

Abstract
The molecular mechanism to regulate energy balance is not completely understood. Here we observed that Egr-1 expression in white adipose tissue (WAT) was highly correlated with dietary-induced obesity and insulin resistance both in mice and humans. Egr-1 null mice were protected from diet-induced obesity and obesity-associated pathologies such as fatty liver, insulin resistance, hyperlipidemia and hyperinsulinemia. This phenotype can be largely explained by the increase of energy expenditure in Egr-1 null mice. Characterization of these mice revealed that the expression of FOXC2 and its target genes were significantly elevated in white adipose tissues, leading to WAT energy expenditure instead of energy storage. Altogether, these studies suggest an important role for Egr-1, which, by repressing FOXC2 expression, promotes energy storage in WAT and favored the development of obesity under high energy intake.
There is a wealth of evidence that energy regulatory and immune pathways are closely linked and interdependent. Many hormones, cytokines, signaling proteins, transcription factors, and bioactive lipids can function in both metabolic and immune regulation1. Recent studies also demonstrate that obesity is an inflammatory condition in which chronic activation of the innate immune system ultimately causes progressive impairment of glucose tolerance2. However, the exact chain of molecular events linking overnutrition, activation of the innate immune system, and impairment of insulin signaling in peripheral tissues remain incompletely understood.
Early growth response-1 (Egr-1) is an inflammatory transcriptional factor. Egr-1 gene is located in “Cytokine Cluster” on 5q31 (cen--(IL4/IL5/IRF1/IL3/CSF2)--TCF7--IL9--EGR1--CD14--FGFA--SPARC--ADRA1—ter). It regulates a large set of inflammatory genes, including immune effector genes such as IL-2 (ref. 3) and TNFα4, cell surface molecules such as IL-2 receptor5, Fas/CD95 (ref. 6) and ICAM-1 (ref. 7), and FasL8. In addition, Egr-1 also regulates growth factors, such as insulin like growth factor-II, basic fibroblast growth factor9, epidermal growth factor receptor, platelet-derived growth factor (PDGF)10 and transforming growth factor (TGF)11, and cell cycle regulators including the retinoblastoma susceptibility gene Rb12, cyclin D1 (ref. 13), as well as hormones such as the luteinizing hormone β (LH-β)14,15. Therefore, Egr-1 not only regulates innate and adaptive immune response, but also plays important roles in cell proliferation, differentiation and apoptosis. Egr-1 is also involved in metabolic diseases. In insulin resistant adipocytes, Egr-1 expression is still induced by insulin16; and thus, Egr-1 mRNA level is highly increased in adipocytes from diabetic animals17. Indeed, Egr-1 can decrease adipocyte insulin sensitivity through tilting PI3K/Akt versus MAPK signal balance18. These studies suggest that Egr-1 may be also involved in metabolic disorders. In contrast to Egr-1, another transcription factor FOXC2 was decreased in type 2 diabetes patients19. Overexpression of FOXC2 in adipocytes leads to a lean and insulin-sensitive phenotype by increasing the sensitivity of the β-adrenergic-cAMP-PKA signaling pathway20.
In this study, we observed that Egr-1 expression in white adipose tissue (WAT) was highly associated with dietary-induced obesity and insulin resistance both in mice and humans. Egr-1 null mice were protected from diet-induced obesity and obesity-associated pathologies such as fatty liver, insulin resistance, hyperlipidemia and hyperinsulinemia. This phenotype can be largely explained by increase of energy expenditure in Egr-1 null mice. Characterization of these mice revealed that the expression of FOXC2 and its target genes was significantly elevated in white adipose tissues, which led WAT to expend instead to store energy. Taken together, these studies suggest an important role for Egr-1, which, by repressing FOXC2 expression, promotes energy storage in WAT and favors the development of obesity under high energy intake.
Results
Egr-1 expression in white adipose tissue is associated with obesity and insulin resistance
As a stress response gene, Egr-1 is often rapidly and transiently activated by a variety of signals, including hypoxia, cytokines, growth factors and hormones21. Dietary-induced obesity is often associated with local hypoxia and inflammatory response in adipose tissues. To examine whether Egr-1 expression in adipose is associated with BMI and insulin resistance, 48 Caucasian non-diabetic men underwent euglycemic-hyperinsulinemic clamp for insulin sensitivity and biopsy of subcutaneous abdominal fat for real-time, quantitative PCR (QPCR). As shown in Fig. 1a&b, Egr-1 mRNA level was highly positively correlated with BMI, but negatively correlated with glucose disposal rate. In another compared experiment, 22 lean persons (BMI = 25 ± 1.6) and 7 obese persons (BMI = 55 ± 8.7) underwent biopsy of subcutaneous abdominal fat for QPCR. As shown in Fig. 1c, Egr-1 mRNA level was 80 folds higher in obese group than that in lean group. To further confirm that dietary obesity induces Egr-1 expression in adipose tissues, we tested the effect of HF diet feeding on the expression of Egr-1 in C57BL/6J mice. As shown in Fig. S1a, the body weight was significantly increased after 20 weeks of HF diet feeding. In the meantime, the fast glucose started to increase at 12 weeks and further increased significantly at 20 weeks of HF diet feeding (Fig. S1b). The level of Egr-1 mRNA in WAT was markedly increased after 12 weeks of high-fat feeding and further increased at 20 weeks of HF diet feeding, which was closely associated with dietary-induced obesity and insulin resistance (Fig. 1d). In contrast, no significant alteration in expression level of Egr-1 mRNA was observed in other tissues that also contribute to energy homeostasis including hypothalamus, liver, skeletal muscle, and adrenal upon high-fat feeding (data not shown). In ob/ob mice, Egr-1 mRNA level was significantly increased in visceral and subcutaneous WAT, but not in brown adipose, compared with control mice (Fig. 1e). Taken together, these results suggest that Egr-1 expression in WAT is highly associated with dietary-induced obesity and insulin resistance.
Figure 1. Egr-1 mRNA levels in WAT are associated with obesity and insulin resistance.

(a&b) Egr-1 mRNA levels in WAT from 48 Caucasian non-diabetic men were measured by QPCR. Glucose disposal rate was measured by hyperinsulinemic-euglycemic clamps and the data on Rd were computed in mg/min/kg of lean body mass for those lean persons (n = 48, BMI, 24.0 ± 0.75). After log transformation, linear regression analysis revealed a strong correlation (r2 = 0.6838, p < 0.0001) between Egr-1 mRNA level in WAT and body mass index of the donor subject (a) and a weak negatively correlation (r2 = 0.2423, P = 0.0023) between Egr-1 mRNA level in WAT and Glucose disposal rate (mg/min/kg) (b). (c) Egr-1 mRNA levels in WAT from lean (n = 22, BMI, 24.0 ± 0.75) and obese (n = 7, BMI, 52 ± 3.1) humans were measured by QPCR. (d). C57BL/6J mice were purchased from Jackson laboratories and divided into two groups fed with chow-diet or high-fat diet, respectively. Egr-1 mRNA level was examined by QPCR both in chow-fed or high-fat fed mice after 4, 12 and 20 weeks of feeding (n = 5). Values were normalized by 18S and fold changes to 4-week, chow-fed group were represented. (e) Egr-1 mRNA levels in adipose tissues from ob/ob and its control mice. Male, 12-week-old mice were sacrificed and RNA was isolated from adipose tissues as indicated. QPCR was performed to measure Egr-1 mRNA levels using the primers as shown in Table S1. Data represent mean ± SD, n = 5.
Loss of Egr-1 protects against diet-induced obesity
To define the functional relevance of dietary obesity-induced adipocyte Egr-1 upregulation in mice, we challenged 9- to 10-week-old wild-type and Egr-1 null mice with a high-fat (HF) diet (60% calories from fat) for 15 weeks. The Egr-1−/− mice showed blunted weight gain in response to this dietary challenge, which was evident after the first 5 weeks of feeding (Fig. 2a). After 15 weeks, the wild-type mice showed 105 ± 13% weight gain, but the Egr-1 null mice gained only one-third as much, 38 ± 20%. The weight gain of the chow-fed Egr-1−/− (26%) mice was also lower than that of wild-type (38%) mice, but the difference was not significant. This phenotype was observed in both male and female mice and in both mid-term feeding (12 weeks) and long-term feeding (20 weeks) (Fig. S2). The body-weight difference was largely due to a decrease in fat mass. Nuclear magnetic resonance (NMR) analysis showed that the fat mass ratio in Egr-1−/− mice was 61% lower than that in wild-type mice under HF diet feeding (Fig. 2b). The fat ratio of chow-fed Egr-1−/− (13%) mice was also lower than that of wild-type (23%) mice, but the difference was not significant. Adipocytes were enlarged in the white fat of the HF fed wild-type mice but were smaller in Egr-1 null mice (Fig. 2c). In brown fat, HF diet induced more lipid accumulation in wild-type mice, but not in Egr-1 null mice. The HF diet also induced fatty liver in the wild-type mice, but the livers of the HF fed Egr-1−/− animals contained almost no visible lipid droplets, which were confirmed by marked differences in Oil Red O staining (Fig. 2c). Consistent with this, hepatic triglyceride and cholesterol contents were significantly lower in Egr-1−/− mice (Fig. 2d&e).
Figure 2. Resistance of Egr-1 null mice to diet-induced obesity.

(a) Growth curves of wild-type and Egr-1−/− mice (n = 3–6) fed chow or high-fat diet for 15 weeks. (b) Body fat mass was evaluated by NMR scanning (n = 3–6) and the ratio of fat mass to body weight was shown. (c) White fat and brown fat pads were collected, fixed, and subjected to hematoxylin and eosin (H&E) staining (n = 2/group). A representative image is shown (10× magnification); Livers were collected, fixed, and subjected to Oil Red O staining (10× magnification). (d&e) Hepatic triglycerides (d) and Cholesterol (e) were measured (n = 3–6). All data are expressed as the mean ± SD; *P < 0.05; **P < 0.01.
Loss of Egr-1 protects from diet-induced insulin resistance and hyperlipidemia
To further characterize the obesity resistant phenotype in Egr-1−/− mice fed a HF diet, carbohydrate and lipid metabolism were analyzed. As expected, the leaner Egr-1−/− mice showed greater insulin sensitivity. Their plasma glucose levels were not significantly different from those in wild-type mice on either chow or HF diet. However, plasma insulin levels were 9% lower in the chow-fed Egr-1−/− mice compared to the wild-type mice, and were markedly lower (65%) in the HF diet-fed Egr-1−/− mice than in HF-fed wild-type mice (Fig. 3a). As an indicator of body fat, consistent with their obesity, leptin level in wild-type mice was increased substantially (3-fold) by HF diet feeding. In comparison, basal level of leptin was lower in Egr-1−/− mice than in wild-type mice and was not increased significantly by HF diet feeding (Fig. 3a). We next performed glucose and insulin tolerance tests. On chow-diet, both wild-type and Egr-1 null mice showed normal response after glucose or insulin perfusion. However, the HF-fed Egr-1−/− mice showed relatively normal responses, while the HF-fed wild-type mice showed insulin intolerance (Fig. 3b) and glucose intolerance (Fig. 3c). As expected, HF diet feeding induced hyperlipidemia in wild-type mice, while this was much attenuated in HF-fed Egr-1 null mice. Lipoprotein analysis revealed that on HF diet, Egr-1−/− mice had substantially lower triglycerides and cholesterol in their VLDL and LDL/HDL fractions respectively compared with wild-type mice (Fig. 3d). These differences were not seen in the mice on chow diet (data not shown). Total plasma triglyceride and free fatty acid levels were not different in the wild-type compared with Egr-1 null mice, indicating that the reduced fat weight was not secondary to decreased lipid availability. However, total plasma cholesterol levels were significantly decreased in Egr-1−/− mice compared with wild-type littermates (Fig. 3e).
Figure 3. Improvement of insulin sensitivity and lipid profiles in Egr-1 null mice.

(a) Plasma glucose, insulin and leptin levels in chow-fed or high fat fed, overnight fasted mice (n = 6). (b) Plasma glucose levels after an acute injection of insulin (0.75 UI/kg) in chow-fed or high-fat fed, overnight fasted mice (n = 5–8). (c) Plasma glucose levels after a glucose load (2 g/kg) in chow fed or high-fat fed, overnight fasted mice (n = 5). (d) Measurements of triglycerides and cholesterol in fractionated lipoproteins were taken from wild-type and Egr-1−/− mice after high-fat feeding (n = 5). (e) Plasma levels of triglycerides, cholesterol and free fatty acids in chow fed or high fat fed, overnight fasted mice (n = 6).
Loss of Egr-1 increases energy expenditure
To examine whether obesity resistance in Egr-1−/− mice was due to alterations in feeding behavior, food intake and fat absorption were measured. Despite of their inability to gain weight on HF diet, Egr-1−/− mice exhibited a food intake per body weight that was comparable to that of wild-type mice fed the same diet (Fig. 4a). When integrated into a formula that takes into consideration food intake, stool output, and lipid content, no significant changes in fat absorption were observed in Egr-1−/− mice compared to wild-type mice both on chow and HF diet (Fig. 4b). These results indicated that resistance to obesity of Egr-1 null mice was not due to decreased energy intake. Thus, their resistance to diet induced obesity should be a consequence of increased in energy expenditure. This was confirmed by both direct and indirect approaches, including analysis of oxygen consumption, CO2 generation and body temperature. The body temperature was higher in the HF fed Egr-1 null mice compared to that in HF fed wild-type mice (Fig. 4c). HF-fed Egr-1−/− mice showed significantly higher O2 consumption (Fig. 4d and Fig. S3a)) and CO2 generation than control wild-type littermates over 24 h (Fig. 4e and Fig. S3b). We also monitored the physical activity during a three-day period and did not observe any difference in the movement rate between Egr-1−/− and wild-type mice (Fig. 4f and Fig. S3c)). Together, these results indicated that resistance to obesity of Egr-1−/− mice was due to increased energy expenditure by increasing metabolic rate.
Figure 4. Increased energy expenditure in Egr-1−/− mice.

(a) Food intake of wild-type and Egr-1−/− mice (n = 6) at first week and ninth week of feeding. (b) Fat absorption of wild-type and Egr-1−/− mice (n = 6) measured on week 9 of feeding. Fat absorption = ([food intake × food lipid content]-[stool output × stool lipid content])/(food intake × food lipid content) × 100. (c) Rectal temperature was measured at week 9 of feeding in chow-fed or high-fat fed mice. (d) Oxygen consumption (VO2) and (e) carbon dioxide production (VCO2) were measured by metabolic cage system (CLAMS, Columbia Instrument) during a period of three days after 16 week of high-fat feeding (n = 5). Values from nighttime and daytime were presented. Values are expressed as the mean ± SD. (*, P < 0.001). (f) Physical activity was measured with CLAMS. The average movement counts during the nighttime and daytime were presented.
Loss of Egr-1 increases FOXC2 expression in white adipose tissues
It was interestingly to note that the phenotype of Egr-1−/− mice resistant to obesity is very similar to that of aP2-FOXC2 transgenic mice20. Thus, we measured FOXC2 mRNA levels in liver, muscle, white and brown fats from wild-type and Egr-1 null mice. At first, we confirmed the Egr-1 expression in adipose from wild-type and Egr-1 null mice by Northern blot (Fig. 5a). Interestingly, FOXC2 was increased by 2 folds in white fat of Egr-1 null mice on chow diet and further increased by 4 folds by HF diet feeding. FOXC2 was not expressed in liver, muscle and brown fat (Fig. 5b). To further confirm the expression changes of FOXC2 in Egr-1−/− white fat, we measured FOXC2 protein levels by Western blotting. As shown in Fig. 5c, FOXC2 protein is expressed in white fat, but not in liver and muscle. Consistent with its mRNA changes, FOXC2 protein was also increased in the white fat of Egr-1−/− mice both on chow diet and HF diet feeding.
Figure 5. FOXC2 expression was increased in WAT from Egr-1−/− mice.
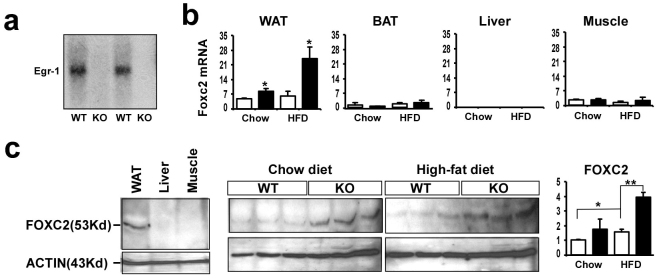
(a) Expression of Egr-1 in WAT from high-fat fed wild-type and Egr-1 null mice was examined by Northern blotting. (b) Expression of FOXC2 in WAT, BAT, liver and muscle from chow-fed or high-fat fed mice was analyzed by QPCR. Values are normalized by 18S. (c) Expression of FOXC2 protein in WAT was analyzed by Western blotting. Left: a 53-Kd band was detected only in WAT, but not in liver and muscle by FOXC2 antibody; Middle: 3 samples of WAT from each group were run on SDS-PAGE and detected by FOXC2 antibody; Right: Fold changes of FOXC2 protein in WAT from chow-fed or high-fat fed mice were determined by densitometry and normalized to β-actin.
Loss of Egr-1 increases UCP1 expression in white adipose tissues
As reported, WAT in mice with an overexpression of FOXC2 acquired certain BAT-like properties with increased expression of PGC1, UCP1 and cAMP pathway proteins such as β1-adrenoceptor (β1-AR)20. So we have measured the expression of β1-AR, PGC-1α, PGC1β and UCP1 in WAT from both wild-type and Egr-1 null mice under chow and HF diet feeding by QPCR. As showed in Fig. 6a, the expression of β1-AR was significantly increased in Egr-1 null WAT compared that in wild-type WAT under both chow and HF diet feeding. Though PGC1α and PGC1β were not changed in WAT under chow diet feeding, they were increased in Egr-1 null WAT under HF diet feeding. UCP1 was not expressed in WAT from both wild-type and Egr-1 null mice under chow diet, but UCP1 expression was higher in Egr-1 null WAT than that in wild-type WAT under HF diet feeding, which was further confirmed by Western blot (Fig. 6b&c). These results suggest the WAT in Egr-1 null mice gains some BAT-like features under HF diet feeding.
Figure 6. Egr-1 null WAT expressed some BAT-specific genes under HF diet feeding.

(a) Expression of β1-AR, PGC1α, PGC1β and UCP1 in WAT from both wild-type (WT) and Egr-1 null mice (KO) under both chow or HF diet feeding was analyzed by QPCR. Values are normalized by 18S. (b) Expression of UCP1 protein in WAT from both wild-type (WT) and Egr-1 null mice (KO) under HF diet feeding was analyzed by Western blotting with UCP1 antibody (Sigma). (c) Fold changes of UCP1 protein in WAT high-fat fed mice were determined by densitometry and normalized to β-actin.
Loss of Egr-1 decreases inflammatory cytokine production in white adipose tissues
It is well documented that obesity is an inflammatory condition leading to chronic activation of the innate immune system and Egr-1 is an inflammatory transcription factor controlling many cytokines expression including TNFα, PAI-1 etc22. Therefore, we measured the plasma levels of these cytokines as well as their mRNA levels. As expected, the production of TNFα was increased after HF diet feeding in wild-type mice, but not in Egr-1−/− mice (Fig. 7a). PAI-1 level was also increased in wild-type mice after HF diet feeding. The PAI-1 level was lower in HF diet-fed Egr-1 null mice, but no statistical significance. The production of adiponectin was not changed between two genotypes under both chow and HF diet feeding. Consistent with this, the TNFα mRNA level was also significantly increased in wild-type WAT after HF diet feeding, but not in Egr-1−/− WAT (Fig. 7b).
Figure 7. Alteration of Adipocytokines in Egr-1−/− mice.

(a) Plasma level of TNFα was measured by an ELISA kit from eBiosense; Plasma levels of PAI-1 and adiponectin were measured by a luminex kit from LINCO (n = 3–6). (b) mRNA levels of TNFα, PAI-1 and adiponectin in WAT from chow-fed or high-fat fed mice were analyzed by QPCR. Values are normalized by 18S rRNA.
Discussion
Through the story of evolution, animals and humans have developed potent and redundant mechanisms that promote the accumulation of energy in fat tissue during periods of “feast”, thus enabling survival during periods of “famine”23. As a consequence, obesity has reached epidemic proportions in the current “obesigenic” environment of readily available high-energy foods and little need for physical activity24. Understanding the regulatory pathways that govern energy storage (i.e., fat accumulation) versus energy expenditure (i.e., fat oxidation) is a key to understanding the diseases of obesity and its associated metabolic syndrome. Here we demonstrate a key role of dietary obesity-induced Egr-1 in adipocytes in the preservation and progression of obesity and obesity-associated pathologies, such as fatty liver, hyperlipidemia, and insulin resistance. Our findings support a model wherein nutrition overload leads to significant expansion of adipose tissue, which further induces Egr-1 expression probably by creating a stress and hypoxic environment. Egr-1 expression not only induces the synthesis and release of adipokines, such as TNFα, to promote insulin resistance, but also suppresses the FOXC2-cAMP-PKA metabolic regulatory system to facilitate energy storage and promote the development of obesity and obesity-associated pathologies (Fig. 8).
Figure 8. Schematic representation of Egr-1 mediated obesity model.
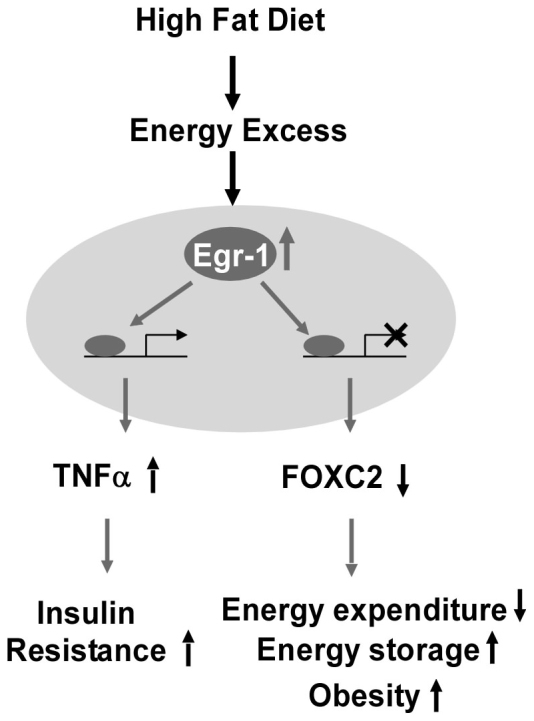
Nutrition overload leads to significant expansion of adipose tissue, which further induces Egr-1 expression probably by creating a stress and hypoxic environment. Egr-1 expression not only induces the synthesis and release of adipokines, such as TNFα, to promote insulin resistance, but also suppresses the FOXC2-cAMP-PKA metabolic regulatory system to facilitate energy storage and promote the development of obesity and obesity-associated pathologies.
As an early response gene, Egr-1 is often rapidly and transiently activated by a variety of signals including stress, growth factors, cytokines and hormones. And as a transcription factor, Egr-1 is able to regulate many genes expression and thus is involved in the regulation of cell growth, differentiation and apoptosis. Here we found sustained expression of Egr-1 in WAT under obese and diabetic conditions both in mice and human. The sustained expression of Egr-1 may be caused by nutrition overload-created stress and hypoxic environment in WAT or by obesity-associated hyperinsulinemia. It was previously reported that Egr-1 remains insulin sensitive in insulin-resistant adipocytes16. We further found that the sustained expression of Egr-1 in adipocytes facilitated energy storage and thus promoted the development of obesity and obesity-associated pathologies, so that loss of Egr-1 led to brown adipose-like change of WAT (Fig. 6), leading to energy expenditure over energy storage and thus protected mice from nutrition overload-induced obesity and obesity-associated pathologies. It is interesting to note that dietary obesity-induced Hif1α in adipocytes also restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system25, suggesting there are some redundant mechanisms to promote energy storage under nutrition overload status. To further examine the specific effect of Egr-1 overexpression in adipocytes, we have generated an adipocyte-specific Egr-1 transgenic mouse line by using aP2 promoter (Fig. S4). Interestingly, forced expression of Egr-1 in adipocytes without nutrition overload did not cause the changes of weight gain and insulin sensitivity, suggesting that Egr-1 participates in the management of energy only under chronic nutrition overload-induced stress.
Mechanistically, we found that the Egr-1-dependent effects on adipocyte lipid catabolism were mediated through regulation of the FOXC2-cAMP-PKA pathway. FOXC2 is a member of forkhead transcription factors and normally expressed only in adipose tissue. FOXC2 promoter polymorphism and abnormal expression had been shown associated with obesity and insulin resistance26,27,28,29. Overexpression of FOXC2 in adipose tissue causes a profound gene expression change including β-adrenergic receptors, PPARs and PGC-1, and BAT-specific gene UCP1 in WAT depot, which drives WAT depot to acquire a brown fat-like histology and to dissipate energy rather than to store energy20,30,31. To address the molecular mechanisms underlying the phenotype of Egr-1−/− mice resistant to diet-induced obesity, we found a significant increase in the expression of FOXC2 in WAT of Egr-1−/− mice on both chow diet and HF diet. Consistently, Egr-1 null WAT expressed some BAT-specific genes such as UCP1 and PGC1 under HF diet feeding. These results suggest that Egr-1 may function as a repressor of FOXC2 gene expression and suppresse FOXC2-mediated energy expenditure program in WAT to facilitate energy storage in WAT under high caloric intake. Further investigation to uncover the mechanisms by which Egr-1 regulates FOXC2 expression will help to understand the regulatory networks underlying obesity and obesity-associated pathologies. A recent report showed that Egr-1 promotes insulin resistance under chronic high-insulin-induced stress by controlling PI3K/Akt and MAPK signal balance via direct induction of the gene expression of PTEN and GGPPS in adipocytes18, suggesting that Egr-1 may regulate glucose and lipid metabolisms through multiple mechanisms.
As the body's largest endocrine organ and one of the most important insulin targeting tissues, adipose tissue has a critical role in insulin sensitivity and glucose homeostasis32,33. Although we cannot at present fully exclude a contribution of tissues beyond WAT to the metabolic alterations in Egr-1−/− mice, several arguments support the concept that WAT play a predominant role. First, thermogenic effectors such as PGC-1 and UCP-1 were increased in WAT but not in BAT and skeletal muscle. Second, the absence of major changes in non-adipose-derived endocrine modulators of adaptive thermogenesis and fat accumulation suggests that these metabolic alterations were not due to neuroendocrine defects. Finally, ingestion of a chronic high-fat diet induced changes in Egr-1 expression only in WAT, but not in other organs involved in metabolism (hypothalamus, adrenal and liver). A recent study also supported this concept18. Yu et al. inhibited Egr-1 transcriptional activity in diabetic db/db mice by injecting adenovirus carrying dominant-negative Egr-1 (dnEgr-1) into epididymal fat pads, which is the predominant fat tissue in mice intra-abdominal cavity. They found that expression of dnEgr-1 epididymal fat was able to significantly improve glucose tolerance and increase insulin sensitivity in db/db mice18.
High-fat feeding causes adipose expansion not only by lipid accumulation but also by adipogenesis. As reported, Egr-2, another member in Egr family, is an important regulator of adipogenesis34. Boyle et al. reported that Egr-1 and Egr-2 exert opposing influence on adipocyte differentiation35. We found that Egr-1, -2 and -3 were all rapidly induced and transiently expressed at early stage during 3T3-L1 adipogenesis. The transient expression of Egr-1 was required for 3T3-L1 adipogenesis as loss of Egr-1 decreased adipogenesis (Fig. S5). So the reduction of adipogenesis in Egr-1 null mice may also contribute to its lean phenotype.
In conclusion, our work identified Egr-1 as a central regulator of adipocyte energy storage. Egr-1 mediates these effects, at least in part, by interfering with the FOXC2-cAMP-PKA pathway, thereby creating a metabolic state permissive for the development of obesity in the face of nutrient overload. Since adipose Egr-1 accumulation in human obese subjects correlates with obesity and insulin resistance, intervention of Egr-1 activity may represent a therapeutic approach for obesity in humans.
Methods
Human study
Volunteers were recruited through public advertisement and were screened for hematological and blood chemistry abnormalities. The study was approved by the Institutional Review Board of the University of Texas Southwestern Medical Center at Dallas. All subjects signed a written informed consent. Subjects with diabetes mellitus and other endocrine disorders, coronary heart disease, and liver function test abnormalities, and those receiving any form of therapies, were excluded from the study. At the time of enrollment, each volunteer was administered a health history questionnaire. Height, weight, and blood pressure measurements were taken on all of the subjects. Hyperinsulinemic-euglycemic clamps was performed as described previously36. The rate of glucose disposal (Rd) was calculated by subtracting the urinary glucose excretion from the Ra and using space correction. The data on Rd were computed in mg/min/kg of lean body mass. Samples of subcutaneous adipose tissue are taken from both the abdomen and right buttock under local anesthesia using a biopsy needle. About 140 mg of subcutaneous adipose tissue is taken from each site and QPCR was performed as described below.
Animal experiments
The generation of the Egr-1 null mouse line has been described previously15. Egr-1+/− breeding pairs was kindly provided by Dr. David Pinsky. Egr-1 null mice are on mixed C57BL/6/129/Sv backgrounds and have been backcrossed to C57BL/6J background more than 10 generations. Age- and sex-matched groups of 8- to 9-week-old wild-type and Egr-1−/− siblings derived from heterozygous intercrosses of Egr-1+/− were used in the experiments. C57BL/6J mice used in the chronic high-fat feeding study were purchased from Jackson Laboratories (Bar Harbor, Maine, USA). All mice were housed in a temperature-controlled environment with 12 hr light/dark cycles. For high-fat diet experiments, one group of mice were fed a chow diet (D12450B, Research Diets, New Brunswick, NJ) containing 10 kcal% fat and the other group were fed a high-fat diet (D12492) containing 60 kcal% fat for 12 to 20 weeks. Body weight was recorded weekly, and food intake and fast blood glucose were measured biweekly. Total body fat mass was analyzed by NMR using the Minispec mq spectrometer (Bruker). Rectal temperature was measured using Thermalert (Physitemp, Clifton, NJ, USA). At the end of the feeding period, mice were anesthetized with halothane and exsanguinated via the ascending vena cava prior to organ harvest. Blood was kept on ice in heparin-coated tubes (Microvette 500 LH; Sarstedt, Inc.) and centrifuged (1500 × g for 15 min at 4°C), and the plasma was stored at −20°C until analysis. Tissues were harvested for analyses as described below. All experiments were approved by the Institutional Animal Care and Research Advisory Committee at the University of Texas Southwestern Medical Center.
Lipid absorption
In week 10 of high-fat diet study, stools were collected from individually housed mice over 72 h. Lipid content of diets and stools was determined gravimetrically and used to calculate the fraction of consumed lipid that was absorbed as described in Figure 1B37.
Liver triglycerides and cholesterol
Cholesterol was extracted from saponified liver (0.3 g) with petroleum ether and quantitated by gas chromatography using stigmasterol as an internal standard. Other lipids were extracted from liver (0.2 g) in chloroform:methanol (2∶1, v/v). Extracts were then washed once in 50 mM NaCl and twice in 0.36 M CaCl2/methanol. The organic phase was separated and brought up to 5 ml with chloroform. Ten microliters of chloroform:Triton X-114 (1∶1, v/v) were added to duplicate 100 μl aliquots of each extract and standards (Sigma Diagnostics), which were then air dried overnight. Colorimetric enzymatic assays were performed using 1 ml triglyceride reagent (ThermoTrace). After 20 min, optical densities of each sample were read at 520 nm and compared to prepared standards.
Plasma analyses and glucose and insulin tolerance
For glucose tolerance, mice were fasted overnight before receiving an intraperitoneal injection of 20% D-glucose (Sigma) (2 g/kg body weight). At 0, 15, 30, 60, 90, and 120 min after injection, 5–10 μl of blood was drawn from the tail and blood glucose levels assayed using an Elite XL Glucometer (Bayer). For insulin tolerance, mice were fasted for 6 h before receiving an intraperitoneal injection of insulin 10 U/ml (sigma) (0.75 U/kg body weight). Then the blood glucose levels were assayed following the procedures above. Different enzymatic assays were used for each analyte, including total cholesterol (Roche Diagnostics), plasma triglycerides (ThermoTrace), free fatty acids (Roche). Plasma lipoprotein levels were analyzed by fast protein liquid chromatography using two serial Superose HR6 columns, followed by enzymatic analysis of their cholesterol and triglyceride contents using the kits described above. Plasma leptin, insulin, adiponectin, tPAI-1 were determined by using Lincoplex Luminex kit (LINCO Research, St. Charles, Missouri, USA). TNFα level was determined using mouse TNFα ELISA Kit (eBioscience, Boston, MA, USA).
Metabolic rate and movement measurements
Metabolic monitoring was performed using a Comprehensive Lab Animal Monitoring System (CLAMS, Columbia Instruments) that simultaneously measures whole-body O2 consumption and physical movements for 16 mice. Mice were acclimated in the monitoring chambers for 2 days before the experiment to minimize the changes in housing environments. Data was collected every 48 min for each mouse over a period of 3 days. Metabolic rate and physical activity were averaged for the whole study period with the exception of the first five data points that tend to be influenced by animal handling at the beginning of studies.
Real-time PCR analysis
Total RNA was extracted from tissues using RNA STAT-60 (Tel-Test, Inc.), treated with DNase I (RNase-free, Roche Molecular Biochemicals), and reversely transcribed into cDNA with random hexamers using the SuperScript II First-Strand Synthesis System (Invitrogen). Primers for each mRNA were designed using Primer Express Software (PerkinElmer Life Sciences) based on sequence data from GenBankTM (Table S1 and ref. 38) and were validated as described39. Real-time PCR reactions contained 10–25 ng cDNA, 150 nM of each primer, and 5 μl 2X-Jump Start SYBR Green PCR Master Mix (Sigma) in 10 μl total volume. PCR reactions were performed in triplicate using an Applied Biosystems Prism 7900 HT instrument. Relative mRNA levels were calculated using either the comparative CT or standard curve methods normalized to cyclophilin mRNA or 18S RNA, respectively.
Western blotting and northern blotting
Proteins were isolated by the kit from Perkin Elmar and resolved on a 12% SDS-polyacrylamide gel, and electrotransferred to a PVDF membrane (Amersham). The membrane was then hybridized with rabbit anti-mouse FOXC2 (Chemicon, Temecula, CA), UCP-1 (Sigma) at a 1∶1000 dilution, followed by secondary antibody incubation at a 1∶1000 dilution. FOXC2 and UCP-1-specific bands were detected by chemiluminescence (ECL kit, Amersham). Northern blotting was performed as described previously40.
Histological procedures
Tissues were dissected and fixed in 4% paraformaldehyde overnight and rinsed with phosphate-buffered saline. White and brown fat were subsequently dehydrated and embedded in plastic (JB-4, Electron Microscopy Sciences) and sectioned at 1.5 μm for H&E staining. For oil red O staining, Liver was incubated in 10% sucrose, paraffin embedded, and sectioned at 4 μm for Oil Red O staining. Sections were examined under a bright-field microscope at 100× magnification.
Statistical analyses
Values are expressed as Mean ± standard error (SD). Significant differences between mean values were evaluated using two-tailed, unpaired Student's t test (when two groups were analyzed) or one-way ANOVA followed by Student Newman-Keuls test (for three or more groups). P < 0.05 was considered significant.
Author Contributions
M.F. and J.Z. designed, performed the experiments and wrote the manuscript; Y.Z. and T.S. helped with the animal experiments; G.F. and S.H. did the UCP1 Western blot; M.C. and N.A. provided the human data; D.F., H.-B. X. and Y.E.C. provided helpful discussion and revised the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Drs. Jian Liang, Angie Bookout, Vicky Lin, Ms. Norma Anderson and Donna Floyd for technical helps on animal experiments. We also thank Drs. David J. Mangelsdorf and Steven A. Kliewer for helpful discussion. M.F. is a recipient of a Junior Faculty Award from American Diabetes Association (ADA). This work was supported by ADA (7-03-JF-18 to M.F.) and National Institutes Health Grants (HL068878 and HL03676 to Y.E.C., DK072158 to N.A. & M.C.).
References
- Wellen K. E. & Hotamisligil G. S. Inflammation, stress, and diabetes. J. Clin. Invest. 115, 1111–1119 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tataranni P. A. & Ortega E. A burning question: does an adipokine-induced activation of the immune system mediate the effect of overnutrition on type 2 diabetes? Diabetes 54, 917–927 (2005). [DOI] [PubMed] [Google Scholar]
- Skerka C., Decker E. L. & Zipfel P. F. A regulatory element in the human interleukin 2 gene promoter is a binding site for zinc finger proteins Sp1 and EGR-1. J. Biol. Chem. 270, 22500–22506 (1995). [DOI] [PubMed] [Google Scholar]
- Krämer B., Meichle A., Hensel G., Charnay P. & Krönke M. Characterization of an Krox-24/Egr-1 responsive element in the human tumor necrosis factor promoter. Biochim. Biophys. Acta 1219, 413–421 (1994). [DOI] [PubMed] [Google Scholar]
- Lin J.-X. & Leonard W. J. The immediate-early gene product Egr-1 regulates the human interleukin-2 receptor b-chain promoter through noncanonical Egr and Sp1 binding sites. Mol. Cell. Biol. 17, 3714–3722 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel A. et al. Transcription factor Egr-1 activity down-regulates Fas and CD23 expression in B cells. J. Immunol. 159, 2678–2684 (1997). [PubMed] [Google Scholar]
- Maltzman J. S., Carmen J. A. & Monroe J. G. Transcriptional regulation of the Icam-1 gene in antigen receptor- and phorbol ester-stimulated B lymphocytes: role for transcription factor EGR-1. J. Exp. Med. 183, 1747–1759 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Weber M., Laur O. & Krammer P. H. Novel Egr/NFAT composite sites mediate activation of the CD95 (APO/Fas) ligand promoter in response to T cell stimulation. Eur. J. Immunol. 29, 3017–3027 (1999). [DOI] [PubMed] [Google Scholar]
- Biesiada E., Razandi M. & Levin E. R. Egr-1 activates basic fibroblast growth factor transcription. J. Biol. Chem. 271, 18576–18581 (1996). [DOI] [PubMed] [Google Scholar]
- Khachigian L. M., Lindner V., Williams A. J. & Collins T. Egr-1-induced endothelial gene expression: a common theme in vascular injury. Science 271, 1427–1430 (1996). [DOI] [PubMed] [Google Scholar]
- Lui C., Adamson E. & Mercola D. Transcription factor EGR-1 suppresses the growth and transformation of human HAT-1080 fibrosarcoma cells by induction of transforming growth factor b1. Proc. Natl Acad. Sci. USA 93, 11831–11836 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M. L., Wu S. & Basler J. W. Prostatic nerve growth factor inducible A gene binds a novel element in the retinoblastoma gene promoter. Cancer Res. 53, 5597–5599 (1993). [PubMed] [Google Scholar]
- Philipp A. et al. Repression of cyclin D1: a novel function of MYC. Mol. Cell. Biol. 14, 4032–4043 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay J. J. & Drouin J. Egr-1 is a downstream effector of GnRH and synergizes by direct interaction with Ptx1 and SF-1 to enhance luteinizing hormone ß gene transcription. Mol. Cell. Biol. 19, 2567–2576 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. L. et al. Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1). Science 273, 1219–1221 (1996). [DOI] [PubMed] [Google Scholar]
- Sartipy P. & Loskutoff D. J. Expression profiling identifies genes that continue to respond to insulin in adipocytes made insulin-resistant by treatment with tumor necrosis factor-alpha. J Biol Chem. 278 (52), 52298–306 (2003). [DOI] [PubMed] [Google Scholar]
- Shen N. et al. An early response transcription factor, Egr-1, enhances insulin resistance in type 2 diabetes with chronic hyperinsulinism. J Biol Chem. 286(16) 14508–15 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X. et al. Egr-1 decreases adipocyte insulin sensitivity by tilting PI3K/Akt and MAPK signal balance in mice. EMBO J. 30 (18), 3754–65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Enerbäck S. & Smith U. Reduced expression of FOXC2 and brown adipogenic genes in human subjects with insulin resistance. Obes Res. 11 (10), 1182–91 (2003). [DOI] [PubMed] [Google Scholar]
- Cederberg A. et al. FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell 106, 563–573 (2001). [DOI] [PubMed] [Google Scholar]
- Gashler A. & Sukhatme V. P. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 50, 191–224 (1995). [DOI] [PubMed] [Google Scholar]
- Decker E. L. et al. Early growth response proteins (EGR) and nuclear factors of activated T cells (NFAT) form heterodimers and regulate proinflammatory cytokine gene expression. Nucleic Acids Res. 31, 911–921 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel J. V. Diabetes mellitus: a "thrifty" genotype rendered detrimental by "progress?". Am. J. Hum. Genet. 14, 353–362 (1962). [PMC free article] [PubMed] [Google Scholar]
- Grundy S. M. Obesity, metabolic syndrome, and cardiovascular disease. J. Clin. Endocrinol. Metab. 89, 2595–2600 (2004). [DOI] [PubMed] [Google Scholar]
- Krishnan J. et al. Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 26 (3), 259–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson E., Almgren P., Hoffstedt J., Groop L. & Ridderstråle M. The FOXC2 C-512T polymorphism is associated with obesity and dyslipidemia. Obes Res. 12 (11), 1738–43 (2004). [DOI] [PubMed] [Google Scholar]
- Di Gregorio G. B., Westergren R., Enerback S., Lu T. & Kern P. A. Expression of FOXC2 in adipose and muscle and its association with whole body insulin sensitivity. Am J Physiol Endocrinol Metab. 287 (4), E799–803 (2004). [DOI] [PubMed] [Google Scholar]
- Yanagisawa K. et al. The FOXC2-512C > T variant is associated with hypertriglyceridaemia and increased serum C-peptide in Danish Caucasian glucose-tolerant subjects. Diabetologia. 46 (11), 1576–80 (2003). [DOI] [PubMed] [Google Scholar]
- Ridderstråle M. et al. FOXC2 mRNA Expression and a 5′ untranslated region polymorphism of the gene are associated with insulin resistance. Diabetes 51(12), 3554–60 (2002). [DOI] [PubMed] [Google Scholar]
- Lidell M. E. et al. The adipocyte-expressed forkhead transcription factor Foxc2 regulates metabolism through altered mitochondrial function. Diabetes 60 (2), 427–35 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Håkansson J., Eliasson B., Smith U. & Enerbäck S. Adipocyte mitochondrial genes and the forkhead factor FOXC2 are decreased in type 2 diabetes patients and normalized in response to rosiglitazone. Diabetol Metab Syndr. 18 (3), 32 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steppan C. M. & Lazar M. A. Resistin and obesity-associated insulin resistance. Trends Endocrinol Metab. 13 (1), 18–23 (2002). [DOI] [PubMed] [Google Scholar]
- Kershaw E. E. & Flier J. S. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 89 (6), 2548–56 (2004). [DOI] [PubMed] [Google Scholar]
- Chen Z., Torrens J. I., Anand A., Spiegelman B. M. & Friedman J. M. Krox20 stimulates adipogenesis via C/EBPbeta-dependent and –independent mechanisms. Cell Metab. 1, 93–106 (2005). [DOI] [PubMed] [Google Scholar]
- Boyle K. B. et al. The transcription factors Egr1 and Egr2 have opposing influences on adipocyte differentiation. Cell Death Differ. 16 (5), 782–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandalia M., Abate N., Garg A., Stray-Gundersen J. & Grundy S. M. Relationship between generalized and upper body obesity to insulin resistance in Asian Indian men. J Clin Endocrinol Metab 84, 2329–2335 (1999). [DOI] [PubMed] [Google Scholar]
- Schwarz M., Russell D. M., Dietschy J. M. & Turley S. D. Marked reduction in bile acid synthesis in cholesterol 7 alpha-hydroxylase-deficient mice does not lead to diminished tissue cholesterol turnover or to hypercholesterolemia. J. Lipid Res. 39, 1833–1843 (1998). [PubMed] [Google Scholar]
- Kalaany N. Y. et al. LXRs regulate the balance between fat storage and oxidation. Cell Metab. 1, 231–244 (2005). [DOI] [PubMed] [Google Scholar]
- Bookout A. L. & Mangelsdorf D. J. A quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. NURSA e-Journal 1 ID 1.11082003.1 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M. et al. Platelet-derived growth factor promotes the expression of peroxisome proliferator-activated receptor γ in vascular smooth muscle cells by a phosphatidylinositol 3-kinase/Akt signaling pathway. Circ Res. 89, 1058–1064 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information