using array CGH. (B) FISH reveals duplication in this region, with and extra copy of the region encoding BRAF. A normal control is shown in the inset. (C) Sequencing of a KIAA1549:BRAF fusion product between exons 16 and 9 of the two genes. (D) PA contain abundant active phospho-ERK. Normal vessels serve as an internal negative control (asterisks).
In two of the papers, the authors demonstrated that 7q34 duplication resulted in expression of a novel fusion transcript between the KIAA1549 locus and BRAF that included the BRAF kinase domain but lacked the inhibitory N-terminal regulatory region of this oncogene (Figure 4C)(28, 29). The fusion showed constitutive kinase activity and was able to transform NIH 3T3 cells (29). BRAF is known to induce signaling in the MAPK pathway, and activated targets including phosphorylated MEK (pMEK) and/or ERK (pERK. Figure 4D) were identified in the majority of PLGA specimens examined in these initial studies (25, 27, 28).
A large number of additional reports have confirmed these exciting findings, and firmly establish KIAA1549:BRAF duplication/fusion as the commonest genetic change in PA (30–39). Specific breakpoints between KIAA1549 and BRAF can vary, but all lead to loss of the BRAF autoregulatory domain (Figure 5). In one recent study of 106 pediatric low grade brain tumors, five types of KIAA1549:BRAF gene fusions were identified, involving exons 1–16/9–18 (49%), 1–15/9–18 (35%), 1–16/11–18 (8%), 1–15/11–18 (6%) and 1–17/10–18 (1%)(39). In this study the 1–16/11–18 fusion was limited to infratentorial sites, and the 1–15/11–18 fusion to supratentorial locations, but it will be necessary to examine larger numbers of cases before firm conclusions can be drawn regarding associations between fusion genotype and tumor phenotype. Genetic mapping of the breakpoints involved have highlighted enrichment for microhomologous DNA sequences, suggesting microhomology-mediated break-induced replication as a possible mechanism for the rearrangements (40).
Figure 5.

Mechanisms of BRAF and RAF1 activation in PLGA
The NF1 gene product acts to inhibit RAS and BRAF activity, and the discovery of fusions activating BRAF therefore links syndromic and sporadic PA to the same oncogenic signaling cascade (Figure 3). Some patients with Noonan syndrome, in which MAPK signaling is activated by mutations in PTPN11, SOS1 and KRAS, have also been reported to have PA (41–43), providing further support for the central role of this pathway. A rare rosette forming glioneuronal tumor of the posterior fossa demonstrating strong pERK immunoreactivity has also been reported in a case of Noonan syndrome (44).
Novel genetic mechanisms driving activation of the MAPK pathway in PLGA continue to be discovered. Two groups identified oncogenic fusions between SRGAP3 and RAF1 predicted to give rise to unregulated kinase activity similar to that seen with KIAA1549:BRAF (Figure 5)(30, 45). Rare 3 base pair insertions at position 599 have also been described in PA (32, 45–47). This alteration (BRAFinsT) results in duplication of a threonine residue, and causes over 6-fold increases in kinase activity in vitro (47). An interstitial deletion causing fusions between FAM131B and BRAF has also been identified (37). However, both the SRGAP3 and FAM131B fusions and the BRAFinsT are much less common than KIAA1549:BRAF fusions in PLGA.
Point Mutations in BRAF, RAF and RAS
MAPK signaling can also sometimes be activated in PLGA by point mutations in various pathway members. Indeed, prior to the discovery of BRAF gene duplications, rare oncogenic mutations in KRAS had been described (48, 49). Subsequent studies have confirmed the presence of occasional KRAS mutations in PA. Forshew and colleagues sequenced HRAS, KRAS and NRAS in 50 PLGA and found an activating KRAS G12A mutation in one cerebellar PA which lacked BRAF and RAF1 fusions (30). Cin and colleagues examined HRAS, KRAS, NRAS, PTP11, and RAF1 in 125 primary PA samples and identified oncogenic KRAS mutations in 2 tumors (37). Thus approximately 2% of PA have mutations activating KRAS. Point mutations in RAF1 may not be selected for due to its lower basal activity as compared to BRAF (45, 50), but it is not clear why mutations in HRAS and NRAS are not present in PLGA.
The commonest point mutation in PLGA occurs in BRAF itself at codon 600, and results in substitution of valine by glutamic acid. The BRAFV600E mutation was first described in extra-CNS tumor cell lines; it is most frequent in melanoma, but can also be found in a range of other neoplasms (51). In PLGA, BRAFV600E mutations were identified in 4/66 (6%) of the tumors examined by Pfister et al. (25), including 3 PA and 1 diffuse astrocytoma. Subsequent studies have also readily identified BRAFV600E mutations in PLGA (28, 30, 32, 37–39, 46, 47), although as discussed below these are most common in tumors other than PA.
BRAF and RAF1 fusions are generally mutually exclusive with other genetic alterations activating MAPK signaling, but some exceptions to this have been reported. Cin and colleagues identified 2 PA patients with concomitant BRAFV600E mutation and BRAF fusion, one of whom also had NF1 syndrome (37). In another study, 6 tumors (out of 198) had both BRAF fusion and BRAFV600E mutation (13).
Genetic Alterations and Tumor Site
Tandem duplications involving BRAF do not occur uniformly in PLGA at all sites in the CNS (Figure 6). The percentage of cerebellar/posterior fossa PA with molecular alterations at 7q34 is particularly high, ranging from 63% to 94% in various reports (27, 28, 30, 31, 37, 52). In one study of 32 posterior fossa PAs, 30 had KIAA1549:BRAF fusions, 1 had a SRGAP3:RAF1 fusion, and 1 had a mutation in KRAS (30). In contrast, the frequency of BRAF duplication/fusion in PA arising in the cerebral cortex is quite low, ranging from 0% to 50% (27, 29, 31–33, 37, 52–54). A total of 72 cerebral cases were reported in these studies, with 18 (25%) showing BRAF duplication or fusion (Figure 6). Our review of published cases yielded percentages of BRAF duplications at other sites higher than cerebral cortex but lower than posterior fossa. This included the optic pathways (42/83, 51%), deep grey matter (17/41, 41%), brainstem (56/95, 59%) and spinal cord (6/11, 55%).
Figure 6.
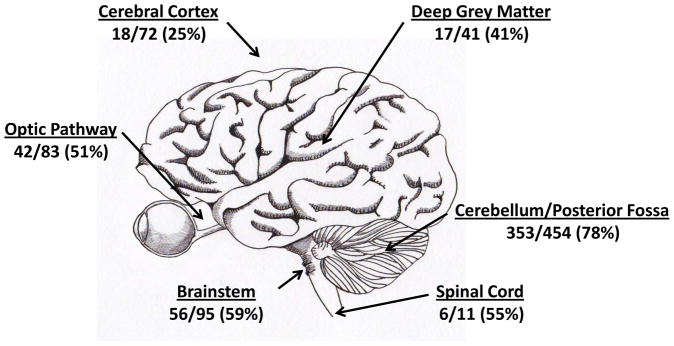
Localization of BRAF duplication/fusion in PA. Summary of tumor localization from published cases, including patients with NF1.
The limited number of other molecular changes in PA makes it harder to definitively assess their spatial distribution. However, it seems that BRAFV600E and KRAS mutations are more common in PA arising outside the posterior fossa. In one series, 10/49 (20%) of non-cerebellar PA had point mutations in one of these two oncogenes, as compared to 3/76 (4%) of cerebellar lesions, a statistically significant difference (37). Schindler and colleagues also found a statistically significant association between BRAFV600E and extracerebellar location of PA (p = 0.009)(46). In contrast, the handful of SRGAP3:RAF1 and FAM131B:BRAF fusions reported to date have largely been in PA arising in the cerebellum (30, 37).
The cause of the increased incidence of BRAF duplication in the posterior fossa is not clear, but it may reflect increased susceptibility of the tumor cell(s) of origin at this site. It has also been suggested that BRAF rearrangements in PA are less common in adult patients (55), which could reflect changes in tumor histogenesis as patients’ age. Regional differences in expression of the fusions could also play a role. While the KIAA1549, SRGAP3 and FAM131B genes are all expressed in the CNS, little is known regarding their precise levels in various regions or cell types. It has recently been demonstrated that KIAA1549:BRAF fusions are expressed at roughly equivalent levels in PA as the endogenous KIAA1549 gene (39).
Senescence in PLGA
One interesting feature of PLGA is the propensity of some tumors, particularly PA, to occasionally spontaneously stop growing or even regress (56, 57). A similar pattern of initial neoplastic proliferation followed by growth arrest is often seen in benign melanocytic nevi of the skin, which also commonly contain genetic alterations in BRAF (58). This process has been termed oncogene induced senescence (OIS), with senescence defined as an irreversible growth arrest. OIS has been shown to result from induction of the p16/Rb, p14ARF/p53 and/or DNA damage response pathways by BRAF and other oncogenes (59, 60). Markers of OIS include enlargement and flattening of cells, expression of p16 and p53, and activation of acidic senescence-associated β-galactosidase (SA-β-Gal). These are frequently found in premalignant lesions, but are essentially absent in malignant tumor cells, suggesting the latter have found ways to bypass or escape senescence. One such mechanism is deletion of p16, which is frequently identified in malignant melanoma (59, 60).
Given the importance of BRAF activation in PA, and their often indolent growth pattern, it is not surprising that several groups have examined the potential role of OIS. In Raabe et al. and Jacob et al., the investigators show that OIS markers including p16, p53 and SA-β-Gal are expressed in both primary PA samples and low passage tumor cultures (61, 62). The groups also found that introducing BRAFV600E into either human neural stem cells, hTERT-immortalized astrocytes or fetal astrocytes results in growth arrest and the induction of p16 and SA-β-Gal. OIS has also been documented in benign cutaneous neurofibromas driven by NF1 loss (63).
The role of p16 in escape from OIS and aggressive tumor growth has attracted particular attention. Jacob and colleagues found that loss of p16 was required for isolation of astrocyte clones stably expressing BRAFV600E. Raabe and colleagues examined p16 expression in 66 PA cases using immunohistochemistry, and found that the 9 patients whose tumors were p16 negative had significantly shorter overall survival. Additional support for a prognostic clinical role comes from FISH studies by two groups. Homozygous p16 deletions were selectively identified in more aggressive anaplastic PA in Rodriguez et al. (52). Horbinski and colleagues examined a large cohort of 198 PLGA, and found that p16 deletion was the second strongest predictor of adverse outcome (after midline location) in the group overall, and also correlated with significantly shorter progression-free survival in tumors with BRAF rearrangement (13). Taken together, these data strongly support the concept that OIS contributes to the sometimes indolent behavior of PA, and that p16 loss can contribute to escape from senescence and clinically aggressive tumor growth (Figure 7). It remains to be seen if OIS also plays a role in the pathobiology of other PLGA.
Figure 7.

Oncogene induced senescence in PA. Senescence-associated β-galactosidase stains of BRAFV600E and vector transduced human neural stem cells are shown in the bottom panels.
Another potential contributor to the slowing or arrest of growth in PLGA is replicative senescence mediated by shortening of telomeres. Using the PCR-based telomeric repeat amplification assay, Chong and colleagues documented telomerase activity in many high grade gliomas but not in the 16 PA and 2 PXA examined (64). Tabori and colleagues subsequently confirmed a lack of telomerase activity in 11 pediatric low grade gliomas (65), as well as a significant decrease in telomere length over time, leading them to propose that lack of telomere maintenance may contribute to growth arrest or regression of PLGA (65, 66). They also found that longer telomere length is inversely correlated with survival, further supporting this concept. A telomerase-independent process known as alternative lengthening of telomeres (ALT) has recently been shown to be active in almost half of pediatric glioblastoma (67). However ALT is very rare or absent in PA (65, 67), although ALT-associated promyelocytic leukemia bodies have been identified in some cases (68).
Neurofibromatosis type 1
NF1 is caused by germline mutations in the gene at 17q11.2 encoding for neurofibromin, a tumor suppressor that works as a GTPase activating protein to deactivate RAS (8). PA are the most frequent NF1-associated CNS tumors, and it is estimated that approximately 15% of pediatric NF1 patients develop optic pathway gliomas (69, 70). Most molecular studies have demonstrated BRAF alterations to be mutually exclusive with NF1 clinical status (29, 32, 39, 53). Conversely, the neurofibromin gene is almost never altered in sporadic PA (71, 72), and NF1-associated and sporadic PA have distinct global gene expression patterns (70, 73). However, in rare instances NF1-associated tumors have been reported with additional activating mutations in BRAF. Such cases reported include KIAA1549:BRAF fusion in a NF1-associated pilomyxoid astrocytoma (30), a BRAFV600E point mutation in a NF1-associated PA(47), and an interesting “triple hit” with concomitant NF1 syndrome, BRAFV600E, and KIAA159-BRAF fusion (37).
While a major molecular consequence of neurofibromin loss is MAPK pathway activation, additional signaling nodes contribute to tumorigenesis in NF1, including mTOR pathway activation (74). Indeed, activation of the PI3K/AKT/mTOR signaling axis is a prominent feature of the rare PA that develop anaplastic change (52), 24% of which are NF1-associated. Recent studies have also highlighted a role for the non-neoplastic stromal microenvironment in optic glioma development in NF1 model systems. Nf1 heterozygous microglia are required for glioma formation in these models, in addition to Nf1 homozygous loss in neoplastic astrocytes. Recent evidence suggests that stromal derived factors (e.g. CXCL12) lead to altered cAMP levels, and facilitate tumor formation in this setting (75).
AKT/mTOR
Another important signaling pathway operating downstream from neurofibromin and RAS is AKT/mTOR, which leads to increased protein translation, cell growth and survival through two multiprotein complexes (mTORC1 and mTORC2) that vary in their sensitivity to rapamycin (76–78). The prototypical low grade glioma in which mTOR activation is an intrinsic molecular property is the tuberous sclerosis-associated SEGA (79). Tuberous sclerosis is characterized by germline mutations in the tumor suppressor genes TSC1 or TSC2, which regulate AKT activation through RHEB, and recent clinical success has been described with mTOR pathway inhibitors (80). Some studies have also highlighted a role for mTOR signaling in NF1-associated tumors, in particular PA (81). Examination of Nf1 deficient mouse models has suggested anatomical variation in neuroglial progenitor proliferation through AKT activation (82). Recent studies have also suggested a possible role for differential mTOR activation in subsets of NF1 associated low grade gliomas difficult to classify by traditional criteria (83).
Outside of the syndrome-associated low grade gliomas, little is known at the present time regarding mTOR pathway activation in tumorigenesis or progression in PLGA. However, immunohistochemical studies suggest the pathway is active in at least some tumors. Of interest, mutations in PIK3CA have been reported in 3 (of 4) rosette-forming glioneuronal tumors, rare low grade neoplasms with a frequent pilocytic astrocytoma-like component (84).
Gene Expression Analysis
Early studies suggested that PA have expression profiles distinct from those of high grade gliomas (85–87), and that PA could also be differentiated from diffuse astrocytoma (88) oligodendroglioma and normal white matter (89). Additional studies identified MBP and Matrilin as potential markers of poor outcome (90, 91). Gene expression analysis has also been used to suggest potential cells of origin for PA, which may be region-specific (73, 92). However, unlike other childhood brain tumors such as medulloblastoma and ependymoma, large-scale studies integrating pathology, clinical factors, and mutations/copy number change with mRNA and miRNA expression have not yet been published. For pediatric medulloblastoma and adult glioblastoma, such correlations have been critical for parsing clinically and genetically meaningful tumor subgroups, potential cells of origin, and generating molecularly relevant classification schemes (93). Hopefully similar integrated data will soon be available for PLGA.
CLINICAL AND THERAPEUTIC IMPLICATIONS
Diagnostic Utility of BRAF Alterations
Distinguishing between the various types of PLGA can sometimes be difficult, thus testing for BRAF alterations could potentially be of great use diagnostically. In the first report in which KIAA1549:BRAF fusions were identified, Jones and colleagues noted that almost all cases were PA (29). A few PLGA not originally diagnosed as PA contained the alteration, but because they were all cerebellar and associated with survival of greater than 12 years the authors suggested that they might represent PA which had been misclassified. A number of subsequent investigations have confirmed a strong association between PA histology and BRAF duplication/fusion (31, 34, 54), but some exceptions exist. In a study including 27 pediatric diffuse astrocytoma, Jacob and colleagues did not detect duplication of 7q34 (31), but the same group later found some diffuse astrocytomas with KIAA1549:BRAF fusions (53). Forshew and colleagues reported a KIAA1549:BRAF fusion in 1 of 11 pediatric diffuse astrocytoma in their study (30), while Sievert and colleagues identified the duplication in 3 of 6 of these tumors (28).
BRAF duplication/fusion events occur fairly frequently in pilomyxoid astrocytoma (30, 39, 53) supporting a recent study suggesting that pilocytic and pilomyxoid astrocytoma may be part of a single disease spectrum (16). However, the few PXA examined to date have not shown these alterations (30, 39). BRAF fusions have also not been identified in high grade pediatric gliomas. Finally, BRAF fusions were recently reported in a few pediatric low grade glioneuronal tumors (39). Taken together, these reports of rare fusions in diffuse astrocytoma and other non-pilocytic lesions suggests that such molecular changes involving BRAF are highly enriched in PA, but not absolutely specific for this diagnosis.
In contrast to BRAF duplication/fusion, point mutations in BRAF are most common in low grade pediatric brain tumors other than PA, and are also found in higher grade gliomas (54). Dougherty and colleagues identified BRAFV600E mutations in 9/18 (50%) of gangliogliomas as well as several PXA (94). A study of 1,320 nervous system tumors found that BRAFV600E was most common in PXA (57/87, 66%) and WHO grade I ganglioglioma (14/77, 18%)(46). Another group also identified common BRAFV600E mutations in PXA (12/20, 60%)(95).
Prognostic Utility of BRAF Alterations in PLGA
Another potential clinical application of molecular testing for alterations in BRAF and other MAPK pathway members is to determine which PLGA are more aggressive, allowing more precise delivery of therapy. Neither our group nor Cin and colleagues found better outcomes in PLGA patients whose tumors contained BRAF fusions (37, 39). Hawkins and colleagues focused on a “clinically relevant” subgroup of PLGA cases defined as non-NF1 patients with non-cerebellar tumor location and subtotal resection. They reported that KIAA1549:BRAF fusions were significantly associated with better outcome in a cohort of 70 PLGA patients meeting these criteria (53). However, when we examined an analogous subgroup within our cohort we did not identify any trend towards better outcome with BRAF fusion (39).
Horbinski and colleagues examined BRAF status in 118 unselected PA with outcome data, but did not identify significantly different outcomes in cases with and without the duplication in their initial study (35). In a subsequent larger study of 198 cases by this group, of which 143 were PA, they found a trend towards improved progression free survival in patients with low grade gliomas whose tumors had BRAF rearrangements (p = 0.06)(13). They also noted that the only patients with BRAF rearrangements who died had midline tumors. In contrast, patients in their cohort with BRAFV600E had a trend towards increased risk of progression (p = 0.07)(13). Given the somewhat conflicting data in these various studies, it seems that while BRAF fusions may portend better outcomes, examination of a larger cohort, preferably from a controlled clinical trial, will be necessary to definitively determine the prognostic role of this alteration.
Clinical Testing for BRAF Alterations
To date, no standard approach to testing for the various alterations affecting MAPK signaling in PLGA has been developed. Because fresh tissue is not always available, assays which can work with formalin fixed, paraffin-embedded (FFPE) specimens will be of greatest practical utility. Given the frequent presence of BRAFV600E point mutations in cutaneous melanoma and other common neoplasms arising outside the CNS, many clinical labs have standard tests for this alteration based on sequencing or hybridization (96). A monoclonal antibody specific for the BRAFV600E protein has also recently been developed, and will be useful in small specimens (97).
A range of approaches to identify BRAF fusions have also been reported. Many laboratories have used duplication of the 7q34 region as a marker for this change. This can be assessed by FISH, which has the advantages of working well in FFPE tissues, spatially localizing the change, and detecting it in small groups of cells. As costs have dropped, array CGH is increasingly being used in a diagnostic capacity in both fresh and FFPE samples, and this technology can detect changes across the genome rather than only those at 7q34, including for example loss of the p16 locus. Direct identification of fusion transcripts using reverse transcription polymerase chain reaction (RT PCR) is highly specific and yields information on fusion breakpoints, but only pre-determined regions can be assessed (39). While this method has traditionally been performed using RNA extracted from frozen tumors, it has recently been shown that 97% sensitivity and 91% specificity can be achieved using fusion transcripts isolated from FFPE tissue (38). Finally, pyrosequencing has been used to detect fusions and changes in BRAF gene dosage in FFPE specimens, and may identify some alterations not found using PCR primers specific for various fusions (98).
Preclinical Testing Models
The development of effective new therapies for PLGA would be greatly assisted by cell or animal based models which accurately reflect the molecular biology and pathology of these tumors. Unfortunately, PLGA models are not as advanced overall as is true for high grade gliomas, although transgenic mice with NF1-associated optic gliomas have been generated and used for preclinical testing (99, 100). Another genetically engineered mouse model which may prove useful in preclinical PLGA testing was recently reported by Gronych and colleagues (101). They found that BRAF activation alone in nestin-positive murine neural progenitors was sufficient to induce the formation of cerebral low grade gliomas, but this was achieved by a combination of V600E mutation and deletion of the negative regulatory region. It therefore still remains to be seen if fusions of the BRAF kinase domain with KIAA1549 or other partners analogous to those in humans will be sufficient to drive PLGA tumorigenesis.
Cultures of human PLGA represent an additional potential platform for preclinical testing. It has been difficult to develop useful lines from PA and other indolent PLGA, but some have been reported (25, 102). The recent description of OIS in low passage cultures may account at least in part for these problems (61, 62). It may be possible to develop more robust cultures by maintaining them as neurospheres in serum-free media, or by manipulating expression of p16 and other OIS factors. Now that signature genetic defects in BRAF have been discovered, it will be critical to demonstrate that any cell lines developed carry the molecular markers of the tumors from which they are derived.
Some preclinical testing has been done with the models currently available. Using their Nf1 mutant optic glioma model, Banerjee and colleagues have explored various rapamycin doses affecting mTOR signaling, and shown that not all pathway biomarkers accurately reflect effective pathway response or changes in tumor growth (74). Proliferation of one human PA culture was inhibited by pharmacological MAPK blockade (25). Finally, murine neurospheres transduced with active BRAF are responsive to Sorafenib (101), and engineered human neural stem and progenitor cell systems (61, 62) could also be used in this fashion.
Therapeutic Possibilities
Given the high frequency of BRAF activation via duplication/fusion and mutation in pilocytic and pilomyxoid astrocytomas, there is considerable interest in targeted inhibition of the MAPK pathway as therapy for these tumors (Figure 7). Sorafenib (Nexavar, Bayer and Onyx Pharmacuticals) is an inhibitor of BRAF which has less potency against BRAFV600E. Currently, Sorafenib is in phase II studies against recurrent and chemotherapy-refractory PLGA (NCT01338857 ClinicalTrials.gov). AZD6244 (Selumetinib, AstraZeneca and Array BioPharma) is a potent inhibitor of MEK also currently in phase I trials against PLGA (NCT01386450, NCT01089101 ClinicalTrials.gov).
BRAFV600E mutations are rare among PLGA overall, but are relatively common in PXA, gangliogliomas and a subset of extra-cerebellar pilocytic astrocytomas (46, 54, 94, 95). Vemurafenib is a competitive small molecule that was designed to bind to and inhibit the ATP binding domain of the BRAFV600Emutant, but not other forms of BRAF (103, 104). Following impressive albeit transient responses of recurrent melanoma to Vemurafenib (104), the United States Food and Drug Administration (FDA) has approved it for the treatment of BRAFV600E mutation positive, inoperable, or metastatic melanoma. It is anticipated that there will be a clinical trial of Vemurafenib against BRAFV600E mutant low-grade gliomas in the near future.
The AKT/mTOR pathway has been implicated in several types of PLGA, including SEGA and PA. Clinical trials have demonstrated that mTOR inhibitors including Sirolimus and Everolimus (RAD-001 or Afinitor; Novartis Pharmaceuticals) have activity against SEGA (105), and Everolimus has received approval by the FDA for the treatment of SEGA that cannot be surgically resected. A recent reported phase I/II study of Sirolimus (Rapamune, Pfizer) and Erlotinib (Tarceva, Genentech) examined 16 patients with recurrent PLGA (106). Of the 7 children with NF1 in this clinical trial, all patients had either stable disease or tumor responses. A phase II study of Everolimus against recurrent and chemotherapy-refractory PLGA has recently been completed (NCT00782626); results of this study are pending. As agents targeting the MAPK and AKT/mTOR pathways are tested, it will be critical to also search for molecular biomarkers which are predictive of response.
CONCLUSIONS
It is now clear that alterations affecting the BRAF oncogene and other members of the MAPK cascade represent the main genetic defects in PLGA. Segmental duplications resulting in fusions between KIAA1549 and BRAF are most common, and result in a novel protein with constitutive kinase activity. Tumors lacking KIAA1549:BRAF fusions often have other changes with similar functional effects, including SRGAP3:RAF1 and FAM131B:BRAF fusions and mutations and insertions activating BRAF. These findings link syndromic and sporadic PA, and suggest that MAPK is the dominant signaling pathway to target therapeutically. The detection of BRAF activation as a cardinal feature of PLGA has also led to the discovery that oncogenic induction of senescence may account for the spontaneous growth arrest or regression of some tumors.
The challenge now is to translate these new discoveries in improved diagnostic, prognostic and predictive markers, and to develop targeted therapies for patients with clinically aggressive tumors. PLGA are a heterogeneous group of lesions, and while BRAF fusions are not entirely specific for one entity they are almost always encountered in PA, thus molecular BRAF testing may help us to distinguish these tumors from other pediatric gliomas. The prognostic role of BRAF alteration is still not clear, and analysis of large, uniformly treated cohorts will likely be required to definitively assess associations with outcome. Finally, it will be critical to determine if specific genetic changes predict response to therapies targeting BRAF or other pathway members. The recent initiation of several clinical trials of MAPK inhibitors in PLGA patients should provide some initial insights into this key question.
Figure 8.

Therapeutic agents targeting BRAF/RAF1 and AKT/mTOR pathways
Acknowledgments
The work of CGE on PLGA has been supported by the Children’s Cancer Foundation and the PLGA foundation. The Childhood Brain Tumor Foundation provided support to FR.
LITERATURE CITED
- 1.Sievert AJ, Fisher MJ. Pediatric low-grade gliomas. J Child Neurol. 2009;24:1397–408. doi: 10.1177/0883073809342005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fisher PG, Tihan T, Goldthwaite PT, Wharam MD, Carson BS, Weingart JD, Repka MX, Cohen KJ, Burger PC. Outcome analysis of childhood low-grade astrocytomas. Pediatr Blood Cancer. 2008;51:245–50. doi: 10.1002/pbc.21563. [DOI] [PubMed] [Google Scholar]
- 3.Pollack IF, Claassen D, al-Shboul Q, Janosky JE, Deutsch M. Low-grade gliomas of the cerebral hemispheres in children: an analysis of 71 cases. J Neurosurg. 1995;82:536–47. doi: 10.3171/jns.1995.82.4.0536. [DOI] [PubMed] [Google Scholar]
- 4.Pencalet P, Maixner W, Sainte-Rose C, Lellouch-Tubiana A, Cinalli G, Zerah M, Pierre-Kahn A, Hoppe-Hirsch E, Bourgeois M, Renier D. Benign cerebellar astrocytomas in children. J Neurosurg. 1999;90:265–73. doi: 10.3171/jns.1999.90.2.0265. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez C, Figarella-Branger D, Girard N, Bouvier-Labit C, Gouvernet J, Paz Paredes A, Lena G. Pilocytic astrocytomas in children: prognostic factors--a retrospective study of 80 cases. Neurosurgery. 2003;53:544–53. doi: 10.1227/01.neu.0000079330.01541.6e. discussion 54–5. [DOI] [PubMed] [Google Scholar]
- 6.Stuer C, Vilz B, Majores M, Becker A, Schramm J, Simon M. Frequent recurrence and progression in pilocytic astrocytoma in adults. Cancer. 2007;110:2799–808. doi: 10.1002/cncr.23148. [DOI] [PubMed] [Google Scholar]
- 7.Takei H, Yogeswaren ST, Wong KK, Mehta V, Chintagumpala M, Dauser RC, Lau CC, Adesina AM. Expression of oligodendroglial differentiation markers in pilocytic astrocytomas identifies two clinical subsets and shows a significant correlation with proliferation index and progression free survival. J Neurooncol. 2008;86:183–90. doi: 10.1007/s11060-007-9455-7. [DOI] [PubMed] [Google Scholar]
- 8.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. LYON: IARC Press; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dirven CM, Koudstaal J, Mooij JJ, Molenaar WM. The proliferative potential of the pilocytic astrocytoma: the relation between MIB-1 labeling and clinical and neuro-radiological follow-up. J Neurooncol. 1998;37:9–16. doi: 10.1023/a:1005905009449. [DOI] [PubMed] [Google Scholar]
- 10.Bowers DC, Gargan L, Kapur P, Reisch JS, Mulne AF, Shapiro KN, Elterman RD, Winick NJ, Margraf LR. Study of the MIB-1 labeling index as a predictor of tumor progression in pilocytic astrocytomas in children and adolescents. J Clin Oncol. 2003;21:2968–73. doi: 10.1200/JCO.2003.01.017. [DOI] [PubMed] [Google Scholar]
- 11.Tibbetts KM, Emnett RJ, Gao F, Perry A, Gutmann DH, Leonard JR. Histopathologic predictors of pilocytic astrocytoma event-free survival. Acta Neuropathol. 2009;117:657–65. doi: 10.1007/s00401-009-0506-3. [DOI] [PubMed] [Google Scholar]
- 12.Fisher BJ, Naumova E, Leighton CC, Naumov GN, Kerklviet N, Fortin D, Macdonald DR, Cairncross JG, Bauman GS, Stitt L. Ki-67: a prognostic factor for low-grade glioma? Int J Radiat Oncol Biol Phys. 2002;52:996–1001. doi: 10.1016/s0360-3016(01)02720-1. [DOI] [PubMed] [Google Scholar]
- 13.Horbinski C, Nikiforova MN, Hagenkord JM, Hamilton RL, Pollack IF. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro Oncol. 2012 doi: 10.1093/neuonc/nos077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez FJ, Scheithauer BW, Burger PC, Jenkins S, Giannini C. Anaplasia in pilocytic astrocytoma predicts aggressive behavior. Am J Surg Pathol. 2010;34:147–60. doi: 10.1097/PAS.0b013e3181c75238. [DOI] [PubMed] [Google Scholar]
- 15.Tihan T, Fisher PG, Kepner JL, Godfraind C, McComb RD, Goldthwaite PT, Burger PC. Pediatric astrocytomas with monomorphous pilomyxoid features and a less favorable outcome. J Neuropathol Exp Neurol. 1999;58:1061–8. doi: 10.1097/00005072-199910000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Johnson MW, Eberhart CG, Perry A, Tihan T, Cohen KJ, Rosenblum MK, Rais-Bahrami S, Goldthwaite P, Burger PC. Spectrum of pilomyxoid astrocytomas: intermediate pilomyxoid tumors. Am J Surg Pathol. 2010;34:1783–91. doi: 10.1097/PAS.0b013e3181fd66c3. [DOI] [PubMed] [Google Scholar]
- 17.Reifenberger G, Kaulich K, Wiestler OD, Blumcke I. Expression of the CD34 antigen in pleomorphic xanthoastrocytomas. Acta Neuropathol. 2003;105:358–64. doi: 10.1007/s00401-002-0652-3. [DOI] [PubMed] [Google Scholar]
- 18.Buccoliero AM, Franchi A, Castiglione F, Gheri CF, Mussa F, Giordano F, Genitori L, Taddei GL. Subependymal giant cell astrocytoma (SEGA): Is it an astrocytoma? Morphological, immunohistochemical and ultrastructural study. Neuropathology. 2009;29:25–30. doi: 10.1111/j.1440-1789.2008.00934.x. [DOI] [PubMed] [Google Scholar]
- 19.Jozwiak J, Jozwiak S, Skopinski P. Immunohistochemical and microscopic studies on giant cells in tuberous sclerosis. Histol Histopathol. 2005;20:1321–6. doi: 10.14670/HH-20.1321. [DOI] [PubMed] [Google Scholar]
- 20.Sharma MC, Ralte AM, Gaekwad S, Santosh V, Shankar SK, Sarkar C. Subependymal giant cell astrocytoma--a clinicopathological study of 23 cases with special emphasis on histogenesis. Pathol Oncol Res. 2004;10:219–24. doi: 10.1007/BF03033764. [DOI] [PubMed] [Google Scholar]
- 21.Cheng Y, Pang JC, Ng HK, Ding M, Zhang SF, Zheng J, Liu DG, Poon WS. Pilocytic astrocytomas do not show most of the genetic changes commonly seen in diffuse astrocytomas. Histopathology. 2000;37:437–44. doi: 10.1046/j.1365-2559.2000.01005.x. [DOI] [PubMed] [Google Scholar]
- 22.Buccoliero AM, Castiglione F, Rossi Degl’innocenti D, Gheri CF, Genitori L, Taddei GL. IDH1 Mutation in Pediatric Gliomas: Has it a Diagnostic and Prognostic Value? Fetal Pediatr Pathol. 2012 doi: 10.3109/15513815.2012.659383. [DOI] [PubMed] [Google Scholar]
- 23.Pollack IF, Hamilton RL, Sobol RW, Nikiforova MN, Lyons-Weiler MA, LaFramboise WA, Burger PC, Brat DJ, Rosenblum MK, Holmes EJ, Zhou T, Jakacki RI. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Childs Nerv Syst. 2011;27:87–94. doi: 10.1007/s00381-010-1264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, Lowe J, Gajjar A, Zhao W, Broniscer A, Ellison DW, Grundy RG, Baker SJ. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28:3061–8. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Kratz C, Olbrich H, Ahmadi R, Thieme B, Joos S, Radlwimmer B, Kulozik A, Pietsch T, Herold-Mende C, Gnekow A, Reifenberger G, Korshunov A, Scheurlen W, Omran H, Lichter P. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118:1739–49. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deshmukh H, Yeh TH, Yu J, Sharma MK, Perry A, Leonard JR, Watson MA, Gutmann DH, Nagarajan R. High-resolution, dual-platform aCGH analysis reveals frequent HIPK2 amplification and increased expression in pilocytic astrocytomas. Oncogene. 2008;27:4745–51. doi: 10.1038/onc.2008.110. [DOI] [PubMed] [Google Scholar]
- 27.Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol. 2008;67:878–87. doi: 10.1097/NEN.0b013e3181845622. [DOI] [PubMed] [Google Scholar]
- 28.Sievert AJ, Jackson EM, Gai X, Hakonarson H, Judkins AR, Resnick AC, Sutton LN, Storm PB, Shaikh TH, Biegel JA. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol. 2009;19:449–58. doi: 10.1111/j.1750-3639.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, Collins VP. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forshew T, Tatevossian RG, Lawson AR, Ma J, Neale G, Ogunkolade BW, Jones TA, Aarum J, Dalton J, Bailey S, Chaplin T, Carter RL, Gajjar A, Broniscer A, Young BD, Ellison DW, Sheer D. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol. 2009;218:172–81. doi: 10.1002/path.2558. [DOI] [PubMed] [Google Scholar]
- 31.Jacob K, Albrecht S, Sollier C, Faury D, Sader E, Montpetit A, Serre D, Hauser P, Garami M, Bognar L, Hanzely Z, Montes JL, Atkinson J, Farmer JP, Bouffet E, Hawkins C, Tabori U, Jabado N. Duplication of 7q34 is specific to juvenile pilocytic astrocytomas and a hallmark of cerebellar and optic pathway tumours. Br J Cancer. 2009;101:722–33. doi: 10.1038/sj.bjc.6605179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu J, Deshmukh H, Gutmann RJ, Emnett RJ, Rodriguez FJ, Watson MA, Nagarajan R, Gutmann DH. Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology. 2009;73:1526–31. doi: 10.1212/WNL.0b013e3181c0664a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H, Pfister S, von Deimling A, Hartmann C. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118:401–5. doi: 10.1007/s00401-009-0550-z. [DOI] [PubMed] [Google Scholar]
- 34.Lawson AR, Tatevossian RG, Phipps KP, Picker SR, Michalski A, Sheer D, Jacques TS, Forshew T. RAF gene fusions are specific to pilocytic astrocytoma in a broad paediatric brain tumour cohort. Acta Neuropathol. 2010;120:271–3. doi: 10.1007/s00401-010-0693-y. [DOI] [PubMed] [Google Scholar]
- 35.Horbinski C, Hamilton RL, Nikiforov Y, Pollack IF. Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta Neuropathol. 2010;119:641–9. doi: 10.1007/s00401-009-0634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tihan T, Ersen A, Qaddoumi I, Sughayer MA, Tolunay S, Al-Hussaini M, Phillips J, Gupta N, Goldhoff P, Baneerjee A. Pathologic characteristics of pediatric intracranial pilocytic astrocytomas and their impact on outcome in 3 countries: a multi-institutional study. Am J Surg Pathol. 2012;36:43–55. doi: 10.1097/PAS.0b013e3182329480. [DOI] [PubMed] [Google Scholar]
- 37.Cin H, Meyer C, Herr R, Janzarik WG, Lambert S, Jones DT, Jacob K, Benner A, Witt H, Remke M, Bender S, Falkenstein F, Van Anh TN, Olbrich H, von Deimling A, Pekrun A, Kulozik AE, Gnekow A, Scheurlen W, Witt O, Omran H, Jabado N, Collins VP, Brummer T, Marschalek R, Lichter P, Korshunov A, Pfister SM. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. 2011;121:763–74. doi: 10.1007/s00401-011-0817-z. [DOI] [PubMed] [Google Scholar]
- 38.Tian Y, Rich BE, Vena N, Craig JM, Macconaill LE, Rajaram V, Goldman S, Taha H, Mahmoud M, Ozek M, Sav A, Longtine JA, Lindeman NI, Garraway LA, Ligon AH, Stiles CD, Santagata S, Chan JA, Kieran MW, Ligon KL. Detection of KIAA1549-BRAF fusion transcripts in formalin-fixed paraffin-embedded pediatric low-grade gliomas. J Mol Diagn. 2011;13:669–77. doi: 10.1016/j.jmoldx.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin A, Rodriguez FJ, Karajannis MA, Williams SC, Legault G, Zagzag D, Burger PC, Allen JC, Eberhart CG, Bar EE. BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549:BRAF fusion variants. J Neuropathol Exp Neurol. 2012;71:66–72. doi: 10.1097/NEN.0b013e31823f2cb0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawson AR, Hindley GF, Forshew T, Tatevossian RG, Jamie GA, Kelly GP, Neale GA, Ma J, Jones TA, Ellison DW, Sheer D. RAF gene fusion breakpoints in pediatric brain tumors are characterized by significant enrichment of sequence microhomology. Genome Res. 2011;21:505–14. doi: 10.1101/gr.115782.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fryssira H, Leventopoulos G, Psoni S, Kitsiou-Tzeli S, Stavrianeas N, Kanavakis E. Tumor development in three patients with Noonan syndrome. Eur J Pediatr. 2008;167:1025–31. doi: 10.1007/s00431-007-0636-3. [DOI] [PubMed] [Google Scholar]
- 42.Sanford RA, Bowman R, Tomita T, De Leon G, Palka P. A 16-year-old male with Noonan’s syndrome develops progressive scoliosis and deteriorating gait. Pediatr Neurosurg. 1999;30:47–52. doi: 10.1159/000028761. [DOI] [PubMed] [Google Scholar]
- 43.Schuettpelz LG, McDonald S, Whitesell K, Desruisseau DM, Grange DK, Gurnett CA, Wilson DB. Pilocytic astrocytoma in a child with Noonan syndrome. Pediatr Blood Cancer. 2009;53:1147–9. doi: 10.1002/pbc.22193. [DOI] [PubMed] [Google Scholar]
- 44.Karafin M, Jallo GI, Ayars M, Eberhart CG, Rodriguez FJ. Rosette forming glioneuronal tumor in association with Noonan syndrome: pathobiological implications. Clin Neuropathol. 2011;30:297–300. doi: 10.5414/NP300374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009;28:2119–23. doi: 10.1038/onc.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, Schmieder K, Wesseling P, Mawrin C, Hasselblatt M, Louis DN, Korshunov A, Pfister S, Hartmann C, Paulus W, Reifenberger G, von Deimling A. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011;121:397–405. doi: 10.1007/s00401-011-0802-6. [DOI] [PubMed] [Google Scholar]
- 47.Eisenhardt AE, Olbrich H, Roring M, Janzarik W, Anh TN, Cin H, Remke M, Witt H, Korshunov A, Pfister SM, Omran H, Brummer T. Functional characterization of a BRAF insertion mutant associated with pilocytic astrocytoma. Int J Cancer. 2010 doi: 10.1002/ijc.25893. [DOI] [PubMed] [Google Scholar]
- 48.Sharma MK, Zehnbauer BA, Watson MA, Gutmann DH. RAS pathway activation and an oncogenic RAS mutation in sporadic pilocytic astrocytoma. Neurology. 2005;65:1335–6. doi: 10.1212/01.wnl.0000180409.78098.d7. [DOI] [PubMed] [Google Scholar]
- 49.Janzarik WG, Kratz CP, Loges NT, Olbrich H, Klein C, Schafer T, Scheurlen W, Roggendorf W, Weiller C, Niemeyer C, Korinthenberg R, Pfister S, Omran H. Further evidence for a somatic KRAS mutation in a pilocytic astrocytoma. Neuropediatrics. 2007;38:61–3. doi: 10.1055/s-2007-984451. [DOI] [PubMed] [Google Scholar]
- 50.Emuss V, Garnett M, Mason C, Marais R. Mutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAF. Cancer Res. 2005;65:9719–26. doi: 10.1158/0008-5472.CAN-05-1683. [DOI] [PubMed] [Google Scholar]
- 51.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez EF, Scheithauer BW, Giannini C, Rynearson A, Cen L, Hoesley B, Gilmer-Flynn H, Sarkaria JN, Jenkins S, Long J, Rodriguez FJ. PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma. Acta Neuropathol. 2011;121:407–20. doi: 10.1007/s00401-010-0784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hawkins C, Walker E, Mohamed N, Zhang C, Jacob K, Shirinian M, Alon N, Kahn D, Fried I, Scheinemann K, Tsangaris E, Dirks P, Tressler R, Bouffet E, Jabado N, Tabori U. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res. 2011;17:4790–8. doi: 10.1158/1078-0432.CCR-11-0034. [DOI] [PubMed] [Google Scholar]
- 54.Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M, Fisher PG, Rowitch DH, Ford JM, Berger MS, Ji H, Gutmann DH, James CD. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010;70:512–9. doi: 10.1158/0008-5472.CAN-09-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hasselblatt M, Riesmeier B, Lechtape B, Brentrup A, Stummer W, Albert FK, Sepehrnia A, Ebel H, Gerss J, Paulus W. BRAF-KIAA1549 fusion transcripts are less frequent in pilocytic astrocytomas diagnosed in adults. Neuropathol Appl Neurobiol. 2011;37:803–6. doi: 10.1111/j.1365-2990.2011.01193.x. [DOI] [PubMed] [Google Scholar]
- 56.Sakai K, Miyahara T, Tsutsumi K, Kaneko T, Fukushima M, Tanaka Y, Hongo K. Spontaneous regression of multicentric pilocytic astrocytoma with CSF dissemination in an adult. Brain Tumor Pathol. 2011;28:151–6. doi: 10.1007/s10014-010-0015-z. [DOI] [PubMed] [Google Scholar]
- 57.Gunny RS, Hayward RD, Phipps KP, Harding BN, Saunders DE. Spontaneous regression of residual low-grade cerebellar pilocytic astrocytomas in children. Pediatr Radiol. 2005;35:1086–91. doi: 10.1007/s00247-005-1546-z. [DOI] [PubMed] [Google Scholar]
- 58.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 59.McDuff FK, Turner SD. Jailbreak: oncogene-induced senescence and its evasion. Cell Signal. 2011;23:6–13. doi: 10.1016/j.cellsig.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 60.Larsson LG. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin Cancer Biol. 2011;21:367–76. doi: 10.1016/j.semcancer.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 61.Raabe EH, Lim KS, Kim JM, Meeker A, Mao XG, Nikkhah G, Maciaczyk J, Kahlert U, Jain D, Bar E, Cohen KJ, Eberhart CG. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17:3590–9. doi: 10.1158/1078-0432.CCR-10-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacob K, Quang-Khuong DA, Jones DT, Witt H, Lambert S, Albrecht S, Witt O, Vezina C, Shirinian M, Faury D, Garami M, Hauser P, Klekner A, Bognar L, Farmer JP, Montes JL, Atkinson J, Hawkins C, Korshunov A, Collins VP, Pfister SM, Tabori U, Jabado N. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res. 2011;17:4650–60. doi: 10.1158/1078-0432.CCR-11-0127. [DOI] [PubMed] [Google Scholar]
- 63.Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein PE, MacCollin M, Cichowski K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10:459–72. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chong EY, Lam PY, Poon WS, Ng HK. Telomerase expression in gliomas including the nonastrocytic tumors. Hum Pathol. 1998;29:599–603. doi: 10.1016/s0046-8177(98)80009-9. [DOI] [PubMed] [Google Scholar]
- 65.Tabori U, Vukovic B, Zielenska M, Hawkins C, Braude I, Rutka J, Bouffet E, Squire J, Malkin D. The role of telomere maintenance in the spontaneous growth arrest of pediatric low-grade gliomas. Neoplasia. 2006;8:136–42. doi: 10.1593/neo.05715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tabori U, Dome JS. Telomere biology of pediatric cancer. Cancer Invest. 2007;25:197–208. doi: 10.1080/07357900701208683. [DOI] [PubMed] [Google Scholar]
- 67.Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E, Netto GJ, Epstein JI, Lotan TL, Westra WH, Shih Ie M, Iacobuzio-Donahue CA, Maitra A, Li QK, Eberhart CG, Taube JM, Rakheja D, Kurman RJ, Wu TC, Roden RB, Argani P, De Marzo AM, Terracciano L, Torbenson M, Meeker AK. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol. 2011;179:1608–15. doi: 10.1016/j.ajpath.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Slatter T, Gifford-Garner J, Wiles A, Tan X, Chen YJ, MacFarlane M, Sullivan M, Royds J, Hung N. Pilocytic astrocytomas have telomere-associated promyelocytic leukemia bodies without alternatively lengthened telomeres. Am J Pathol. 2010;177:2694–700. doi: 10.2353/ajpath.2010.100468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Listernick R, Charrow J, Greenwald MJ, Esterly NB. Optic gliomas in children with neurofibromatosis type 1. The Journal of pediatrics. 1989;114:788–92. doi: 10.1016/s0022-3476(89)80137-4. [DOI] [PubMed] [Google Scholar]
- 70.Rodriguez FJ, Giannini C, Asmann YW, Sharma MK, Perry A, Tibbetts KM, Jenkins RB, Scheithauer BW, Anant S, Jenkins S, Eberhart CG, Sarkaria JN, Gutmann DH. Gene expression profiling of NF-1-associated and sporadic pilocytic astrocytoma identifies aldehyde dehydrogenase 1 family member L1 (ALDH1L1) as an underexpressed candidate biomarker in aggressive subtypes. J Neuropathol Exp Neurol. 2008;67:1194–204. doi: 10.1097/NEN.0b013e31818fbe1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kluwe L, Hagel C, Tatagiba M, Thomas S, Stavrou D, Ostertag H, von Deimling A, Mautner VF. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. Journal of neuropathology and experimental neurology. 2001;60:917–20. doi: 10.1093/jnen/60.9.917. [DOI] [PubMed] [Google Scholar]
- 72.Wimmer K, Eckart M, Meyer-Puttlitz B, Fonatsch C, Pietsch T. Mutational and expression analysis of the NF1 gene argues against a role as tumor suppressor in sporadic pilocytic astrocytomas. Journal of neuropathology and experimental neurology. 2002;61:896–902. doi: 10.1093/jnen/61.10.896. [DOI] [PubMed] [Google Scholar]
- 73.Sharma MK, Mansur DB, Reifenberger G, Perry A, Leonard JR, Aldape KD, Albin MG, Emnett RJ, Loeser S, Watson MA, Nagarajan R, Gutmann DH. Distinct genetic signatures among pilocytic astrocytomas relate to their brain region origin. Cancer Res. 2007;67:890–900. doi: 10.1158/0008-5472.CAN-06-0973. [DOI] [PubMed] [Google Scholar]
- 74.Banerjee S, Gianino SM, Gao F, Christians U, Gutmann DH. Interpreting mammalian target of rapamycin and cell growth inhibition in a genetically engineered mouse model of Nf1-deficient astrocytes. Mol Cancer Ther. 2011;10:279–91. doi: 10.1158/1535-7163.MCT-10-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Warrington NM, Woerner BM, Daginakatte GC, Dasgupta B, Perry A, Gutmann DH, Rubin JB. Spatiotemporal differences in CXCL12 expression and cyclic AMP underlie the unique pattern of optic glioma growth in neurofibromatosis type 1. Cancer research. 2007;67:8588–95. doi: 10.1158/0008-5472.CAN-06-2220. [DOI] [PubMed] [Google Scholar]
- 76.Babcock JT, Quilliam LA. Rheb/mTOR activation and regulation in cancer: novel treatment strategies beyond rapamycin. Curr Drug Targets. 2011;12:1223–31. doi: 10.2174/138945011795906589. [DOI] [PubMed] [Google Scholar]
- 77.Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36:320–8. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J. 2012;441:1–21. doi: 10.1042/BJ20110892. [DOI] [PubMed] [Google Scholar]
- 79.Chan JA, Zhang H, Roberts PS, Jozwiak S, Wieslawa G, Lewin-Kowalik J, Kotulska K, Kwiatkowski DJ. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol. 2004;63:1236–42. doi: 10.1093/jnen/63.12.1236. [DOI] [PubMed] [Google Scholar]
- 80.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–11. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 81.Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005;65:2755–60. doi: 10.1158/0008-5472.CAN-04-4058. [DOI] [PubMed] [Google Scholar]
- 82.Lee da Y, Yeh TH, Emnett RJ, White CR, Gutmann DH. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev. 2010;24:2317–29. doi: 10.1101/gad.1957110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jentoft M, Giannini C, Cen L, Scheithauer BW, Hoesley B, Sarkaria JN, Abell-Aleff PC, Rodriguez EF, Li Y, Rodriguez FJ. Phenotypic variations in NF1-associated low grade astrocytomas: possible role for increased mTOR activation in a subset. Int J Clin Exp Pathol. 2010;4:43–57. [PMC free article] [PubMed] [Google Scholar]
- 84.Ellezam B, Theeler BJ, Luthra R, Adesina AM, Aldape KD, Gilbert MR. Recurrent PIK3CA mutations in rosette-forming glioneuronal tumor. Acta Neuropathol. 2012;123:285–7. doi: 10.1007/s00401-011-0886-z. [DOI] [PubMed] [Google Scholar]
- 85.Rickman DS, Bobek MP, Misek DE, Kuick R, Blaivas M, Kurnit DM, Taylor J, Hanash SM. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res. 2001;61:6885–91. [PubMed] [Google Scholar]
- 86.Colin C, Baeza N, Bartoli C, Fina F, Eudes N, Nanni I, Martin PM, Ouafik L, Figarella-Branger D. Identification of genes differentially expressed in glioblastoma versus pilocytic astrocytoma using Suppression Subtractive Hybridization. Oncogene. 2006;25:2818–26. doi: 10.1038/sj.onc.1209305. [DOI] [PubMed] [Google Scholar]
- 87.Hunter S, Young A, Olson J, Brat DJ, Bowers G, Wilcox JN, Jaye D, Mendrinos S, Neish A. Differential expression between pilocytic and anaplastic astrocytomas: identification of apolipoprotein D as a marker for low-grade, non-infiltrating primary CNS neoplasms. J Neuropathol Exp Neurol. 2002;61:275–81. doi: 10.1093/jnen/61.3.275. [DOI] [PubMed] [Google Scholar]
- 88.Rorive S, Maris C, Debeir O, Sandras F, Vidaud M, Bieche I, Salmon I, Decaestecker C. Exploring the distinctive biological characteristics of pilocytic and low-grade diffuse astrocytomas using microarray gene expression profiles. J Neuropathol Exp Neurol. 2006;65:794–807. doi: 10.1097/01.jnen.0000228203.12292.a1. [DOI] [PubMed] [Google Scholar]
- 89.Gutmann DH, Hedrick NM, Li J, Nagarajan R, Perry A, Watson MA. Comparative gene expression profile analysis of neurofibromatosis 1-associated and sporadic pilocytic astrocytomas. Cancer Res. 2002;62:2085–91. [PubMed] [Google Scholar]
- 90.Wong KK, Chang YM, Tsang YT, Perlaky L, Su J, Adesina A, Armstrong DL, Bhattacharjee M, Dauser R, Blaney SM, Chintagumpala M, Lau CC. Expression analysis of juvenile pilocytic astrocytomas by oligonucleotide microarray reveals two potential subgroups. Cancer Res. 2005;65:76–84. [PubMed] [Google Scholar]
- 91.Sharma MK, Watson MA, Lyman M, Perry A, Aldape KD, Deak F, Gutmann DH. Matrilin-2 expression distinguishes clinically relevant subsets of pilocytic astrocytoma. Neurology. 2006;66:127–30. doi: 10.1212/01.wnl.0000188667.66646.1c. [DOI] [PubMed] [Google Scholar]
- 92.Tchoghandjian A, Fernandez C, Colin C, El Ayachi I, Voutsinos-Porche B, Fina F, Scavarda D, Piercecchi-Marti MD, Intagliata D, Ouafik L, Fraslon-Vanhulle C, Figarella-Branger D. Pilocytic astrocytoma of the optic pathway: a tumour deriving from radial glia cells with a specific gene signature. Brain. 2009;132:1523–35. doi: 10.1093/brain/awp048. [DOI] [PubMed] [Google Scholar]
- 93.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–72. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ, Storm PB, Biegel JA. Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro Oncol. 2010;12:621–30. doi: 10.1093/neuonc/noq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dias-Santagata D, Lam Q, Vernovsky K, Vena N, Lennerz JK, Borger DR, Batchelor TT, Ligon KL, Iafrate AJ, Ligon AH, Louis DN, Santagata S. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One. 2011;6:e17948. doi: 10.1371/journal.pone.0017948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sharma SG, Gulley ML. BRAF mutation testing in colorectal cancer. Arch Pathol Lab Med. 2010;134:1225–8. doi: 10.5858/2009-0232-RS.1. [DOI] [PubMed] [Google Scholar]
- 97.Capper D, Preusser M, Habel A, Sahm F, Ackermann U, Schindler G, Pusch S, Mechtersheimer G, Zentgraf H, von Deimling A. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol. 2011;122:11–9. doi: 10.1007/s00401-011-0841-z. [DOI] [PubMed] [Google Scholar]
- 98.Setty P, Gessi M, Waha A, Hammes J, El-Maarri O, Pietsch T. Sensitive determination of BRAF copy number in clinical samples by pyrosequencing. Diagn Mol Pathol. 2011;20:148–57. doi: 10.1097/PDM.0b013e3182143817. [DOI] [PubMed] [Google Scholar]
- 99.Hegedus B, Banerjee D, Yeh TH, Rothermich S, Perry A, Rubin JB, Garbow JR, Gutmann DH. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res. 2008;68:1520–8. doi: 10.1158/0008-5472.CAN-07-5916. [DOI] [PubMed] [Google Scholar]
- 100.Bajenaru ML, Garbow JR, Perry A, Hernandez MR, Gutmann DH. Natural history of neurofibromatosis 1-associated optic nerve glioma in mice. Ann Neurol. 2005;57:119–27. doi: 10.1002/ana.20337. [DOI] [PubMed] [Google Scholar]
- 101.Gronych J, Korshunov A, Bageritz J, Milde T, Jugold M, Hambardzumyan D, Remke M, Hartmann C, Witt H, Jones DT, Witt O, Heiland S, Bendszus M, Holland EC, Pfister S, Lichter P. An activated mutant BRAF kinase domain is sufficient to induce pilocytic astrocytoma in mice. J Clin Invest. 2011;121:1344–8. doi: 10.1172/JCI44656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bax DA, Little SE, Gaspar N, Perryman L, Marshall L, Viana-Pereira M, Jones TA, Williams RD, Grigoriadis A, Vassal G, Workman P, Sheer D, Reis RM, Pearson AD, Hargrave D, Jones C. Molecular and phenotypic characterisation of paediatric glioma cell lines as models for preclinical drug development. PLoS One. 2009;4:e5209. doi: 10.1371/journal.pone.0005209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shahabi V, Whitney G, Hamid O, Schmidt H, Chasalow S, Alaparthy S, Jackson J. Assessment of association between BRAF-V600E mutation status in melanomas and clinical response to ipilimumab. Cancer Immunology, Immunotherapy. :1–5. doi: 10.1007/s00262-012-1227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AMM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. New England Journal of Medicine. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopoulos A, Thomas G, Crone KR. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Annals of Neurology. 2006;59:490–98. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- 106.Packer RJ, Yalon M, Rood BR, Chao M, Miller MM, McCowage G, Vezina G. Phase I/II Study of Tarceva/Rapamcin for Recurrent Pediatric Low-Grade Gliomas (LGG) Neuro-Oncology. 2010;12:ii20. [Google Scholar]