

Abstract
Nucleoside analogues are a significant class of anticancer agent. As prodrugs, they terminate the DNA synthesis upon transforming to their active triphosphate metabolites. We have encapsulated a biologically activate nucleotide analogue (i.e. gemcitabine triphosphate (GTP)), instead of the nucleoside (i.e. gemcitabine) derivative, into a novel Lipid/Calcium/Phosphate nanoparticle (LCP) platform. The therapeutic efficacy of LCP-formulated GTP was evaluated in a panel of human non-small-cell lung cancer (NSCLC) and human pancreatic cancer models after systemic administrations. GTP-loaded LCPs induced cell death and arrested the cell cycle in the S phase. In vivo efficacy studies showed that intravenously injected GTP-loaded LCPs triggered effective apoptosis of tumor cells, significant reduction of tumor cell proliferation and cell cycle progression, leading to dramatic inhibition of tumor growth, with little in vivo toxicity. Broadly speaking, the current study offers preclinical proof-of-principle that many active nucleotide or phosphorylated nucleoside analogues could be encapsulated in the LCP nanoplatform and delivered systemically for a wide variety of therapeutic applications.
Keywords: LCP nanoparticle, gemcitabine triphosphate, apoptosis, proliferation, cell cycle progression
Introduction
Cytotoxic nucleoside analogues have received considerable attention as clinical cancer treatments, including treatments for hematological malignancies and solid tumors [1]. About 20% of small molecule therapeutics that have been approved by the FDA for cancer treatment are nucleoside analogues or related compounds [2]. These drugs act as antimetabolites by interfering with nucleic acid synthesis and enzymes of nucleotide metabolism [3].
Gemcitabine (2′, 2′-difluoro 2′-deoxycytidine) (Gem) is a nucleoside analogue widely used in chemotherapy as a first-line treatment of various carcinomas, including non-small cell lung cancer (NSCLC) and pancreatic cancer [4]. As a prodrug, Gem relies on nucleoside transporters to enter into cells. The overall drug uptake depends on the balance of the nucleoside transporters and drug efflux proteins presented on the cell membrane. Once in the cells, Gem undergoes sequential phosphorylation into 5′-triphosphates by deoxycytidine kinase (dCK) before it can be incorporated into DNA and cause DNA synthesis inhibition, which leads to DNA damage and cell apoptosis. However, processes of mutational resistance readily occur in the nucleoside activation process, which makes Gem less efficacious. Examples of mutational resistance include down-regulation of nucleoside influx transporters (e.g. ENT1, ENT2, CNT1, CNT3), up-regulation of drug efflux proteins (e.g. MRP, BCRP), and inactivation of dCK [5]. In addition, Gem is rapidly metabolized to 2′,2′-difluorodeoxyuridine by the deoxycytidine deaminase and quickly excreted by the kidney following in vivo administration.
There have been extensive research efforts to overcome gemcitabine resistance, of which nanoparticle delivery systems and lipophilic gemcitabine prodrugs are major approaches [6, 7]. Although they have enabled the circumvention of the nucleoside transporter barriers and the deaminase inactivation, dCK or cellular kinases are still required for its conversion to the bioactive triphosphate derivatives after cellular uptake.
In order to enhance the in vivo delivery efficiency of Gem and overcome the aforementioned delivery challenges, our strategy is to entrap the bioactive form of Gem, gemcitabine triphosphate (GTP), into a cell-specific, targeted Lipid/Calcium/Phosphate nanoparticle (LCP). In this way, the metabolism mechanisms of Gem will be bypassed and the bioactive drug will be delivered directly into the cell through receptor-mediated endocytosis. Furthermore, LCPs can protect GTP from enzymatic breakdown, preventing inactivation within circulation and rapid excretion by the kidney.
Nanoparticles (NPs) are prone to lysosomal degradation after endocytosis. To avoid the degradation of entrapped drugs, NPs must be able to escape from endosomes before trafficking to lysosomes. In order to achieve this goal, we used calcium phosphate (CaP) precipitate to entrap GTP. The CaP core of the LCPs is dissolvable in endosomes due to its low pH [8]. Increase in osmotic pressure in the endosome leads to endosome swelling and rupture, releasing the entrapped GTP into the cytoplasm and allowing it to enter into the nucleus. A lipid bilayer membrane surrounds the GTP-entrapped CaP core and a high density of polyethylene glycol (PEG), either with or without a tethered targeting ligand anisamide (AA), was grafted onto the surface. These characteristics increased the tumor accumulation of the drug through the enhanced permeability and retention (EPR) effect [9]. They also allowed tumor specific targeting and reduced reticuloendothelial system (RES) uptake [10].
In this study, the feasibility of using the novel LCPs as a nanocarrier for the delivery of biologically active GTP was evaluated on human NSCLC and pancreatic cancer models. The small molecule ligand anisamide (AA) was modified on the LCP for specific tumor targeting via the sigma receptors that are overexpressed in many human cancer cells. Since cytidine triphosphate (CTP) has a similar structure to GTP and does not have any therapeutic activity, CTP-loaded LCPs were prepared as control nanoparticles. The cytotoxic activity of LCP-formulated GTP was compared with free GTP and its prodrug, Gem, both in vitro and in vivo.
Materials and Methods
Materials
Gemcitabine triphosphate tris(triethylammonium) salt was synthesized by Bioduro company (Beijing, China). Gemcitabine was purchased from HDH Pharma, Inc. (Research Triangle Park, NC). 1, 2-Dioleoyl-3-trimethylammonium-propane chloride salt (DOTAP), dioleoyl phosphatydic acid (DOPA), and 1,2-distearoryl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol-2000) ammonium salt (DSPE-PEG2000) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). DSPE-PEG-anisamide (AA) was synthesized in our lab as described previously [11]. Other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Cell culture
H460 human NSCLC cells and BxPC-3 human pancreatic cancer cells, originally obtained from American Type Culture Collection (ATCC), were cultured in a RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). Cells were cultivated in a humidified incubator at 37°C and 5% CO2.
Experimental animals
Female nude mice that were 6–8 weeks of age were used in all studies. Female nude mice were purchased from National Cancer Institute (Bethesda, MD) and bred in Division of Laboratory Animal Medicine (DLAM) at University of North Carolina at Chapel Hill. To establish the xenograft models, 5×106 cells in 100 μL of PBS were injected subcutaneously into the right flank of the mice. All work performed on animals was approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill.
Preparation of GTP-loaded LCPs
The LCP cores were prepared by water-in-oil micro-emulsions in the oil phase containing cyclohexane/Igepal CO-520 solution (71/29, v/v) [12]. One hundred and eighty μL of 20 mM GTP was mixed with 12.5 mM Na2HPO4 (pH=9.0) to the total volume of 600 μL before the addition of a 20 mL oil phase with continuous stirring. The calcium micro-emulsion was prepared by adding 600 μL of 2.5 M CaCl2 to a separate 20 mL oil phase. Four hundred μL of 20 mM DOPA in chloroform was added to the phosphate phase and then the two separate micro-emulsions were mixed. After stirring for 5 min, another 400 μL of 20 mM DOPA was added into the emulsion. The emulsion was stirred continuously for 20 min before 40 mL of absolute ethanol was added. The ethanol emulsion mixture was centrifuged at 9,000 g for 15 min to pellet the LCP core and the supernatant was discarded. The LCP core was washed twice with absolute ethanol and dried under N2. The LCP core pellets were dissolved in 2 mL chloroform and stored in a glass vial at −20°C for future use. To prepare the final GTP-loaded LCPs (GTP-LCP-PEG) with an outer lipid coating, 330 μL LCP core in chloroform was mixed with 38.7 μL of 10 mg/ml cholesterol, 28 μL of 25 mg/ml DOTAP, 96 μL of 25 mg/ml DSPE-PEG. The final molar ratios for cholesterol, DOTAP and DSPE-PEG were 7:7:6. After evaporating the chloroform, the residual lipids were dissolved in 30 μL THF followed by 50 μL absolute ethanol, and then suspended in 160 μL water. After brief sonication, the solution was dialyzed in distilled water to remove THF and ethanol. The AA-targeted GTP-loaded LCPs (GTP-LCP-PEG-AA) were prepared in the same way, except that 20% DSPE-PEG was replaced by an equal molar amount of DSPE-PEG-AA.
Characterization of GTP-loaded LCPs
The particle size and zeta potential of LCPs were determined by Dynamic light scattering (DLS) using a Malvern ZetaSizer Nano series (Westborough, MA). GTP encapsulation efficiency was measured by a UV spectrophotometer (Beckman Coulter Inc., DU 800 spectrophotometer). Transmission electron microscope (TEM) images of GTP-loaded LCPs were acquired through the use of JEOL 100CX II TEM (Tokyo, Japan). Briefly, 4 μl of LCP solution was dropped onto a 300 mesh carbon coated copper grid (Ted Pella, Inc., Redding, CA) for 2 min. Excess fluid was removed with filter paper, and the copper grid was dried before observation with TEM.
In vitro cellular uptake
In the preparation of LCPs, small amount of NBD-labeled phosphatydic acid (one percent of the total dioleoyl phosphatydic acid added) was used to label the LCP cores. H460 cells were seeded at a density of 1×105 per well in 24-well plates (Corning Inc., Corning, NY) with cover glass. Twenty-four h later, cells were treated with NBD labeled LCP formulations at 37°C for 1 h. After washing twice with PBS, cells were fixed with 4% paraformaldehyde at room temperature for 10 min, and stained the nuclei with DAPI (Vector Laboratories, Inc., Burlingame, CA) before imaging with a Leica SP2 Confocal Laser Scanning Microscope (Germany). In the receptor competitive inhibition experiment, cells were pre-incubated with 50 μM haloperidol (a sigma receptor agonist) for 1 h prior to the incubation with NBD-labeled GTP-LCP-PEG-AA for 1 h [13]. To quantify the cellular uptake efficiency, H460 cells were seeded in 6-well plates (3×105 cells per well) 24 h before the treatment of NBD labeled LCPs for 1 h. Then cells were harvested and washed with PBS, and the NBD fluorescence intensity in the cells was detected by a FACS Canto flow cytometry (BD Biosciences). Data from a total of 10,000 events were acquired for each sample and the mean fluorescence intensity of NBD in the cells was analyzed with FACS Diva software (BD Biosciences).
In vitro cell viability assay
In vitro cell viability of GTP-loaded LCPs and free drugs (GTP, Gem) was determined using the 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide (MTT) assay. H460 and BxPC-3 cells were seeded at a density of 1×104 per well in 96-well plates. When cells reached 30% confluence, different concentrations of GTP-LCP-PEG-AA, GTP-LCP-PEG, GTP, Gem, and CTP-LCP-PEG-AA were added to the medium. After 48 h exposure to the LCPs and free drugs at 37°C, MTT (Biosynth Inc.) was added to each well with a final concentration of 0.5 mg/mL, and incubated with cells at 37°C for 4 h. The resulting formazan crystals were solubilized by adding 150 μL DMSO to each well. The absorbance at a wavelength of 570 nm was measured with a microplate reader. Cell viability was calculated as the percentage of the absorbance of the treated cells to that of untreated cells. The IC50 concentrations for each of the treatments (GTP-LCP-PEG-AA, GTP-LCP-PEG, GTP, Gem) were listed in Supplementary Table 1.
Cell cycle analysis
H460 and BxPC-3 Cells (2×105) were seeded in 6-well plates, and GTP-LCP-PEG-AA, GTP-LCP-PEG, GTP, Gem, and CTP-LCP-PEG-AA were added the following day, resulting in a final concentration of 1.8 μM. After 24 h incubation, cells were trypsinized and washed with PBS followed by fixation in pre-cooled 70% ethanol at −20°C for at least 1 h. Fixed cells were washed with PBS staining buffer (BD Pharmingen, San Diego, CA) and incubated with RNAase (final concentration 75 μg/mL) at 37°C for 30 min, followed by incubation with 10 μg propidium iodide (PI) at room temperature for 30 min. Finally, cells were washed and suspended in PBS, and analyzed with a FACS Canto flow cytometry (BD Biosciences) to measure the PI intensity, which correlates with the DNA content in the cell cycle. A total of 10,000 events were acquired for each sample and data were analyzed with FACS Diva software (BD Biosciences).
Caspase activation
H460 and BxPC-3 cells (2×105) were seeded in 6-well plates, and treated with GTP-LCP-PEG-AA, GTP-LCP-PEG, CTP-LCP-PEG-AA, GTP, and Gem with a dose of 1.8 μM for 48 h. For the in vivo caspase activation study, H460 and BxPC-3 tumor-bearing mice (n=3) were given 3 daily intravenous (IV) injections of GTP-LCP-PEG-AA, GTP-LCP-PEG, CTP-LCP-PEG-AA, GTP, and Gem with a dose of 7.5 μmol/Kg (1.98 mg/Kg in terms of Gem). Equivalent molar dose of GTP-loaded LCPs, Gem and GTP were used in the following studies. Twenty-four hours after the last injection, mice were sacrificed and tumors were harvested. The cells and tumor tissues were lysed with a radioimmunoprecipitation assay (RIPA) buffer that was supplemented with a protease inhibitor cocktail (Promega, Madison, WI). Tumor tissues were lysed by brief homogenization and sonication. The protein lysates were collected by centrifugation at 14,000 rpm for 10 min at 4°C. Protein concentrations were determined using the BCA assay kit (Pierce Biotechnology) following the manufacturer’s recommendations. Forty μg protein of each sample was used to detect caspase-3/7 activity of the cell or tumor lysates according to the manufacturer’s instructions (Promega).
Western blot analysis
Twenty-four hours after three daily IV injections, H460 and BxPC-3 tumor-bearing mice were sacrificed and tumor lysates were prepared as previously described. Forty μg of protein per lane was separated by 4%–12% SDS-PAGE electrophoresis (Invitrogen) before being transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad). The membranes were blocked for 1 h with 5% silk milk at room temperature and then incubated with mouse monoclonal poly(ADP-ribose) polymerase-1 (PARP-1) antibodies and rabbit polyclonal cyclin D1 antibodies (1:500 dilution; Santa Cruz biotechnology, Inc.) overnight at 4°C. β-actin antibody (1:4000 dilution; Santa Cruz biotechnology, Inc.) was probed as the loading control. The membranes were washed 3 times and then incubated with a secondary antibody (1:4000 dilution; Santa Cruz biotechnology, Inc.) at room temperature for 1 h. Goat anti-mouse secondary antibody was used for PARP and β-actin primary antibody, and goat anti-rabbit secondary antibody was used for cyclin D1 primary antibody. Finally, the membranes were washed 4 times and developed by an enhanced chemiluminescence system according to the manufacturer’s instructions (Thermo scientific).
Immunostaining
In vivo tumor cell apoptosis after systemic administration was determined by the TdT-mediated dUTP Nick-End Labeling (TUNEL) assay. H460 and BxPC-3 tumor-bearing mice were given three daily IV injections of GTP-LCP-PEG-AA, GTP-LCP-PEG, CTP-LCP-PEG-AA, GTP, and Gem with a GTP dose of 7.5 μmol/Kg (n=3). Twenty-four h after the final injection, mice were sacrificed and tumors were fixed in 10% formalin for 24 h before being embedded in paraffin and sectioned at a thickness of 5 μm. The TUNEL staining was performed as recommended by the manufacturer (Promega). DAPI mounting medium (Vector Laboratories, Inc., Burlingame, CA) was dropped on the sections for nucleus staining. Images of TUNEL-stained tumor sections were captured with a fluorescence microscope (Nikon Corp., Tokyo, Japan). The percentage of apoptotic cells was obtained by dividing the number of apoptotic cells (TUNEL positive cells shown as green dots in Fig. 5) from the number of total cells (blue nuclei stained by DAPI, not shown) in each microscopic field, and 10 representative microscopic fields were randomly selected in each treatment group (n=3) for this analysis.
Fig. 5.
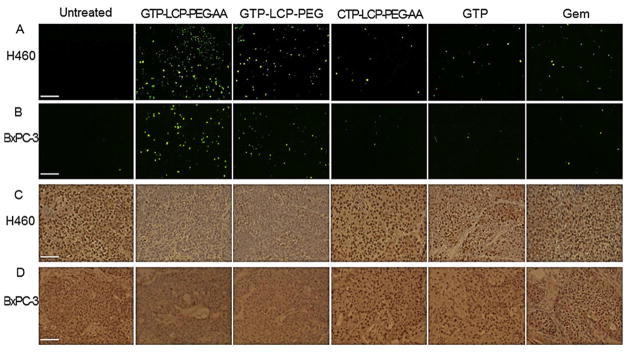

GTP-loaded LCPs triggered tumor cell apoptosis and inhibited tumor cell proliferation effectively in vivo in H460 and BxPC-3 xenografts. Mice were given 3 daily IV injections. Twenty-four h after the last injection, mice were sacrificed and tumor tissues were sectioned for TUNEL assay (A, B) and PCNA immunohistochemistry (C, D). The immunostaining results of H460 tumors (A, C), BxPC-3 tumors (B, D) were shown. Scale bar, 100 μm. (E–F) Percentage of apoptotic cells in H460 (E) and BxPC-3 (F) xenograft tumors after the treatment of different formulations. (G–H) Percentage of PCNA positive cells in H460 (G) and BxPC-3 (H) xenograft tumors after the treatment of different formulations. Statistics were as follows: (E) *p<0.01: GTP-LCP-PEG-AA vs. Untreated; ‡p<0.05: GTP-LCP-PEG vs. Untreated. (F) *p<0.01: GTP-LCP-PEG-AA vs. Untreated; ‡p<0.05: GTP-LCP-PEG vs. Untreated. (G) **p<0.0005 vs. Untreated. (H) **p<0.0005 vs. Untreated.
Proliferation of tumor cells after the aforementioned treatments and dosing schedule was detected by immunohistochemistry, using an antibody against proliferating cell nuclear antigen (PCNA) (1:200 dilution, Santa Cruz). The immunohistochemistry was performed using a mouse-specific HRP/DAB detection IHC kit as recommended by the manufacturer (Abcam, Cambridge, MA). The percentage of proliferation cells was obtained by dividing the number of PCNA positive cells (shown as brown dots) from the number of total cells (blue nuclei stained by hematoxylin) in each microscopic field, and 10 representative microscopic fields were randomly selected in each treatment group (n=3) for counting.
In addition to H460 and BxPC-3 xenograft models, we also tested the therapeutic response of GTP-LCP-PEG-AA on a NSCLC A549 xenograft model and a panel of pancreatic xenograft models, including Mia PaCa-2, AsPC-1, Capan-1, CFPAC-1, PANC-1 and SW1990 in vivo. To establish the pancreatic xenograft models, female nude mice were implanted with 5×106 pancreatic cells which were obtained from American Type Culture Collection (ATCC). Results were obtained for TUNEL assay and PCNA immunohistochemistry.
Pharmacokinetic profile of GTP-loaded LCPs
In the preparation of GTP-loaded LCPs, we mixed a trace amount of radioactive cytidine 5′ triphosphate (CTP) [5-3H] disodium salt (American Radiolabeled Chemicals, Inc., 1 mCi/mL) with GTP. 3H-labeled CTP served as a marker for the entrapped GTP. Thus, by measuring the 3H radioactivity, the percentage of GTP-loaded LCPs or free GTP circulating in the blood can be calculated. Nude mice bearing H460 tumors were IV injected with 3H-labeled, GTP-loaded LCPs or free GTP. At 20 min, 1 h, 2 h, 4 h, 8 h, 24 h post injection time point, 30 μL blood was drawn from the venous plexus of the eyes of the mice, and was mixed with 4 mL scintillation cocktail (Thermo Fisher Scientific Inc.). In order to avoid color interference with the scintillation counting, 120 μL of hydrogen peroxide (30% in water) was added to samples and vortexed to bleach the blood color before measurement. The 3H radioactivity in blood samples was counted using a liquid scintillation analyzer (Packard BioScience Co. TRI-CARB 2900TR). The percentage of the injected dose (%ID) in the blood was calculated based on the 3H radioactivity recovered in the blood samples. The initial concentration was set as 100% of the injected dose (ID). The post injection time (t) and %ID were fitted with a non-compartment model using the WinNonlin program and the key PK parameters were calculated.
Tumor growth inhibition and toxicity study
A tumor growth inhibition study was completed on H460 and BxPC-3 subcutaneous xenograft mouse models. Mice were inoculated with 5×106 H460 or BxPC-3 cells by subcutaneous injection. Treatment was started 12 days after tumor cell implantation. When the tumor volumes reached about 150–200 mm3, the mice were randomly assigned into 6 treatment groups (n=5), and intravenously injected different formulations, including GTP-LCP-PEG-AA, GTP-LCP-PEG, CTP-LCP-PEG-AA, GTP, and Gem. IV injections were performed every other day for a total of 4 injections at a GTP dose of 7.5 μmol/Kg. Tumor sizes were measured every other day with calipers across their two perpendicular diameters, and the tumor volume was calculated using the following formula: , where V = tumor volume, W = the smaller perpendicular diameter and L = the larger perpendicular diameter. Body weight of each mouse was recorded every other day. Humane sacrifice of mice was performed when tumors reached 20 mm in one dimension.
To test whether GTP-loaded LCPs would induce hepatic and renal dysfunction after frequent multiple dosing, mice were given three daily IV injections, and 24 h after the final injection, blood was drawn from the venous plexus of the eyes of the mice. Blood samples were immediately centrifuged at 3,000 g for 5 min at 4°C, and the supernatant blood serums were collected for hematological analysis. Blood urine nitrogen (BUN), creatinine, aspartate aminotransferase (AST), alanine aminotransferase (ALT) values were measured at UNC Animal Clinical Laboratory Core Facility by using Vitros 350 Chemistry System (Ortho Clinical Diagnostics Company, Rochester, NY), as indications of hepatic and renal functions.
Statistical analysis
Results were expressed as a mean ± standard deviation (SD). Student’s t-test and one-way analysis of variance (ANOVA) test were used to evaluate statistical significance. A p value of p<0.05 was considered to indicate statistical significance.
Results
Characterization of GTP-loaded LCPs
The GTP used in this study is gemcitabine triphosphate tris(triethylammonium) salt (Fig. 1A) which is synthesized as previously reported [14]. The final concentration of GTP in LCPs was 0.5 mM. The particle sizes and zeta potentials of different LCP formulations were shown in Table 1. The average sizes of GTP-loaded LCPs were around 40 nm. The higher zeta potential of the targeted LCPs than that of non-targeted LCPs was due to the presence of the positively charged AA ligand. The GTP-loaded LCPs were homogeneous and spherical in shape, as shown in the TEM micrograph (Fig. 1B). The encapsulation efficiency of GTP in LCPs was around 25%, as measured by UV spectrometry at a wavelength of 288 nm (data not shown). The GTP release profile of LCP nanoparticles were shown in Supplementary Figure 1. About 70–80% GTP was released from the particles in 48 h.
Fig. 1.
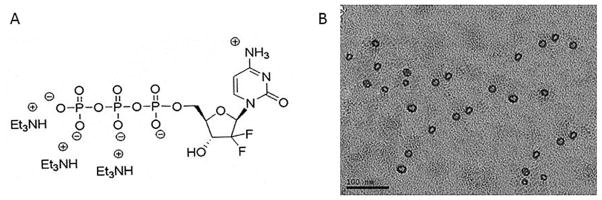
(A) The chemical structure of GTP (Gemcitabine triphosphate tris(triethylammonium) salt) used in this study. (B) TEM image of GTP-loaded LCPs.
Table 1.
Particle size and zeta potential of LCPs.
| Size (nm) | PDI | Zeta potential (mV) | |
|---|---|---|---|
| GTP-LCP-PEG-AA | 42.0 ± 9.0 | 0.17 ± 0.05 | 25.4 ± 5.7 |
| GTP-LCP-PEG | 37.7 ± 7.2 | 0.16 ± 0.03 | 7.2 ± 3.1 |
| CTP-LCP-PEG-AA | 33.4 ± 7.7 | 0.16 ± 0.01 | 18.9 ± 5.4 |
In vitro specific tumor cell targeting
The cellular uptake of GTP-LCP-PEG-AA and GTP-LCP-PEG at 37°C is shown in Figure 2. It is clear that the cellular endocytosis of AA-modified LCP was higher than that of untargeted LCP as shown by confocal images (Fig. 2A). The quantitative flow cytometry also confirmed that the AA ligand increased the intracellular uptake of LCPs (Fig. 2B–2C). The addition of haloperidol, a known agonist for sigma receptors, significantly reduced the cellular uptake of AA-modified LCP, suggesting AA modification on the LCP induced sigma receptor-mediated endocytosis.
Fig. 2.

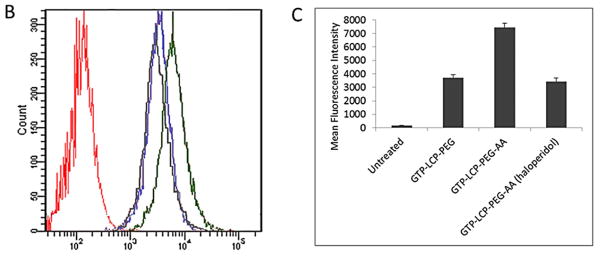
The cellular uptake of GTP-LCP-PEG and GTP-LCP-PEG-AA at 37°C for 1 h on H460 cells. (A) Confocal images for intracellular uptake. Green: NBD labeled LCP nanoparticles; Blue: DAPI stained nuclei. Scale bar, 15 μm. (B) Flow cytometry study of cellular uptake. Red: untreated control cells; Blue: GTP-LCP-PEG; Green: GTP-LCP-PEG-AA; Gray: GTP-LCP-PEG-AA after haloperidol (50 μM) incubation. (C) Quantitative results from flow cytometry by measuring intracellular mean fluorescence intensity.
GTP-loaded LCPs reduced the cell viability
The MTT assay was carried out to determine the cell viabilities after the treatment of GTP-loaded LCPs and free drugs. After 48 h incubation, the GTP-loaded LCPs caused a dramatic reduction of viable H460 (Fig. 3A) and BxPC-3 (Fig. 3B) cells in a dose-dependent manner. Free Gem and GTP also decreased the cell viability in vitro. This observation is explained by the facts that Gem can be taken up into cells through nucleoside transporters and GTP can be metabolized to Gem by the alkaline phosphatase on the cell surface before entering into cells [15]. CTP-loaded LCPs had little effect on cell viability.
Fig. 3.
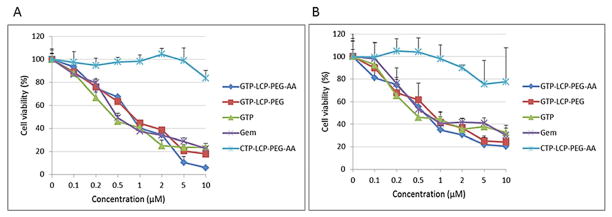
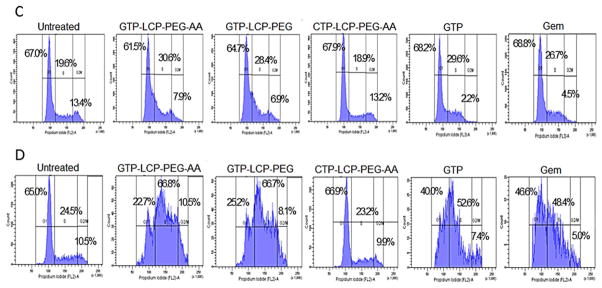
(A–B) GTP-loaded LCPs decreased cell viability in vitro on H460 (A) and BxPC-3 (B) cells. The cell viabilities of H460 (A) and BxPC-3 (B) cells were measured by MTT assay after 48 h exposure to GTP-loaded LCPs, CTP-loaded LCPs and free drugs (GTP, Gem). Data are mean ± S.D. (n=3). (C–D) Cell cycle arrest on H460 (C) and BxPC-3 (D) cells after 24 h treatment with different formulations and free drugs were analyzed by flow cytometry. The cell population of each sample was gated into three cell cycle phases: G1, S, G2/M. The percentage of H460 or BxPC-3 cells in different cell cycle phases after treatments was shown in the graph, based on the results from FACS software.
GTP-loaded LCPs arrested the cell cycle in the S phase
We next evaluated the effect of GTP-loaded LCPs on the cell cycle progression. The mechanism of GTP cell-killing is its competition with cytidine triphosphate during DNA replication, which results in the inhibition of chain elongation. The termination of DNA synthesis impedes the cell cycle progression, leading to cell death. After 24 h incubation with GTP-loaded LCPs, the percentage of cells in the S phase increases in H460 and BxPC-3 cells (Fig. 3C–3D), indicating the cell cycles were arrested in the S phase. GTP-loaded LCPs inhibited DNA synthesis by interfering with the progress toward the completion of S-phase in the cell cycle [16] because the process of incorporating nucleotides generally occurs during DNA replication in the S phase of the cell cycle [17]. CTP-loaded LCPs had no effect on cell cycle alteration in both cell lines (Fig. 3C–3D). The statistical analysis of the cell cycle result is listed in the Supplementary Figure 2.
GTP-loaded LCPs induced caspase activation in vitro and in vivo
Caspases are activated in cells when cells undergo the apoptotic process. Among them, caspase-3 and caspase-7 are primarily responsible for the proteolytic cleavage of a large number of substrates during apoptosis [18]. As shown in Fig. 4, the in vitro caspase-3/7 activities of GTP-loaded LCPs and free drug (GTP, Gem) treatments were significantly higher than the control in H460 (Fig. 4A) and BxPC-3 (Fig. 4B) cells. In vivo caspase activation was tested on H460 and BxPC-3 xenograft models. Twenty-four h after three daily IV injections, the caspase-3/7 activity in tumors after the treatment of GTP-loaded LCPs was significantly higher than that of free drugs in H460 (Fig. 4C) and BxPC-3 (Fig. 4D) xenografts, indicating that the LCPs greatly improved the in vivo delivery efficiency of GTP to the tumors. Our data suggests that free GTP and Gem were able to activate caspase signaling events only in vitro and did not elicit caspase activity in vivo.
Fig. 4.

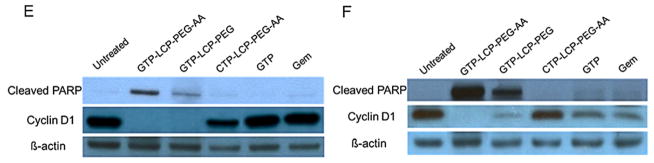
(A–D) GTP-loaded LCPs induced caspase activation in vitro (A, B) and in vivo (C, D). (A–B) In vitro caspase activation in H460 (A) and BxPC-3 (B) cells after 48 h incubation with different formulations. (C–D) In vivo caspase activation in tumors in H460 (C) and BxPC-3 (D) xenografts after 3 daily IV injections. Statistics were as follows: **p<0.005 vs. Untreated, *p<0.01 vs. Untreated. (E–F) GTP-loaded LCPs induced PARP cleavage and reduced cyclin D1 expression level in tumors after 3 daily IV injections in H460 (E) and BxPC-3 (F) xenografts. Twenty-four h after the last injection, tumor lysates were extracted and analyzed by western blot analysis for cleaved PARP, cyclin D1 and β-actin (loading control).
GTP-loaded LCPs triggered PARP cleavage and inhibited cell cycle progression in vivo
PARP is mainly involved in DNA repair induction and apoptosis. During the execution phase of apoptosis, intact PARP (116 kDa) is cleaved to its significant segment (85 kDa) by members of the caspase family, mainly caspase-3 and caspase-7 [18], reflecting an immediate cellular response to DNA damage. Therefore, the cleavage of PARP serves as a reliable marker of apoptosis [19]. From the western blots data (Fig. 4E and 4F), the cleaved PARP was significantly elevated after the treatment of GTP-loaded LCPs in NSCLC and pancreatic tumors. The cleaved PARP expression level of AA-targeted GTP-loaded LCPs was higher than that of non-targeted LCPs, due to the specific targeting of tumor cells. Free GTP and Gem did not induce PARP elevation and did not trigger apoptosis in vivo. This was most likely due to the rapid inactivation and excretion of free GTP and Gem after systemic administration, which results in little cellular apoptotic response. The high expression level of the cleaved PARP coincided with the high caspase-3/7 activity after the treatment of GTP-loaded LCPs in vivo, suggesting that the induction of apoptosis by GTP-loaded LCPs was due to the activation of the caspase-dependent pathway [20].
Cyclin D1 is involved in regulating cell cycle progression. GTP-loaded LCPs dramatically reduced the cyclin D1 expression (Fig. 4E and 4F), while free drugs did not show any cell cycle alteration after in vivo treatment. CTP-loaded LCPs did not elicit any cellular response in apoptosis induction or cell cycle alteration.
GTP-loaded LCPs triggered tumor cell apoptosis and inhibited tumor cell proliferation in vivo
Apoptotic signaling cascades result in DNA fragmentation (DNA damage) that can be detected by TUNEL assay. After 3 daily injections, AA-targeted GTP-loaded LCPs elicited the most effective killing effects in vivo and triggered a dramatic increase in apoptotic cells in H460 (Fig. 5A and 5E) and BxPC-3 (Fig. 5B and 5F) xenograft tumors when compared with the control. Non-targeted GTP-loaded LCPs showed moderate cell-killing effects. AA-targeted GTP-loaded LCPs displayed higher efficacy than the non-targeted group, suggesting that the receptor-mediated endocytosis was involved in the in vivo cellular uptake of LCPs. Free drugs caused few apoptotic cells in vivo after systemic administration, probably because the majority of the free drugs were metabolized and cleared before they encountered the tumor cells. These in vivo apoptosis results also agree with the caspase activation and PARP cleavage in tumors shown in Fig. 4.
Next, we detected the inhibition of tumor cell proliferation following different treatments. PCNA is expressed in the cell nuclei during DNA synthesis and can be used as a marker for cell proliferation [21]. From the results, GTP-loaded LCPs significantly decreased the number of PCNA positive cells in H460 (Fig. 5C and 5G) and BxPC-3 (Fig. 5D and 5H) xenograft tumors compared to the control. However, free drugs and CTP-loaded control LCPs had few anti-proliferative effects. This dramatic reduction of the tumor cell proliferation also related to the reduced cell cycle progression as illustrated by the decreased cyclin D1 protein expression in tumors shown in Fig. 4E and 4F.
We also tested the anti-tumor potency of AA-targeted GTP-loaded LCPs on a panel of NSCLC (A549) and pancreatic cancer (Mia PaCa-2, AsPC-1, Capan-1, CFPAC-1, PANC-1 and SW1990) xenograft models by TUNEL assay and PCNA immunohistochemistry (Fig. 6). These NSCLC and pancreatic cancer cells have been reported to overexpress sigma receptors [22–24], as confirmed by the western blot analysis (Supplementary Figure 3). Our data suggest that systemic treatment of GTP-LCP-PEG-AA induced significant number of apoptotic cells (~4% to 12%, Fig. 6B) and decreased the number of proliferating cells (2 to 4 folds lower, Fig. 6C) in tumors compared to the corresponding untreated tumors in all NSCLC and pancreatic cancer models tested. These data suggest that GTP-loaded LCP delivery platform was efficacious in many tumor malignancies.
Fig. 6.

AA-targeted and GTP-loaded LCPs triggered tumor cell apoptosis and inhibited tumor cell proliferation effectively in vivo in NSCLC A549 tumor and a panel of pancreatic tumor xenografts. (A) Twenty-four h after 3 daily IV injections, mice were sacrificed and tumor tissues were sectioned for TUNEL assay and PCNA immunohistochemistry. Scale bar, 100 μm. (B) Percentage of apoptotic cells in tumors after the treatment of GTP-LCP-PEG-AA. (C) Percentage of PCNA positive cells in tumors after the treatment of GTP-LCP-PEG-AA. Statistics were as follows: (B) *p<0.01 vs. Untreated; ‡p<0.05 vs. Untreated. (C) **p<0.0005 vs. Untreated; *p<0.001 vs. Untreated; ‡p<0.01 vs. Untreated.
GTP-loaded LCPs prolonged blood circulation time in vivo
The pharmacokinetics (PK) experiment (Fig. 7) indicated a prolonged blood circulation time for the GTP-loaded LCPs compared to the free GTP. After bolus administration, about 35–40% of the LCPs were retained in the blood circulation after 2 h, while the free GTP was rapidly cleared within 20 min. The PK data were fitted with a non-compartment model using the WinNonlin program and the key PK parameters were calculated (Table 2). There was no significant difference between the PK profiles of AA-targeted and non-targeted LCPs. The results indicated that GTP-loaded LCPs increased the area under the curve (AUC) for more than 25-fold and decreased the clearance more than 25-fold compared to the free GTP. The mean residence time (MRT) increased about 7-fold, the steady state volume of distribution (Vss) decreased about 4-fold. Thus, compared to free GTP, LCP-formulated GTP had less chance to distribute into tissues, leading to an increase in plasma concentration profile. Taken together, incorporation GTP in LCPs increased the drug retention in the circulation, and reduced the drug distribution and clearance, which should result in the effective therapeutic response occurring at a lower dose.
Fig. 7.
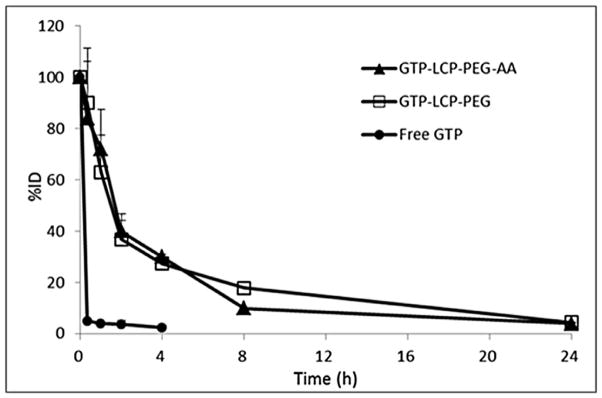
Percentage of the GTP injected dose (%ID) in blood after intravenous bolus administration of GTP-loaded LCPs and free GTP in H460 tumor-bearing mice.
Table 2.
The key pharmacokinetic parameters of GTP-loaded LCPs and free GTP in H460 xenograft model.
| t1/2 (h) | AUC (h·%) | AUMC (h2·%) | MRT (h) | CL (1/h) | Vss | |
|---|---|---|---|---|---|---|
| GTP-LCP-PEG-AA | 5.36 | 400.74 | 2122.30 | 5.30 | 0.25 | 1.33 |
| GTP-LCP-PET | 7.75 | 468.10 | 2812.46 | 6.01 | 0.21 | 1.26 |
| Free GTP | - | 14.89 | 25.34 | 0.83 | 6.72 | 5.58 |
Abbreviations: t1/2, elimination half-life; AUC: area under the curve; MRT: mean residence time; CL: clearance; Vss: steady state of volume distribution.
Tumor growth inhibition study
The tumor growth inhibition was evaluated in nude mice bearing either H460 or BxPC-3 subcutaneous tumors. As shown in Fig. 8A and 8B, GTP-LCP-PEG-AA treatment group exhibited the most efficient tumor growth inhibition after 4 systemic injections, and GTP-LCP-PEG also suppressed tumor growth effectively compared to the control and CTP-loaded LCPs. In GTP-LCP-PEG-AA treatment group, the final tumor loads were dramatically suppressed by 80% and 50% compared to the control group in H460 and BxPC-3 xenograft models, respectively. Free GTP and Gem showed limited therapeutic effects as compared to GTP-loaded LCPs. No serious toxicity, indicated by body weight changes, was observed (Fig. 8C and 8D).
Fig. 8.

GTP-loaded LCPs inhibited the tumor growth on H460 (A) and BxPC-3 (B) xenografts. The relative body weight changes of H460 (C) and BxPC-3 (D) xenografts were calculated. Data are mean ± S.D. (n=5). Statistics were as follows: **p<0.0005 vs. Untreated, ‡p<0.001 vs. Untreated, *p<0.005 vs. Untreated.
Hematological parameters were measured 24 h after 3 daily IV treatments (Table 3). There was little elevation of BUN, creatinine and liver enzymes (AST, ALT) in blood serum compared to the control, and all parameter values were in the normal range. Thus, GTP-loaded LCPs, at the therapeutic dose, did not elicit any toxicity in kidney and liver and there were no significant safety issues.
Table 3.
Serum levels of BUN, creatinine, AST, ALT after 3 daily IV injections.
| BUN mg/dl | Creatinine mg/dl | AST U/L | ALT U/L | |
|---|---|---|---|---|
| Control | 24 | 0.3 | 111.0 ± 4.2 | 56.0 ± 2.8 |
| GTP-LCP-PEG-AA | 24.7 ± 1.2 | 0.3 | 144.7 ± 12.9 | 69.3 ± 22.0 |
| GTP-LCP-PEG | 27.0 ± 2.8 | 0.3 | 148.5 ± 82.0 | 74.2 ± 25.5 |
| CTP-LCP-PEG-AA | 27.0 ± 1.4 | 0.3 | 134.0 ± 8.5 | 73.0 ± 12.7 |
|
| ||||
| Normal Range | 18--33.7 | 0.2--0.9 | 110.5--247 | 33.4--132 |
Discussion
All nucleoside analogues require transport into the cell and phosphorylation to the triphosphate form to be biologically active. Specific nucleoside transporters are responsible for the cellular uptake of the principal therapeutic nucleosides (i.e. gemcitabine) [17]. It is reported that high dose therapy with nucleoside analogs can overcome the mutational deficiency of nucleoside transporters and limiting rates of cellular entry by enhancing passive accumulation of the drug within the cell [25]. Additionally, due to the rapid enzyme deamination and renal clearance, Gem has a very short plasma half-life [26]. In order to achieve therapeutic drug levels, Gem has to be administered at a high dose (1,000 mg/m2) by intravenous infusion [26] which causes toxicity, particularly kidney and liver dysfunctions. The extensive usage of nucleoside analogs also results in the development of drug resistance [3].
In order to improve the therapeutic efficiency of nucleoside analogues, some prodrug and drug delivery approaches have been developed. Liposomes were used to load nucleoside drugs due to their biphasic (lipophilic/hydrophilic) character. But most nucleoside analogues are located exclusively in the aqueous compartment of liposomes because of their relatively high solubilities. However, they readily diffuse through the liposome bilayer, which limits their stability on storage and reduces the drug concentration accumulated inside the tumors [27]. The fabrication of lipophilic nucleoside derivatives increased the cell membrane permeability and was expected to bypass the barriers of nucleoside transporters [28]. Other studies focused on entrapping the modified lipophilic nucleoside derivatives into liposome’s lipid bilayer [29] or micelle’s hydrophobic inner core [30]. Though these nanocarriers can alleviate the entrapped nucleoside drugs from enzymatic degradation, their therapeutic potency is still dependent on the conversion of the lipophilic prodrug to the nucleoside drug inside the tumor cells. Besides, the lipophilic nucleosides cannot overcome the resistance mutations in the kinases [31]. Thus, there is an unmet medical need for the development of new technologies allowing a more effective systemic delivery of nucleoside analogues for the treatment of cancer patients.
In this study, we report a novel systemic delivery platform of nucleoside analogs. Gemcitabine triphosphate (GTP), the bioactive form of Gem, was encapsulated into a nanoparticle system containing a calcium phosphate precipitate core, i.e. LCP. Such that the cellular uptake deficiency (i.e. deficiency in nucleoside transporters) and the following requirement of nucleoside kinase activation process of Gem (i.e. inactivation of dCK) can be avoided. Due to the triphosphate structure, GTP can be readily co-precipitated in the CaP core of LCP. A lipid bilayer was used to wrap around the LCP core to allow a high density of DSPE-PEG being grafted onto the surface. This nanoparticle design can protect the GTP from enzymatic degradation and renal clearance in vivo. The surface modification with PEG helps shield the cationic charge of the lipid bilayer and may minimize the interaction with circulating blood components [32].
With AA as a tumor specific targeting ligand, LCPs can be more effectively taken up into tumor cells via sigma-receptor-mediated endocytosis. When LCPs are delivered to acidic endosomes, the CaP core of LCPs rapidly dissolves to increase osmotic pressure, eventually bursting the endosomes and enabling the entrapped GTP to escape [8]. The cationic lipid DOTAP wrapping the LCP core may also promote the release of GTP from endosomes to the cytosol by destabilizing the anionic endosome membrane [33]. Altogether, the selection of a right drug formulated in a “smart” delivery system is likely to decrease the therapeutic dose required and corresponding dose-dependent adverse effects.
NSCLC and pancreatic cancers are among the top 3 leading causes of cancer death worldwide, and pancreatic cancer is particularly deadly. AA-targeted GTP-loaded LCPs showed a potent anti-cancer efficacy in NSCLC H460 and pancreatic BxPC-3 models at a low dose. In order to further prove the potential therapeutic applications of GTP-loaded LCPs in extensive tumor malignancies, we randomly selected 6 additional human pancreatic adenocarcinoma cell lines, and established the xenograft tumor models in nude mice. All these pancreatic tumor xenografts responded well to systemic treatment with AA-targeted GTP-loaded LCPs (Fig. 6). In addition, Gem is prone to elicit potent clinical efficacy in rapidly proliferating cancer cells with high metabolic activity due to its impediment of DNA replication [3]. This is probably the reason that H460 xenograft with fast growing tumor showed more significant apoptosis induction and proliferation inhibition than other cancer models. The different cell-killing effects may also be attributed to the extent of the tumors’ vascular abnormalities, which alter the EPR effect. The effectiveness of this LCP formulation to gemcitabine-resistant cancer models will under further investigation. Based on our extensive study on multiple NSCLC and pancreatic cancer models, we conclude that the targeted GTP-loaded LCPs elicit significant anti-cancer efficacy in vivo. This targeted LCP-formulated GTP represents a new class of improved nucleoside anticancer drug which should be further investigated for potential clinical trials.
Conclusion
Encapsulating the bioactive GTP in LCPs holds the advantages of overcoming chemoresistance, targeting specifically to tumor, lowering dosages and toxicities. We have shown that AA-targeted GTP-loaded LCPs can induce effective cell death and enhanced S-phase cell cycle arrest in NSCLC and pancreatic cell lines in vitro. In vivo, AA-targeted GTP-loaded LCPs effectively delayed the tumor growth of human NSCLC H460 and pancreatic BxPC-3 cancer xenografts, with little in vivo toxicity. Molecular analysis indicated that this high efficacy of targeted GTP-loaded LCPs was associated with its ability to induce dramatic apoptosis of tumor cells, as shown in the TUNEL assay, caspase-3/7 activation and the cleaved PARP overexpression in tumor tissues; as well as significantly inhibit tumor cell proliferation and cell cycle progression, as indicated in the PCNA immunohistochemistry and the cyclin D1 down-regulation in tumor tissues. To the best of our knowledge, this is the first report using gemcitabine bioactive form, GTP, instead of any forms of gemcitabine pro-drug as the delivery cargo for cancer therapy. Further, we predicted that many chemodrugs with phosphate group(s) could be entrapped into LCPs and delivered systemically, so that some of the chemodrugs will no longer rely on the relative kinase phosphorylation to be bioactive.
Supplementary Material
Supplementary Figure 1. In vitro drug release of GTP from LCP nanoparticles in PBS at 37 °C. The error bars were obtained from triplicate samples.
Supplementary Figure 2. Statistical analysis of cell cycle on (A) H460 and (B) BxPC-3 cells. *p<0.05, vs Untreated.
Supplementary Figure 3. Sigma receptor expression levels in NSCLC and pancreatic cell lines.
Supplementary Table 1. IC50 values in each treatment group in MTT assay on H460 and BxPC-3 cells (concentration unit: μM).
Acknowledgments
We thank Dr. Srinivas Ramishetti for testing GTP compound, Dr. Jinfeng Du for data analysis and Kelly Racette for editing the manuscript. Research was supported by NIH grants CA151652 and CA149363.
List of Abbreviations
- NSCLC
Non-Small-Cell-Lung Cancer
- GTP
Gemcitabine Triphosphate
- LCP
Lipid Calcium Phosphate nanoparticle
- Gem
Gemcitabine
- dCK
deoxycytidine kinase
- AA
anisamide
- CTP
Cytidine Triphosphate
- PEG
Polyethylene glycol
- TEM
Transmission Electron Microscope
- TUNEL
TdT-mediated dUTP Nick-End Labeling
- PCNA
Proliferating Cell Nuclear Antigen
- BUN
Blood Urine Nitrogen
- AST
Aspartate aminotransferase
- ALT
Alanine aminotransferase
- CaP
Calcium Phosphate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Achanta G, Pelicano H, Feng L, Plunkett W, Huang P. Interaction of p53 and DNA-PK in response to nucleoside analogues: potential role as a sensor complex for DNA damage. Cancer Res. 2001;61(24):8723–9. [PubMed] [Google Scholar]
- 2.Parker WB. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev. 2009;109(7):2880–93. doi: 10.1021/cr900028p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Senanayake TH, Warren G, Vinogradov SV. Novel anticancer polymeric conjugates of activated nucleoside analogues. Bioconjug Chem. 2011;22(10):1983–93. doi: 10.1021/bc200173e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Moorsel CJ, Peters GJ, Pinedo HM. Gemcitabine: Future Prospects of Single-Agent and Combination Studies. Oncologist. 1997;2(3):127–34. [PubMed] [Google Scholar]
- 5.Hung SW, Mody HR, Govindarajan R. Overcoming nucleoside analog chemoresistance of pancreatic cancer: a therapeutic challenge. Cancer Lett. 2012;320(2):138–49. doi: 10.1016/j.canlet.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allain V, Bourgaux C, Couvreur P. Self-assembled nucleolipids: from supramolecular structure to soft nucleic acid and drug delivery devices. Nucleic Acids Res. 2012;40(5):1891–903. doi: 10.1093/nar/gkr681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rejiba S, Reddy LH, Bigand C, Parmentier C, Couvreur P, Hajri A. Squalenoyl gemcitabine nanomedicine overcomes the low efficacy of gemcitabine therapy in pancreatic cancer. Nanomedicine. 2011;7(6):841–9. doi: 10.1016/j.nano.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Chen YC, Tseng YC, Mozumdar S, Huang L. Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery. J Control Release. 2010;142(3):416–21. doi: 10.1016/j.jconrel.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–92. [PubMed] [Google Scholar]
- 10.Li SD, Huang L. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochim Biophys Acta. 2009;1788(10):2259–66. doi: 10.1016/j.bbamem.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banerjee R, Tyagi P, Li S, Huang L. Anisamide-targeted stealth liposomes: a potent carrier for targeting doxorubicin to human prostate cancer cells. Int J Cancer. 2004;112(4):693–700. doi: 10.1002/ijc.20452. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Yang Y, Huang L. Calcium phosphate nanoparticles with an asymmetric lipid bilayer coating for siRNA delivery to the tumor. J Control Release. 2012;158(1):108–14. doi: 10.1016/j.jconrel.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Wang X, Wang J, Zhang X, Zhang Q. Octreotide-modified polymeric micelles as potential carriers for targeted docetaxel delivery to somatostatin receptor overexpressing tumor cells. Pharm Res. 2011;28(5):1167–78. doi: 10.1007/s11095-011-0381-1. [DOI] [PubMed] [Google Scholar]
- 14.Risbood PA, Kane CT, Jr, Hossain MT, Vadapalli S, Chadda SK. Synthesis of gemcitabine triphosphate (dFdCTP) as a tris(triethylammonium) salt. Bioorg Med Chem Lett. 2008;18(9):2957–8. doi: 10.1016/j.bmcl.2008.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karl DM, Craven DB. Effects of alkaline phosphatase activity on nucleotide measurements in aquatic microbial communities. Appl Environ Microbiol. 1980;40(3):549–61. doi: 10.1128/aem.40.3.549-561.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim IY, Kang YS, Lee DS, et al. Antitumor activity of EGFR targeted pH-sensitive immunoliposomes encapsulating gemcitabine in A549 xenograft nude mice. J Control Release. 2009;140(1):55–60. doi: 10.1016/j.jconrel.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Johnson SA. Clinical pharmacokinetics of nucleoside analogues: focus on haematological malignancies. Clin Pharmacokinet. 2000;39(1):5–26. doi: 10.2165/00003088-200039010-00002. [DOI] [PubMed] [Google Scholar]
- 18.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326 (Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arya G, Vandana M, Acharya S, Sahoo SK. Enhanced antiproliferative activity of Herceptin (HER2)-conjugated gemcitabine-loaded chitosan nanoparticle in pancreatic cancer therapy. Nanomedicine. 2011;7(6):859–70. doi: 10.1016/j.nano.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 20.Tang SN, Fu J, Shankar S, Srivastava RK. EGCG Enhances the Therapeutic Potential of Gemcitabine and CP690550 by Inhibiting STAT3 Signaling Pathway in Human Pancreatic Cancer. PLoS One. 2012;7(2):e31067. doi: 10.1371/journal.pone.0031067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andreeva ER, Orekhov AN. Evaluation of cell proliferation in human atherosclerotic lesions. Methods Mol Med. 2001;52:213–8. doi: 10.1385/1-59259-073-X:213. [DOI] [PubMed] [Google Scholar]
- 22.Hornick JR, Spitzer D, Goedegebuure P, Mach RH, Hawkins WG. Therapeutic targeting of pancreatic cancer utilizing sigma-2 ligands. Surgery. 2012;152(3 Suppl 1):S152–6. doi: 10.1016/j.surg.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riganas S, Papanastasiou I, Foscolos GB, et al. Synthesis, sigma(1), sigma(2)-receptors binding affinity and antiproliferative action of new C1-substituted adamantanes. Bioorg Med Chem. 2012;20(10):3323–31. doi: 10.1016/j.bmc.2012.03.038. [DOI] [PubMed] [Google Scholar]
- 24.Everaert H, Flamen P, Franken PR, Verhaeghe W, Bossuyt A. Sigma-receptor imaging by means of I123-IDAB scintigraphy: clinical application in melanoma and non-small cell lung cancer. Anticancer Res. 1997;17(3B):1577–82. [PubMed] [Google Scholar]
- 25.White JC, Rathmell JP, Capizzi RL. Membrane transport influences the rate of accumulation of cytosine arabinoside in human leukemia cells. J Clin Invest. 1987;79(2):380–7. doi: 10.1172/JCI112823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brusa P, Immordino ML, Rocco F, Cattel L. Antitumor activity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. Anticancer Res. 2007;27(1A):195–9. [PubMed] [Google Scholar]
- 27.Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Control Release. 2004;100(3):331–46. doi: 10.1016/j.jconrel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 28.Lansakara PD, Rodriguez BL, Cui Z. Synthesis and in vitro evaluation of novel lipophilic monophosphorylated gemcitabine derivatives and their nanoparticles. Int J Pharm. 2012;429(1–2):123–34. doi: 10.1016/j.ijpharm.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarpietro MG, Ottimo S, Giuffrida MC, Rocco F, Ceruti M, Castelli F. Synthesis of n-squalenoyl cytarabine and evaluation of its affinity with phospholipid bilayers and monolayers. Int J Pharm. 2011;406(1–2):69–77. doi: 10.1016/j.ijpharm.2010.12.038. [DOI] [PubMed] [Google Scholar]
- 30.Li Q, Du YZ, Yuan H, et al. Synthesis of lamivudine stearate and antiviral activity of stearic acid-g-chitosan oligosaccharide polymeric micelles delivery system. Eur J Pharm Sci. 2010;41(3–4):498–507. doi: 10.1016/j.ejps.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 31.Meier C, Balzarini J. Application of the cycloSal-prodrug approach for improving the biological potential of phosphorylated biomolecules. Antiviral Res. 2006;71(2–3):282–92. doi: 10.1016/j.antiviral.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Satterlee A, Huang L. In Vivo Gene Delivery by Nonviral Vectors: Overcoming Hurdles? Mol Ther. 2012 doi: 10.1038/mt.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Y, Szoka FC., Jr Mechanism of DNA release from cationic liposome/DNA complexes used in cell transfection. Biochemistry. 1996;35(18):5616–23. doi: 10.1021/bi9602019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. In vitro drug release of GTP from LCP nanoparticles in PBS at 37 °C. The error bars were obtained from triplicate samples.
Supplementary Figure 2. Statistical analysis of cell cycle on (A) H460 and (B) BxPC-3 cells. *p<0.05, vs Untreated.
Supplementary Figure 3. Sigma receptor expression levels in NSCLC and pancreatic cell lines.
Supplementary Table 1. IC50 values in each treatment group in MTT assay on H460 and BxPC-3 cells (concentration unit: μM).