

Abstract
Congenital hyperinsulinism (CHI) occurs as a consequence of unregulated insulin secretion from the pancreatic beta-cells. Severe recessive mutations and milder dominant mutations have been described in the ABCC8 and KCNJ11 genes encoding SUR1 and Kir6.2 subunits of the beta-cell ATP-sensitive K(+) channel. Here we report two patients with CHI unresponsive to medical therapy with diazoxide. Sequencing analysis identified a compound heterozygous mutation in ABCC8 in both patients. The first one is a carrier for the known mild dominant mutation p.Glu1506Lys jointly with the novel mutation p.Glu1323Lys. The second carries the p.Glu1323Lys mutation and a second novel mutation, p.Met1394Arg. Functional studies of both novel alleles showed reduced or null cell surface expression, typical of recessive mutations. Compound heterozygous mutations in congenital hyperinsulinism result in complex interactions. Studying these mechanisms can improve the knowledge of this disease and modify its therapy.
Keywords: Congenital hyperinsulinism, Mutation, ABCC8, Functional study
1. Introduction
Congenital hyperinsulinism (CHI) refers to a group of inherited disorders caused by dysregulated insulin secretion (Stanley, 2002). Until now, the disorder has been associated with several genes, including ABCC8, KCNJ11, GLUD1, GCK, HADH, UCP2, SLC16A1, and HNF4A. Related disorders include the type 1B congenital disorder of glycosylation, and Beckwith–Wiedemann syndrome (Kapoor et al., 2009). The most common genetic causes of CHI are inactivating mutations on the ABCC8 and KCNJ11 genes coding for the two subunits of the ATP-sensitive K(+) (KATP) channel in the beta-cell of the pancreas. The related proteins are respectively a high-affinity sulfonylurea receptor (SUR1) and an inwardly rectifying potassium channel (Kir6.2).
Histologically, two types of CHI have been mainly described, focal and diffuse. The focal form, localized to one region of the pancreas, has been associated with paternal isodisomy for a KATP mutation in b-cells, whereas the diffuse form is associated with autosomal recessive and dominant mutations in the ABCC8 and KCNJ11 genes (Saint-Martin et al., 2011). Despite the fact that focal and diffuse forms have similar clinical presentations, they differ in management. While the focal form can be treated with surgical excision with complete healing of the patient, the diffuse form would require total or near total pancreatectomy with many complications (James et al., 2009).
Here we report two patients with bi-allelic mutations of ABCC8 gene causing a phenotype unresponsive to medical treatment.
2. Materials and methods
2.1. Patients and clinical evaluation
Patient 1 is a 4-year-old boy, the first-born from unrelated Italian parents. He was born at 37 weeks gestation. Birth weight was 3.6 kg (50th–75th percentile) and length was 51 cm (50th–75th percentile). During pregnancy, the mother had a positive oral glucose tolerance test showing gestational diabetes, which resolved after delivery. The family history revealed type 2 diabetes in several relatives on both maternal and paternal sides. The proband’s father, as well as the paternal aunt reported some episodes of faintness after meals which were associated with low glucose level (<50 mg/dL) without any other medical cause. In addition, at 53 years the proband’s paternal grandmother was diagnosed as having type 2 diabetes and was treated with oral agents. Recently, she stopped therapy and the latest glycemia was up to 400 mg/dL preprandial. From the first day of life, patient 1 was hypoglycemic (blood glucose 35 mg/dL) with elevated levels of insulin (25 μU/mL). He was poorly responsive to diazoxide (15 mg/kg/day) which failed to prevent recurrent relapses of hypoglycemia. Octreotide (15 μg/kg/day) was more effective, but failed to fully normalize glucose levels. A 18F-L-dopa PET revealed a diffuse form of CHI.
Patient 2 is a 3-year-old boy, first born of unrelated Italian parents. He was born at 39 weeks gestation. Birth weight was 4.4 kg (>90th percentile) and the length was 53 cm (75th–90th percentile). The parents had no history of hypoglycemia and no information was available about the grandparents. At birth, the baby had severe hypoglycemia (blood glucose 20 mg/dL) with hyperinsulinemia (60 μU/mL). Therapy with diazoxide (20 mg/kg/day) as well as octreotide (15 μg/kg/day) failed to control the hypoglycemia. The 18F-L-dopa PET scan detected a diffuse form of CHI. Subsequently, the patient was treated with diet and octreotide with poor control of glycemia. Written informed consent was obtained from each patient.
2.2. Genes sequencing and mutation data handling
Molecular analysis of ABCC8 and KCNJ11, by direct sequencing of all coding regions and exon-intron boundaries, was conducted on genomic DNA using standard techniques. To evaluate new variants, Single Nucleotide Polymorphism Database (http://www.ncbi.nlm.nih.gov/snp), 1000 Genomes Project (http://www.1000genomes.org/) and Exome Variant Server (http://evs.gs.washington.edu/EVS/) were used. Polymorphism Phenotyping (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/) and Sorting Intolerant From Tolerant (SIFT; http://sift.jcvi.org) were used for the prediction of potential functional impact of amino acid substitutions (Hon et al., 2009; Ramensky et al., 2002; Sunyaev et al., 2001). The new variants were tested in 100 healthy Italian controls.
2.3. Functional studies
A functional study was carried out introducing point mutations on SUR1 into hamster ABCC8 cDNA in the pECE plasmid using the QuikChange site-directed mutagenesis kit (Stratagene, Santa Clara, CA) as previously described (Huopio et al., 2000). Hamster cDNA was used because the expression level is higher. Mutations were confirmed by DNA sequencing. Kir6.2 cDNA was in pCDNAI. COSm6 cells were plated onto 35 mm culture dishes and transfected with wild-type (WT) or mutant SUR1 and WT Kir6.2 cDNA using FuGene6 (Promega Corporation, Madison, WI). Cells were used for experiments ~36–72 h post-transfection.
Western blot analysis of whole cell lysate was performed to assess the processing efficiency of mutant channels. Briefly, proteins from cell lysates were separated by SDS-PAGE and transferred to nitrocellulose membrane. The membrane was probed with an anti-SUR1 antibody followed by incubation with horseradish peroxidase-conjugated secondary antibody, and visualized by enhanced chemiluminescence (Yan et al., 2007).
A quantitative chemiluminescence assay as previously described (Yan et al., 2007) was used to directly measure mutant channel expression at the cell surface. For these experiments, a SUR1 tagged with a FLAG-epitope at the N-terminus was used. The FLAG-epitope tag has shown not to affect channel expression and functional properties in prior studies (Cartier et al., 2001).
Functional properties of channels were studied using inside-out patch-clamp recordings as previously described by Yan et al. (2007). Briefly, COSm6 cells were transfected with cDNA encoding WT or mutant channel proteins, as well as cDNA for the green fluorescent protein (GFP) for identification of transfected cells. Patch-clamp recordings were made 36–72 h post-transfection. Research methods are available on request.
3. Results
3.1. Genetic analysis
In patient 1, molecular analysis of ABCC8 (NP_000343; NM_000352) revealed two heterozygous missense mutations (Fig. 1). The first one was the known dominant mutation p.Glu1506Lys; c.4516G>A (Huopio et al., 2002). It was also identified in the proband’s father, in the paternal aunt and grandmother. The second heterozygous mutation was the p.Glu1323Lys; c.3967G>A. This mutation was also found in the patient’s mother and in the maternal grandmother. The p.Glu1323Lys change is a novel mutation, not included in either online database as a polymorphism or as a known disease-causing mutation. The p.Glu1506Lys mutation has been reported in several families and causes a dominant CHI well controlled by diazoxide (Huopio et al., 2003). Although there is still controversy on this issue, p.Glu1506Lys has been suggested to predispose to the development of insulin deficiency and type 2 diabetes (Huopio et al., 2002). The worst condition of the patient 1 compared to other patients with the p.Glu1506Lys mutation could be due to the simultaneous inheritance of this and the p.Glu1323Lys mutation.
Fig. 1.
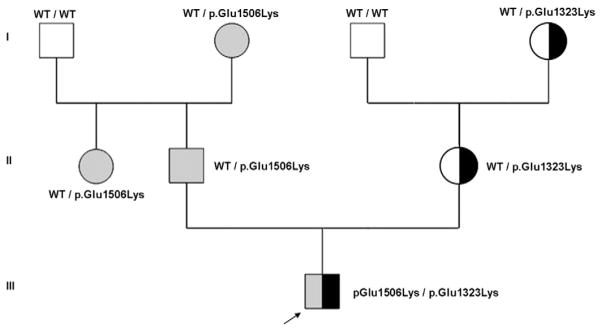
A pedigree of family and compound heterozygosity of the patient 1, formed by the p.Glu1506Lys mutation (in gray), inherited from his father and also present in the paternal aunt and paternal grandmother, and the p.Glu1323Lys mutation (in black), inherited from the mother and maternal grandmother.
Patient 2 was heterozygous for the same novel p.Glu1323Lys mutation. In addition he has a novel variant, p.Met1394Arg (c.4181T>G). In this family, the carriers for one of these mutations appeared to be unaffected, consistent with a recessive inheritance. Both variants are highly conserved across species and were absent in 200 control alleles. PolyPhen2 indicated that both variants are possibly/probably damaging and SIFT analysis predicted them to be disease-causing. Functional analysis was previously performed on the p.Glu1506Lys mutation (Vieira et al., 2010).
3.2. Expression studies
To test the effect of the two novel missense mutations we performed an expression study in COSm6 cells (Fig. 2). As Fig. 2A shows neither the E1323K nor the M1394R mutants are glycosylated appropriately in comparison to the WT, indicating that these mutants probably cause trafficking defects and are likely retained in the endoplasmic reticulum. Although the whole-cell lysate Western blot result indicates poor processing efficiency of both mutants, it does not report directly the surface expression level of the mutant channel. To measure surface expression directly, we used a quantitative chemiluminescence assay as described in the Materials and methods section. The results show that both the mutants have markedly reduced surface expression when compared to WT channels (Fig. 2B). The E1323K mutant is expressed at the surface at a low level (16.98±4.87%) but the presence of the M1394R mutant is nearly obliterated at the cell surface (Fig. 2B).
Fig. 2.
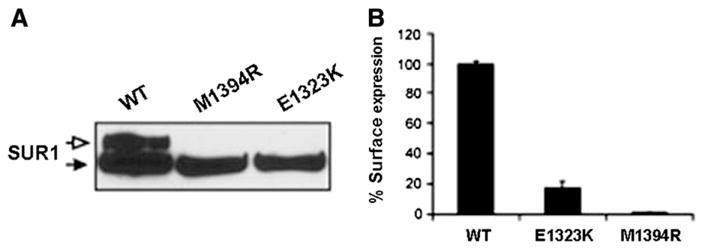
(A) Analysis of mutant flagSUR1 proteins. COSm6 cells were co-transfected with cDNAs for Kir6.2 and WT-flagSUR1, the M1394R-flagSUR1 or the E1323K-flagSUR1. Both mutants exhibit the lower core glycosylated band (solid arrow). However, neither mutant exhibits the upper complex glycosylated band (open arrow) as seen in WT, indicating that the mutant proteins are not processed and trafficked along the secretory pathway like the WT protein. (B) Both mutants show reduced cell surface expression, although the E1323K mutant retains some surface expression (16.98±4.87%; n=3).
In inside-out patch-clamp electrophysiological recordings, small currents were detected in cells expressing the E1323K mutant (relative to WT; see Figs. 3A and B and note the current amplitude scale difference), but no currents were observed in cells expressing the M1394R mutant after more than 10 patches. Moreover, channel responses to the metabolic regulator magnesium adenosine diphosphate (MgADP) (Fig. 3A) and the channel agonist diazoxide (Fig. 3B) were evaluated for the E1323K mutant. The E1323K mutant exhibited similar responses to both MgADP and diazoxide as WT channels in the presence of inhibitory ATP (0.1 mM).
Fig. 3.

(A) The E1323K mutant shows similar MgADP response (69.16±5.42%; n=8) as WT channels (76.87±6.80%; n=7). Currents were measured at −50 mV in symmetrical K-INT solution, and inward currents are shown as upward deflections. Patches were exposed to differing concentrations of ATP and ADP, as indicated by the bars above the records. Free Mg2+ concentration was maintained at 1 mM in all ATP-containing solutions. (B) Same as in (A) except that channel response to diazoxide was compared. The percent of current for WT and E1323K was (70.31±8.62%; n=8) and (54.12±10.71%; n=5), respectively.
4. Discussion
Patient 1 had a dominant mutation previously associated with a mild hypoglycemia. In patients carrying only this mutation, although the mutant protein forms a non functioning hetero-octameric KATP channel, the WT allele forms a 1/16 of channels totally working, resulting in a hyperinsulinemic hypoglycemia responsive to the therapy (Pinney et al., 2008). In our patient the second mutation seems to have features of recessive mutations, including defective trafficking and the presence in healthy mother and maternal grandmother. Since this trafficking defect it could be possible that most of the KATP channels expressed at the cell surface contain only the E1506K SUR1 mutant. We hypothesize that the combined effect of these two mutations could explain the worst phenotype compared to others with the only p.Glu1506Lys.
In patient 2, who is compound heterozygous for the p.[Glu1323Lys]: [Met1394Arg] mutations, both defects seem to have a recessive inheritance and impair the channel trafficking to the plasma membrane, even though E1323K does have normal gating properties. Obviously, more definitive and specific functional studies (and larger confirmation populations) are required to obtain evidences about the causality of these mutations.
Finally, mutations of the KATP channel are different in patterns of inheritance and functional consequence and compound heterozygous results in complex interactions modifying the potential pathogenesis. Molecular analysis in these patients, including functional assays of the mutant channels, may have implications both for understanding the pathophysiological mechanism and treatment of the disease.
Acknowledgments
The authors wish to thank the patients and their parents who participated in the study for their continuous support.
Funding for this work was provided by Centro Regionale Malattie Metaboliche Ereditarie Regione Veneto, Italy. D.G.R.V. n 3578.
This work was supported in part by grants from the NIH: R37 DK056268 and R01 DK53012 (to C.A.S.); R01 DK057699 and R01 DK066485 (to S-L.S.); and UL1 RR 024135.
Abbreviations
- CHI
congenital hyperinsulinism
- KATP
ATP-sensitive K(+)
- SUR1
sulfonylurea receptor
- Kir6.2
inwardly rectifying potassium channel
References
- Cartier EA, et al. Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc Natl Acad Sci U S A. 2001;98:2882–2887. doi: 10.1073/pnas.051499698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon LS, et al. Computational prediction of the functional effects of amino acid substitutions in signal peptides using a model-based approach. Hum Mutat. 2009;30:99–106. doi: 10.1002/humu.20798. [DOI] [PubMed] [Google Scholar]
- Huopio H, et al. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest. 2000;106:897–906. doi: 10.1172/JCI9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huopio H, et al. K(ATP) channels and insulin secretion disorders. Am J Physiol Endocrinol Metab. 2002;283:E207–216. doi: 10.1152/ajpendo.00047.2002. [DOI] [PubMed] [Google Scholar]
- Huopio H, et al. A new subtype of autosomal dominant diabetes attributable to a mutation on the gene for sulfonylurea receptor 1. Lancet. 2003;361:301–307. doi: 10.1016/S0140-6736(03)12325-2. [DOI] [PubMed] [Google Scholar]
- James C, et al. The genetic basis of congenital hyperinsulinism. J Med Genet. 2009;46:289–299. doi: 10.1136/jmg.2008.064337. [DOI] [PubMed] [Google Scholar]
- Kapoor RR, et al. Hyperinsulinism in developmental syndromes. Endocr Dev. 2009;14:95–113. doi: 10.1159/000207480. [DOI] [PubMed] [Google Scholar]
- Pinney SE, et al. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest. 2008;118:2877–2886. doi: 10.1172/JCI35414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky V, et al. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Martin C, et al. KATP channel mutations in congenital hyperinsulinism. Semin Pediatr Surg. 2011;20:18–22. doi: 10.1053/j.sempedsurg.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Stanley CA. Advances in diagnosis and treatment of hyperinsulinism in infants and children. J Clin Endocrinol Metab. 2002;11:4857–4859. doi: 10.1210/jc.2002-021403. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, et al. Prediction of deleterious human alleles. Hum Mol Genet. 2001;15:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- Vieira TC, et al. Hyperinsulinemic hypoglycemia evolving to gestational diabetes and diabetes mellitus in a family carrying the inactivating ABCC8 E1506K mutation. Pediatr Diabetes. 2010;11:505–508. doi: 10.1111/j.1399-5448.2009.00626.x. [DOI] [PubMed] [Google Scholar]
- Yan FF, et al. Congenital hyperinsulinism associated ABCC8 mutations that cause defective trafficking of ATP-sensitive K+ channels: identification and rescue. Diabetes. 2007;56:2339–2348. doi: 10.2337/db07-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]