

Abstract

An efficient and simple route to biologically and pharmaceutically important o-hydroxyaryl ketones, xanthones, 4-chromanones, and flavones has been developed utilizing readily available carboxylic acids and commercially available o-(trimethylsilyl)aryl triflates.
1. Introduction
Aryne insertions into C-N,1,2 C-C,3 C-Si,4 C-Sn,5 and C-Hal6 bonds and the C=O bond of aldehydes7 have been well documented in the literature. The formal insertion of a benzyne ring into the C-O bond of a carboxylic acid would provide a powerful synthetic transformation to access a range of biologically important molecules. In our previous work, we have reported that the reaction of arenecarboxylic acids with arynes in MeCN provides the corresponding aryl esters in excellent yields, i.e. the product is formed by formal benzyne insertion into the O-H bond of the starting material (Scheme 1).8 Following up on this chemistry, we found and reported in an earlier communication reaction conditions, which allow one to obtain good to excellent yields of the corresponding C-O insertion products.9 Later, Ma and coworkers reported an interesting cascade sequence that starts from 2,3-allenoic acids and affords chromones in good to excellent yields using KF/18-crown-6 in THF.10 The present manuscript provides a complete account of the scope and limitations of the synthesis of o-hydroxyaryl ketones starting from carboxylic acids, as well as its applications to the synthesis of xanthones, 4-chromanones and flavones.
Scheme 1.

Known Reaction of Arenecarboxylic Acids and Arynes in MeCN.
2. Results and Discussion
2.1. Synthesis of o-Hydroxyaryl Ketones
We allowed a simple aliphatic carboxylic acid (butyric acid, 1) to react with the Kobayashi benzyne precursor o-(trimethylsilyl)phenyl triflate11 (2) in the presence of CsF in acetonitrile (Table 1). At both room temperature and at 65 °C, as expected,8 the O-arylation product, ester 4, was predominately formed (entries 1 and 2). To avoid the formation of the latter product, we switched to a far less acidic solvent, THF. To our delight, together with the monoarylation product 4, the o-hydroxyaryl ketone 3 was formed in a 26% yield (entry 3).
Table 1.
Optimization of Benzyne Insertion into the C-O bond of an Aliphatic Carboxylic Acid.a

| |||||
|---|---|---|---|---|---|
| entry | equiv of 2 | fluoride source (equiv) | solvent, mL | temp. (°C) | % yield of 3b |
| 1 | 1.0 | CsF (3) | MeCN, 5 | rt | 0c |
| 2 | 1.0 | CsF (3) | MeCN, 5 | 65 | 0c |
| 3 | 1.0 | CsF (3) | THF, 5 | 65 | 26 |
| 4 | 1.0 | CsF (3) | THF, 5 | 125 | 43 |
| 5 | 1.0 | CsF (3) | DME, 5 | 65 | 20c |
| 6 | 1.0 | CsF (3) | DME, 5 | 125 | 27c |
| 7d | 1.0 | CsF (3) | THF, 5 | 125 | 14c |
| 8e | 1.0 | CsF (3) | THF, 5 | 125 | 4c |
| 9 | 1.2 | CsF (3) | THF, 15 | 125 | 50 |
| 10 | 1.5 | CsF (4) | THF, 15 | 125 | 77 |
| 11 | 1.75 | CsF (4.7) | THF, 15 | 125 | 70 |
| 12 | 2 | CsF (6) | THF, 15 | 125 | 34 |
| 13 | 1.5 | TBAT (2) | Tol, 5 | 50 | 34c |
All reactions were carried out on a 0.25 mmol scale in 5 mL of solvent during a 24 h period.
Isolated yields, unless stated otherwise.
1H NMR spectroscopic yields using an internal standard.
1 Equiv of K2CO3 was added to the reaction mixture.
1 Equiv of Cs2CO3 was added to the reaction mixture.
A closer examination of the reaction mixture revealed that additional products inseparable by chromatography, namely tertiary alcohol 5 and xanthene 6, both bis-aryne insertion products, are produced (Scheme 2). Based on our findings, we propose the following mechanism for the formation of products 3-6. The aryl anion intermediate 8, formed after nucleophilic attack of the carboxylate group of the carboxylic acid on the highly electrophilic benzyne (7) generated in situ, undergoes a formal anionic Fries rearrangement to the phenoxide 10 by a four-membered ring intermediate 9.12 This results in formation of the desired o-hydroxyaryl ketone 3 after proton abstraction from the reaction media. The monoarylation product 4 can be formed by protonation of the aryl anion 8. The by-product 5 is likely formed from reaction of the phenoxide 10 with an additional benzyne intermediate 7,13 which, followed by dehydration, leads to formation of the xanthene 6. The proposed 4-membered ring intermediate 9 is consistent with similar intermediates reported recently by Greaney’s group in a related study of primary aromatic amides.2
Scheme 2.

Proposed Mechanism for the Reaction of Butyric Acid and Benzyne.
Isolation of the C-O insertion product 3 prompted us to pursue this project, as it constitutes an expeditious route to o-hydroxyaryl ketones, often used as precursors for the synthesis of biologically important flavones and chalcones,14 starting from cheap and easily accessible carboxylic acids. Most traditional synthetic approaches to o-hydroxyaryl ketones ultimately proceed by the Friedel-Crafts acylation of phenols with acyl chlorides in the presence of Lewis acids,15 or the Fries rearrangement of suitable phenyl esters.16 Both of these procedures suffer from regioselectivity issues and proceed in the presence of strong Lewis acids, which limits the potential of this transformation. It is noteworthy that o-hydroxyaryl ketones are an important class of biologically interesting structures,17 and some of them, such as cotoin and hydrocotoin, have been found in nature.18
Unexpectedly, running the reaction at higher temperatures allowed us to achieve higher yields of the desired hydroxyaryl ketone, at the same time lowering the relative ratios of the major side-products 4-6. Running the reaction at 125 °C (sealed vial) allowed us to isolate the desired hydroxyaryl ketone 3 in a 43% yield (Table 1, entry 4). Changing the solvent to 1,2-dimethoxyethane (DME) resulted in lower yields of the desired product 3 and higher ratios of the arylation product 4 at both 65 °C and 125 °C (entries 5 and 6). To test the possibility that the undesired protonation of the intermediate 8 is caused by the proton of the starting acid, we attempted to run the reaction in the presence of a stoichoimetric amount of different bases. Unfortunately, the use of K2CO3 as an additive resulted in a lower (14%) yield, which might be caused by the poor solubility of the butyrate formed (entry 7). Running the reaction in the presence of Cs2CO3 resulted in a still lower (4%) yield of the desired product with significant amounts of the ester 4 and the alkene 6 being detected (entry 8). Other bases (e.g., NaH, Na2CO3, KOPiv, sym-collidine) failed to improve the yield of the desired hydroxyaryl ketone. Using the sodium salt of the butyric acid or its TMS-ether resulted in lower yields of the desired product 3 and higher relative ratios of the alkene 6.
Increasing the amount of the benzyne precursor 1 from 1.0 to 1.2 equiv and diluting the reaction mixture resulted in an increased (50%) yield of the desired product (entry 9). A further increase in the amount of the benzyne precursor (up to 2 equiv) revealed that the highest yield (entry 10, 77%) is achieved with a 1.5-fold excess of the benzyne precursor to the starting butyric acid (entries 9-12). A further increase in the amount of benzyne results in higher ratios of the bis-arylated products 5 and 6, thus lowering the yield of the desired product 3. Extending the time of the reaction and using more CsF does not have any significant effect on the yield of the desired ketone 3. We also attempted to run the reaction under Greaney’s conditions, tetrabutylammonium triphenyldifluorosilicate (TBAT) in toluene at 50 °C.2 However, only a 34% yield of the product 3 was observed in this case, while the undesired ester 4 was detected in a 54% yield (entry 13). We subsequently applied our optimized conditions (Table 1, entry 10) to other carboxylic acids (Table 2).
Table 2.
Reaction of Carboxylic Acids with Arynes.a

| |||
|---|---|---|---|
| entry | R | product | yieldb (%) |
| 1 |
|
3 | 77 |
| 2 |
|
12 | 68 |
| 3 |
|
13 | 62 |
| 4 |
|
14 | 72 |
| 5 |

|
15 | 58 |
| 6 |

|
16 | 39 |
| 7 |

|
17 | 30 |
| 8 |

|
18 | 44 |
Reaction conditions: 0.25 mmol of acid, 1.5 equiv of benzyne precursor and 4.0 equiv of CsF in 15 mL of THF were heated in a closed vial at 125 °C for 24 h.
Isolated yield.
This methodology tolerates cycloalkyl and benzyl groups. The corresponding products 12 and 13 were obtained in 68% and 62% yields, respectively (entries 2 and 3). The alkene-containing product 14 was successfully isolated in a 72% yield (entry 4). The ester- and ketone-containing carboxylic acids provided the desired products 15 and 16 in lower 58% and 39% yields, respectively (entries 5 and 6).
A number of α-substituted carboxylic acids, such as N-methylindoleacetic acid, N-acetylglycine, succinimidoacetic acid, and α-bromophenylacetic acid were examined under our optimized reaction conditions. Unfortunately, presumably due to either the increased acidity of the protons next to the reacting carboxylic acid functionality and/or the low solubility of some of these substrates in THF, most of these reactions resulted in the formation of inseparable mixtures of by-products with the undesired O-arylation products often being formed in considerable amounts. One of the more successful examples of this type of carboxylic acid is 3-thiopheneacetic acid, which resulted in formation of the hydroxyaryl ketone 17 in a 30% yield (entry 7).
In the case of arenecarboxylic acids, perhaps due to the increased acidity of the resulting hydroxyaryl ketones, over-arylation products were a major concern not only because of decreased yields of the desired insertion products, but also because of complications during their isolation. Thus, in the case of p-nitrobenzoic acid, the corresponding tertiary alcohol analogous to intermediate 5 could be isolated in a 54% yield and none of the desired hydroxyaryl ketone was detected. Changing the electronic properties of the substituents on the phenyl ring had little effect on the outcome of the reaction. The desired products of p-methoxy- and p-methylbenzoic acids were formed in less than 30% yields (as measured by 1H NMR spectroscopic analysis using an internal standard). Unfortunately, none of these products could be isolated due to a number of side-products with similar polarities. One of the more successful reactions was with β-naphthoic acid, which afforded the corresponding o-hydroxyaryl ketone 18 in a 44% yield (entry 8).
Allowing butyric acid to react with an excess (3 equiv) of the benzyne precursor in DME, the bis-aryne insertion xanthene product 6 was obtained in a 50% yield (Scheme 3).19 Xanthenes are a pharmaceutically important scaffold with many members shown to possess antimalarial,20 antitrypanosomal,21 antileishmanial,21 and antitumor22 activities.
Scheme 3.

Synthesis of a Xanthene.
We also examined the reaction of butyric acid with the unsymmetrical dimethoxy-substituted benzyne precursor 19. 23 We were delighted to observe the regioselective formation of the desired isomer 20 in a 49% yield (Scheme 4). The regioselectivity is consistent with that reported by the Stoltz group.23 The product 20 is a natural product that has been found in an Indian shrub Dysophylla stellata Benth.24
Scheme 4.

Synthesis of a Naturally-occurring Hydroxyaryl Ketone.
2.2. Synthesis of Xanthones
While the low efficiency of the reaction of arenecarboxylic acids with arynes is likely due to the high reactivity of the phenolic OH group of the resulting o-hydroxyaryl ketones towards arynes, trapping the phenol by an intramolecular SNAr reaction could provide an interesting route to xanthones and analogues, which are very important ring systems in biology and pharmacy.25
Indeed, a close examination of the reaction between o-methoxybenzoic acid and benzyne revealed that about 58% of the starting material has been converted to the xanthone 21 and only 12% could be assigned as the expected o-hydroxyaryl ketone. When o-halobenzoic acids are allowed to react with the benzyne precursor and CsF under our previously optimized conditions, we were delighted to see formation of the xanthone 21 (Table 3). While o-chloro and o-iodobenzoic acids provided poor yields (30% and 38%, respectively), the o-fluorobenzoic acid afforded the desired xanthone in an 80% yield (entry 1).
Table 3.
Reaction of o-Haloarenecarboxylic Acids with Arynes.a

| |||
|---|---|---|---|
| entry | Hal | product | yieldb (%) |
| 1 | F |
 21 |
80 |
| 2 | F |
 22 |
79 |
| 3 | Cl |
 23 |
22 |
| 4 | Cl |
 24 |
87c |
| 5 | Br |
 25 |
55c |
| 6 | Br |
 26 |
59c,d |
| 7 | Br |
 27 |
49c,d,e |
| 8 | F |
 28 |
47f |
| 9 | F |
 29 |
71g |
Reaction conditions: 0.25 mmol of the carboxylic acid, 1.5 equiv of the benzyne precursor and 4.0 equiv of CsF in 15 mL of THF were heated in a closed vial at 125 °C for 24 h.
Isolated yield.
Reaction conditions: 0.25 mmol of the carboxylic acid, 1.5 equiv of the aryne precursor, and 2.0 equiv of TBAT in 5 mL of toluene were heated at 60 °C for 24 h.
The isolated o-hydroxyaryl ketone was heated in MeCN in the presence of 2 equiv of K2CO3 at 100 °C for 24 h to provide the desired xanthone.
3,5-Dimethoxy-2-(trimethylsilyl)phenyl triflate was used as the aryne precursor.
4,5-Dimethyl-2-(trimethylsilyl)phenyl triflate was used as the aryne precursor.
3-Methoxy-2-(trimethylsilyl)phenyl triflate was used as the aryne precursor.
Our xanthone process tolerates other halides present in the benzoic acid moiety. Thus, the bromo-substituted xanthone 22 was isolated in a 79% yield (entry 2). The presence of the bromide functionality provides a useful handle for further diversification of the system if a combinatorial library of these compounds is desired.26
We could obtain 4-aza-xanthone (23), albeit in only a 22% yield (entry 3), starting from 2-chloronicotinic acid. The poor yield in this reaction can be attributed to the high reactivity of the nucleophilic pyridine nitrogen toward benzyne, an often reported process in the literature.27 The reaction of the electron-poor nitro-substituted carboxylic acid under our optimized reaction conditions provided the desired xanthone 24 in only a 48% yield, with complications during isolation of the desired product. However, running this reaction under reaction conditions reported by the Greaney group for a related insertion into the C-N bond of aromatic amides2 afforded the xanthone 24 in a high 87% yield (entry 4). Running the reaction of 1-bromo-2-naphthoic acid under analogous conditions afforded the tetracyclic xanthone 25 in a 55% yield (entry 5).
The reaction of electron-rich 2-bromo-4,5-dimethoxybenzoic acid with benzyne afforded only trace amounts of the desired product 26 in both THF and DME. Employing the reaction conditions reported by the Greaney group, in this case provided the uncyclized o-hydroxyaryl ketone that upon cyclization in acetonitrile at elevated temperatures in the presence of K2CO3 afforded the desired xanthone 26 in a 59% overall yield (entry 6). Interestingly, the bis-demethylated version of xanthone 26 has been shown to significantly inhibit the growth of melanoma cancer cells and the mitogenic response of human lymphocytes to a common mitogen PHA.28
Our standard xanthone protocol has also been used for the reaction of 2-bromo-4,5-dimethoxybenzoic acid with the unsymmetrical dimethoxybenzyne precursor 19. After the benzyne insertion and subsequent induced SNAr reaction, the final tetraoxygenated xanthone 27 was isolated in a 49% overall yield (entry 7). It is noteworthy that the compound 27 is found in nature, as well as partially demethylated analogues. 29 The tetrademethylated version, norathyriol, has been found in at least 19 different natural sources and has been shown to possess hypotensive activity.30 It is also an effective inhibitor of cutaneous plasma extravasation31 and a monoamine oxidase inhibitor.32
Reacting o-fluorobenzoic acid with 4,5-dimethylbenzyne and unsymmetrical 3-methoxybenzyne under our CsF/THF optimized conditions afforded the corresponding xanthones 28 and 29 in 47% and 71% yields, respectively (entries 8 and 9). The low yield in the former case may be attributed to the poor solubility (and hence isolation complications) of the desired product 28. The unsymmetrical benzyne precursor provided the product 29 as a single regioisomer.33
2.3. Synthesis of 4-Chromanones and Flavones
The phenolate anion (see intermediate 10 in Scheme 2) formed in situ could also be potentially trapped with a Michael acceptor in an intramolecular fashion. This would open up a novel route to biologically interesting 4-chromanones, flavones, and chromones from readily available acrylic and propiolic acids.34
Indeed, in the reaction of methacrylic acid with benzyne under the optimized reaction conditions used in the synthesis of o-hydroxyaryl ketones, the 4-chromanone 30 was formed in a moderate 58% yield. Surprisingly, none of the dihydrocoumarin product that could result from the ring closure of an intermediate like 8 was detected, which suggests that the formation of the 4-membered ring intermediate is a more favorable pathway. None of the uncyclized hydroxyaryl ketone product was detected either, suggesting a very fast rate of cyclization for the final Michael addition reaction. Running the methacrylic acid reaction in the presence of various bases (e.g. Cs2CO3, Et3N, iPr2NEt, sym-collidine) failed to improve the yield of the reaction. Substituting the substrate with either the TES- or TBDMS-esters of methacrylic acid both afforded the desired product in lower yields. Running the reaction at decreased temperatures and/or switching the solvent to DME resulted in a lower yield of the desired product. After additional optimization, we have found that diluting the reaction mixture helps to improve the yield of the 4-chromanone 30 by ~10%. Apparently, one of the side reactions involves an intermolecular process. One of the significant challenges observed in this transformation is the poor reactivity of the starting carboxylic acid. As a result, running the reaction with <1.5 equiv of the benzyne precursor does not lead to complete conversion of the starting carboxylic acid.35 Running the reaction with >2.0 equiv of the precursor 2 allows the carboxylic acid to react completely. However, it also results in increased amounts of overarylation products (e.g. an alcohol analogous to intermediate 5 in Scheme 2). Running the reaction under dilute conditions with 1.75 equiv of the benzyne precursor provides an 80% yield of the desired product 30. Finally, adding the benzyne precursor and CsF in two separate portions allowed us to improve the yield up to 84%.36 The latter reaction conditions were applied to a number of other 2-alkenoic acids (Table 4).
Table 4.
Reaction of Acrylic Acids with Arynes.a

| |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | product | yieldb (%) |
| 1 | Me | H | H | 30 | 84 |
| 2 | H |

|
H | 31 | 85 |
| 3 | H | nPr | H | 32 | 82 |
| 4 | H | NC(CH2)3 | H | 33 | 71 |
| 5 | H | Me | Me | 34 | 77 |
| 6 | H |

|
Me | 35 | 64 |
| 7 | Ph | H | H | 36 | 56 |
| 8 | H | Ph | H | 37 | 74c |
| 9 | Me | Me | H | 38 | 76d |
| 10 | Me | Ph | H | 39 | 67e |
| 11 |

|
H | 40 | 78 | |
| 12 | H | CO2Me | H | 41 | 0 |
| 13 | H | Me | Me |
 42 |
53f |
| 14 | H | Ph | H |
 43 |
29c,g |
Reaction conditions: 0.25 mmol of the carboxylic acid, 1.5 equiv of aryne precursor and 4.0 equiv of CsF in 15 mL of THF were heated in a closed vial at 125 °C for 18 h. Then an additional 0.5 equiv of the aryne precursor and 1.0 equiv of CsF were added and the heating continued at 125 °C for 6 h.
Isolated yield.
The yield includes the product obtained after base-induced cyclization of the o-hydroxyaryl ketone (see the Supporting Information).
The E/Z ratio is ~1.8/1.
The E/Z ratio is ~5.1/1.
4,5-Dimethoxy-2-(trimethylsilyl)phenyl triflate was used as the aryne precursor.
3,5-Dimethoxy-2-(trimethylsilyl)phenyl triflate was used as the aryne precursor.
3-Alkyl-substituted acrylic acids successfully provided the corresponding cyclohexyl-substituted and propyl-substituted 4-chromanones 31 and 32 in 85% and 82% yields, respectively (entries 2 and 3). The carboxylic acid with a nitrile substituent at the end of an alkyl chain provided the 4-chromanone 33 in a 71% yield (entry 4). 3,3-Dialkyl-substituted acrylic acids (3-methyl-2-butenoic acid and geranic acid) provided the corresponding chromanones 34 and 35 in good yields (77% and 64%, respectively) (entries 5 and 6). α-Phenylacrylic acid provided the cyclized product 36 in a 56% yield (entry 7). Interestingly, in the case of cinnamic acid, a 60% yield of the desired product 37 was obtained along with the uncyclized o-hydroxyaryl chalcone. Treating the reaction mixture with a base (piperidine/THF/H2O)37 cyclized the hydroxyaryl ketone to the desired flavanone 37 in a 74% overall yield (entry 8). 2,3-Disubstituted acrylic acids provided diastereomeric mixtures of chromanones 38 and 39 in 76% and 67% yields, respectively (entries 9 and 10). Cyclopentene-1-carboxylic acid afforded the tricyclic product 40 in a 78% yield with exclusively a cis configuration at the 5,6-ring junction (entry 11). Unfortunately, monomethyl fumarate (as well as monomethyl maleate), bearing an ester group on the carbon-carbon double bond, did not provide any of the expected chromanone product (entry 12). One possible explanation might be the decreased nucleophilicity of the carboxylic group toward the benzyne caused by the presence of the two electron-withdrawing groups. Other carboxylic acids that provided low yields (less than 30%) of the expected products include 2,4-alkadienoic (2,4-hexadienoic and 5-phenyl-2,4-pentadienoic) acids and 3-heteroaryl-substituted (2-thienyl and 3-indolyl) acrylic acids. These substrates could potentially engage in Diels-Alder reactions with the dienophilic benzyne at such high temperatures.
We have also examined the reaction of alkenoic acids with other benzyne precursors. The symmetrical 4,5-dimethoxybenzyne, when allowed to react with 3,3-dimethylacrylic acid, provided the cyclized product 42 in a 53% yield.38 It is noteworthy that an analogue of this compound, precocene II, exhibits significant anti-juvenile hormone activity in insects.39
When cinnamic acid was allowed to react with the unsymmetrical dimethoxy-benzyne precursor 19, at first the uncyclized o-hydroxychalcone was isolated in a 68% yield. Subjecting the latter compound to a piperidine-promoted intramolecular Michael addition reaction resulted in formation of the desired chromanone 43 in a 48% yield, along with 42% of the unreacted starting material. A chiral version of compound 43 is a natural product found in Caesalpinia pulcherrima,40 and the monodemethylated version of the latter, known as pinostrobin, is reported to be an effective aromatase inhibitor.41
The reaction of itaconic acid methyl ester 44 with benzyne under our optimized conditions did not result in formation of the expected flavanone. Instead, the 3-coumaranone derivative 45 was isolated in a 79% yield (Scheme 5). Since monomethyl fumarate did not provide any isolable product under identical reaction conditions (Table 4, entry 12), this reaction is best explained mechanistally as follows: a) the carboxylic acid undergoes the expected insertion with benzyne to provide intermediate 46; b) the latter isomerizes to a more stable isomer 47 under the reaction conditions; c) intramolecular Michael addition in the substrate 47 results in formation of the 5-membered ring product 45.
Scheme 5.

Mechanism for the Formation of Coumaranone 45.
We have also examined the reaction of phenylpropiolic acid with benzyne in THF under our optimized reaction conditions. Unfortunately, due to the high temperature of the reaction, the major product observed was the decarboxylated starting material. To minimize the side reaction observed in the reaction of phenylpropiolic acid, we attempted to decrease the temperature to 90 °C. Gratifyingly, the yield of the product increased to 48%. If we employed TBAT, the desired product 48 could be isolated in a similar 49% yield, while running the reaction at only 65 °C. We then replaced THF with toluene, essentially employing reaction conditions analogous to those reported by Greaney’s group using amides.2 Running the reaction in toluene at 60 °C allowed us to obtain the flavone 48 in increased yields, with the optimal conditions employing 1.5 equiv of the benzyne precursor (56% yield; Table 5, entry 1).
Table 5.
Reaction of 2-Alkynoic Acids with Arynes.a

Reaction conditions: 0.25 mmol of the carboxylic acid, 1.5 equiv of the aryne precursor and 2.0 equiv of TBAT in 5 mL of toluene were heated at 60 °C for 24 h.
Isolated yield.
Reaction conditions: 0.25 mmol of the carboxylic acid, 1.5 equiv of the aryne precursor and 2.0 equiv of TBAT in 15 mL of THF were heated at 65 °C for 24 h.
Using these optimized reaction conditions, we have examined the reaction of several 2-alkynoic acids with benzyne. 2-Butynoic acid provided the chromone 49 in a 64% yield (entry 2). The reaction of the unsubstituted propiolic acid with CsF in THF did not provide any recognizable products. However, running the reaction in the presence of TBAT provided the desired chromone 50 in a 71% yield (entry 3). Unfortunately, acetylenedicarboxylic acid provided mixtures of unidentified products using either the CsF- or TBAT-mediated protocols.
It is noteworthy that we can also obtain chromones in the reaction of 2- and 3-haloalkenoic acids with benzyne. Apparently, the chromanones produced with 2-bromoacrylic and 2-bromocyclohexenoic acids undergo an elimination reaction furnishing the chromones 50 and 53 in 18% and 85% yields, respectively (Scheme 6). The low yield of the former product might be due to a less favorable dehydrohalogenation reaction, as well as the possible low stability of the starting material under the reaction conditions that we have employed.
Scheme 6.

Reaction of Haloalkenoic Acids with Benzyne.
Further Mechanistic Considerations
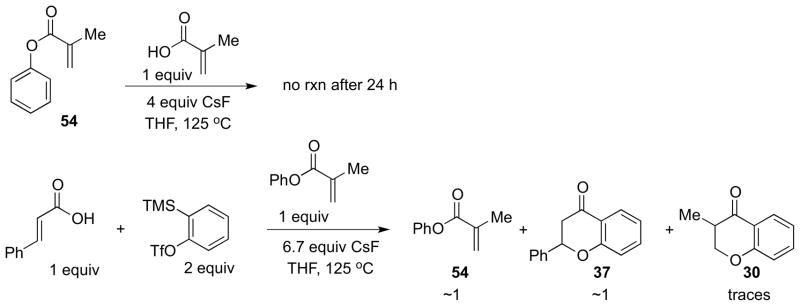
Some further mechanistic considerations follow. An acid-catalyzed Fries rearrangement leading from ester 4 (see Scheme 2) to an o-hydroxyaryl ketone (analogous to compound 3) has been considered as a mechanistic possibility for our overall transformation. Based on literature accounts, however, strong acidic media is absolutely necessary for such a rearrangement to occur.42 Nevertheless, we allowed phenyl methacrylate (54) to react with methacrylic acid in THF in the presence of CsF at 120 °C (Scheme 7). As expected, no 4-chromanone was detected under these reaction conditions after 24 hours. Replacing the basic CsF media with 1 equiv of BF3·Et2O did not result in the formation of the target compound 30 either. However, the benzyne intermediate or molecules that result from its generation might play the role of a Lewis acid in this transformation. In a crossover experiment, we have allowed cinnamic acid to react with 2 equiv of the benzyne precursor in the presence of CsF and phenyl methacrylate at 125 °C. Unexpectedly, the chromanone 30, that could only be formed from the ester 54, was detected in the 1H NMR spectrum of the crude reaction mixture, albeit in only trace amounts. This suggests that the cationic Fries mechanism is, at the most, a very minor pathway for the transformation of acrylic acids into the corresponding chromanones.
Scheme 7.
Mechanistic Investigation of the Possible Cationic Fries Rearrangement.
We have also examined the possibility that methyl esters can engage in a similar C-O insertion process with a benzyne. While no traces of any reaction were observed in the case of methyl benzoate, methyl methacrylate unexpectedly resulted in the formation of two stable products in ~10% yields. Once the structures of these products were established, it became evident that no C-O insertion had taken place in the starting ester 55. Instead, because of the favorable configuration of the methacrylate unit, a set of two ene-reactions apparently occurs to produce the two ester products 56 and 57 (Scheme 8).
Scheme 8.

Mechanism of the Double Ene Reaction.
After optimization studies, it was found that running the methyl methacrylate reaction in acetonitrile at room temperature in the presence of an excess of the benzyne (3 equiv), the diarylated product 57 can be isolated in an 80% yield.
Other methods leading to stereodefined trisubstituted alkenes usually utilize multiple synthetic steps and require the use of expensive rhodium catalysts.43 We therefore examined the scope of our aryne ene reaction. Unfortunately, allylic systems similar to methyl methacrylate, such as methacrylonitrile, methyl trans-2-butenoate, 3-methyl-2-cyclohexenone, allyl and benzyl cyanides, resulted in only a very low conversion of the starting material or in the formation of mixtures of unidentified products. While a few ene reactions with a benzyne intermediate have been observed before,43 our reaction of methyl methacrylate proceeds under very mild conditions (MeCN, room temperature) and allows one to obtain the diarylated product 57 in a high yield.
3. Conclusions
In conclusion, we have developed an efficient and simple route to o-hydroxyaryl ketones, xanthones, 4-chromanones, flavones and their analogues by aryne incorporation into the C-O bond of easily accessible carboxylic acids and subsequent reactions. Various functional groups have proven to be compatible with the reaction conditions. The method should prove useful and reliable for accessing these biologically and pharmaceutically important heterocyclic structures.
4. Experimental section
4.1. General
The 1H NMR spectra were recorded at 300, 400, and 600 MHz as specified for particular compounds. The 13C NMR spectra were recorded at 75, 100, and 150 MHz. Chemical shifts are reported in δ units (ppm) by assigning the TMS resonance in the 1H NMR spectrum as 0.00 ppm and the CDCl3 resonance in the 13C NMR spectrum as 77.23 ppm. All coupling constants (J) are reported in Hertz (Hz). Thin layer chromatography was performed using 60 mesh silica gel plates, and visualization was effected by short wavelength UV light (254 nm). All melting points are uncorrected. High resolution mass spectra (HRMS) were recorded using EI at 70 eV or using a QTOF mass spectrometer (APCI at a voltage of 70 eV). All reagents were used directly as obtained commercially, unless otherwise noted.
The characterization of compounds 3, 6, 12-15, 18, 21, 23, 29, 30, 32, 34, 36-40, 42, 48, and 49 can be found in our earlier report.9
4.2. General procedure for the reaction of carboxylic acids with arynes
The aryne precursor (1.5 equiv) was added to a mixture of the carboxylic acid (0.25 mmol) and CsF (4.0 equiv) in 15 mL of freshly distilled THF, and the reaction mixture was then stirred in a closed vial at 125 °C for 18 h. After the reaction mixture was allowed to cool to room temperature, it was eluted through a plug of silica gel with ethyl acetate and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc as the eluent to afford the desired o-hydroxyaryl ketone.
1-(2-Hydroxyphenyl)-4-phenylbutane-1,4-dione (16)
This compound was obtained as a white solid in a 39% yield: mp 107–108 °C 1 H NMR (400 MHz, CDCl3) δ 3.43–3.54 (m, 4H), 6.94 (t, J = 7.6 Hz, 1H), 6.99 (d, J = 8.1 Hz, 1H), 7.46–7.53 (m, 3H), 7.60 (t, J = 7.3 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 7.5 Hz, 2H), 12.12 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 32.3 (×2), 118.7, 119.2, 119.5, 128.3, 128.9, 130.1, 133.5, 136.6, 136.8, 162.5, 198.4, 204.8; HRMS (APCI) calcd for [M+H]+ C16H15O3 255.1016, found 255.1014.
1-(2-Hydroxyphenyl)-2-(thiophen-3-yl)ethanone (17)
This compound was obtained as a yellow semisolid in a 30% yield: 1H NMR (400 MHz, CDCl3) δ 4.34 (s, 2H), 6.91 (t, J = 7.6 Hz, 1H), 7.00 (d, J = 8.8 Hz, 1H), 7.03 (d, J = 5.7 Hz, 1H), 7.15 (s, 1H), 7.33 (dd, J = 4.9, 3.0 Hz, 1H), 7.48 (t, J = 8.4 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 12.20 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 39.8, 118.7, 118.9, 119.0, 123.1, 126.1, 128.5, 130.3, 133.4, 136.6, 162.9, 203.3; HRMS (EI) calcd for C12H10O2S 218.0396, found 218.0391.
1-(2-Hydroxy-4,6-dimethoxyphenyl)butan-1-one (20)
This compound was obtained as a white solid in a 49% yield: mp 69–72 °C (lit.44 mp 70 °C); 1H NMR (400 MHz, CDCl3) δ 0.98 (t, J = 7.4 Hz, 3H), 1.59–1.65 (m, 2H), 2.96 (t, J = 7.3 Hz, 2H), 3.81 (s, 3H), 3.85 (s, 3H), 5.92 (d, J = 2.4 Hz, 1H), 6.06 (d, J = 2.5 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.0, 18.1, 46.1, 55.5 (×2), 90.7, 93.6, 105.8, 162.7, 165.7, 167.6, 205.8; HRMS (APCI) calcd for [M+H]+ C12H17O4 225.1121, found 225.1122.
2-Bromoxanthone (22)
This compound was obtained as a white solid in a 79% yield: mp 148–150 °C (lit.45 mp 150 °C); 1H NMR (400 MHz, CDCl3) δ 7.33–7.42 (m, 2H), 7.47 (d, J = 8.5 Hz, 1H), 7.69–7.79 (m, 2H), 8.29 (dd, J = 7.9, 1.7 Hz, 1H), 8.42 (d, J = 2.5 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 117.3, 118.3, 120.2, 121.7, 123.3, 124.5, 127.0, 129.4, 135.4, 137.9, 155.1, 156.2, 176.2; HRMS (APCI) calcd for [M+H]+ C13H8BrO2 274.9702, found 274.9706. The 1H and 13C NMR spectral data are in good agreement with the literature data.13
2,3-Dimethylxanthone (28)
This compound was obtained as a white solid in a 47% yield: mp 156–158 °C; 1H NMR (400 MHz, CDCl3) δ 2.35 (s, 3H), 2.38 (s, 3H), 7.24 (s, 1H), 7.34 (t, J = 7.5 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 8.04 (s, 1H), 8.32 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 19.5, 20.8, 118.1, 118.3, 119.9, 122.1, 123.8, 126.5, 126.9, 133.3, 134.6, 145.7, 154.9, 156.3, 177.3. HRMS (APCI) calcd for [M+H]+ C15H13O2 225.0910, found 225.0911. The 1H and 13C NMR spectral data are in good agreement with the literature data.13
Synthesis of 2-nitroxanthone (24)
o-(Trimethylsilyl)phenyl triflate (1.5 equiv) was added to a mixture of 2-chloro-5-nitrobenzoic acid (0.25 mmol) and TBAT (2.0 equiv) in 5 mL of toluene, and the reaction mixture was then stirred at 60 °C for 24 h. After allowing the reaction mixture to cool to room temperature, the mixture was extracted with EtOAc (20 mL × 2) from the brine solution (40 mL), the organic fractions were combined, and the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc as the eluent to afford the desired xanthone 24 as a pale brown solid in an 87% yield: mp 203–205 °C (lit. mp46 202–203 °C); 1H NMR (400 MHz, CDCl3) δ 7.47 (t, J = 7.6 Hz, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.64 (d, J = 9.2 Hz, 1H), 7.81 (ddd, J = 8.7, 7.1, 1.7 Hz, 1H), 8.33 (dd, J = 7.9, 1.7 Hz, 1H), 8.54 (dd, J = 9.2, 2.8 Hz, 1H), 9.19 (d, J = 2.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 118.2, 119.7, 121.3, 121.6, 123.5, 125.3, 126.9, 129.0, 135.9, 155.8, 159.1, 175.7; HRMS (APCI) calcd for [M+H]+ C13H8NO4 242.0448, found 242.0452. The 1H and 13C NMR spectral data are in good agreement with the literature data.47
Synthesis of benzo[c]xanthone (25)
o-(Trimethylsilyl)phenyl triflate (1.5 equiv) was added to a mixture of 1-bromo-2-naphthoic acid (0.25 mmol) and TBAT (2.0 equiv) in 5 mL of toluene, and the reaction mixture was then stirred at 60 °C for 24 h. After allowing the reaction mixture to cool to room temperature, the mixture was extracted with EtOAc (20 mL × 2) from the brine solution (40 mL), the organic fractions were combined, and the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc as the eluent to afford the desired xanthone 25 as a white solid in a 55% yield: mp 149–151 °C (lit. mp48 147–149 °C); 1H NMR (600 MHz, CDCl3) δ 7.43 (s, 1H), 7.61–7.80 (m, 5H), 7.90–7.94 (m, 1H), 8.27 (d, J = 8.2 Hz, 1H), 8.40 (d, J = 6.4 Hz, 1H), 8.66 (d, J = 6.5 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 117.6, 118.1, 121.5, 122.5, 122.9, 124.0, 124.1, 124.4, 126.6, 126.9, 128.1, 129.6, 134.4, 136.6, 153.7, 155.8, 176.9; HRMS (APCI) calcd for [M+H]+ C17H11O2 247.0754, found 247.0751. The 1H and 13C NMR spectral data are in good agreement with the literature data.13
Synthesis of 2,3-dimethoxyxanthone (26)
o-(Trimethylsilyl)phenyl triflate (1.5 equiv) was added to a mixture of 2-bromo-4,5-dimethoxybenzoic acid (0.25 mmol) and TBAT (2.0 equiv) in 5 mL of toluene, and the reaction mixture was then stirred at 60 °C for 24 h. After allowing the reaction mixture to cool to room temperature, the mixture was extracted with EtOAc (20 mL × 2) from the brine solution (40 mL), the organic fractions were combined, and the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc as the eluent to afford the corresponding hydroxyaryl ketone. The latter was dissolved in 4 mL of MeCN and heated in the presence of K2CO3 (2 equiv) at 100 °C for 24 h. After allowing the reaction mixture to cool to room temperature, the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc as the eluent to afford the desired xanthone 26 as a light brown solid in a 59% yield: mp 161–162 °C (lit.49 mp 163–164 °C); 1H NMR (400 MHz, CDCl3) δ 3.99 (s, 3H), 4.01 (s, 3H), 6.90 (s, 1H), 7.36 (t, J = 7.6 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.63–7.70 (m, 2H), 8.30–8.35 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 56.3, 56.5, 99.6, 105.3, 114.9, 117.6, 121.5, 123.7, 126.5, 133.9, 146.7, 152.4, 155.4, 156.0, 176.0; HRMS (APCI) calcd for [M+H]+ C15H13O4 257.0808, found 257.0809. The 1H and 13C NMR spectral data are in good agreement with the literature data.13
Synthesis of 1,3,6,7-tetramethoxyxanthone (27)
The corresponding dimethoxyaryne precursor (1.5 equiv) was added to a mixture of 2-bromo-4,5-dimethoxybenzoic acid (0.25 mmol) and TBAT (2.0 equiv) in 5 mL of toluene, and the reaction mixture was then stirred at 60 °C for 24 h. After allowing the reaction mixture to cool to room temperature, the mixture was extracted with EtOAc (20 mL × 2) from the brine solution (40 mL), the organic fractions were combined and the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc as the eluent to afford the corresponding hydroxyaryl ketone. The latter was dissolved in 4 mL of MeCN and heated in the presence of K2CO3 (2 equiv) at 100 °C for 24 h. After allowing the reaction mixture to cool to room temperature, the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc as the eluent to afford the desired xanthone 27 as a light brown solid in a 49% yield: mp 202–203 °C (lit.50 mp 202 °C); 1H NMR (400 MHz, CDCl3) δ 3.86 (s, 3H), 3.92–3.95 (m, 9H), 6.29 (d, J = 2.1 Hz, 1H), 6.40 (d, J = 2.1 Hz, 1H), 6.74 (s, 1H), 7.60 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 55.6, 56.2, 56.3, 56.3, 92.5, 95.0, 98.9, 105.7, 106.9, 116.0, 146.4, 150.7, 154.3, 159.7, 161.8, 164.2, 174.6; HRMS (APCI) calcd for [M+H]+ C17H17O6 317.1020, found 317.1023. The 1H and 13C NMR spectral data are in good agreement with the literature data.51
4.3. General procedure for the reaction of 2-alkenoic acids with arynes
The aryne precursor (1.5 equiv) was added to a mixture of the 2-alkenoic acid (0.25 mmol) and CsF (4.0 equiv) in 15 mL of freshly distilled THF, and the reaction mixture was then stirred in a closed vial at 125 °C for 18 h. After allowing the reaction mixture to cool, additional aryne precursor (0.5 equiv) and CsF (1.0 equiv) were quickly added and heating was continued at 125 °C for 6 h. After the reaction mixture was allowed to cool to room temperature, it was eluted through a plug of silica gel with ethyl acetate and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc as the eluent to afford the desired 4-chromanones.
2-Cyclohexylchroman-4-one (31)
This compound was obtained as a pale yellow oil in an 85% yield: 1H NMR (400 MHz, CDCl3) δ 1.06–1.34 (m, 5H), 1.64–1.85 (m, 5H), 1.98 (d, J = 12.3 Hz, 1H), 2.60–2.77 (m, 2H), 4.14–4.24 (m, 1H), 6.94–7.01 (m, 2H), 7.46 (ddd, J = 8.7, 7.2, 1.8 Hz, 1H), 7.86 (dd, J = 7.8, 1.8 Hz, 1H); 13 C NMR (150 MHz, CDCl3) δ 25.9, 26.0, 26.3, 28.2, 28.3, 40.3, 41.8, 82.0, 117.9, 121.0, 126.9, 135.9, 161.9, 193.2 (doubled signals at 25.9–26.0 and 28.2–28.3 are likely due to different electronic environments of the corresponding carbons); HRMS (APCI) calcd for [M+H]+ C15H19O2 231.1380, found 231.1382. The 1H and 13C NMR spectral data are in good agreement with the literature data.52
4-(4-Oxochroman-2-yl)butanenitrile (33)
This compound was obtained as a pale yellow oil in a 71% yield: 1H NMR (400 MHz, CDCl3) δ 1.82–2.08 (m, 4H), 2.48 (t, J = 6.6 Hz, 2H), 2.63–2.78 (m, 2H), 4.41–4.51 (m, 1H), 6.96 (d, J = 8.4 Hz, 1H), 7.02 (t, J = 7.5 Hz, 1H), 7.48 (t, J = 7.8 Hz, 1H), 7.87 (d, J = 7.9 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 17.1, 21.3, 33.7, 42.9, 76.9, 117.9, 119.2, 120.9, 121.6, 127.1, 136.2, 161.2, 191.8; HRMS (APCI) calcd for [M+H]+ C13H14NO2 216.1019, found 216.1021.
2-Methyl-2-(4-methylpent-3-en-1-yl)chroman-4-one (35)
This compound was obtained as a colorless oil in a 64% yield: 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 3H), 1.57 (s, 3H), 1.63–1.73 (m, 4H), 1.75–1.85 (m, 1H), 2.05–2.17 (m, 2H), 2.66 (d, J = 16.4 Hz, 1H), 2.79 (d, J = 16.4 Hz, 1H), 5.02–5.10 (m, 1H), 6.89–7.01 (m, 2H), 7.46 (ddd, J = 8.6, 7.2, 1.8 Hz, 1H), 7.84 (dd, J = 7.8, 1.8 Hz, 1H); 13 C NMR (100 MHz, CDCl3) δ 17.8, 22.5, 24.2, 25.9, 39.5, 47.7, 81.3, 118.5, 120.6, 120.8, 123.5, 126.6, 132.5, 136.3, 160.0, 192.9; HRMS (APCI) calcd for [M+H]+ C16H21O2 245.1536, found 245.1541.
Synthesis of 5,7-dimethoxy-2-phenylchroman-4-one (43)
After the reaction between cinnamic acid and the unsymmetrical dimethoxy-substituted benzyne precursor 19, the uncyclized o-hydroxyaryl ketone was isolated in a 68% yield as a yellow solid: mp 71–74 °C: 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 3.92 (s, 3H), 5.96 (s, 1H), 6.11 (s, 1H), 7.33–7.47 (m, 4H), 7.58–7.65 (m, 3H), 7.78 (d, J = 15.6 Hz, 1H), 7.91 (d, J = 15.6 Hz, 1H); HRMS (APCI) calcd for [M+H]+ C17H17O4 285.1121, found 285.1129. The latter was dissolved in 5 mL of THF, then 10 mL of water and 10 μL of piperidine were added, and the mixture was stirred for 2 h at 40 °C. After allowing the reaction mixture to cool to room temperature, the mixture was extracted with EtOAc (20 mL × 2), the organic fractions were combined, and the solvent was evaporated under reduced pressure. Flash chromatography on silica gel using hexanes/EtOAc as the eluent afforded 42% of the unreacted starting material and 48% of the desired 4-chromanone 43 as a white solid: mp 144–146 °C (lit.53 mp 145–146 °C); 1H NMR (400 MHz, CDCl3) δ 2.80 (dd, J = 16.4, 2.7 Hz, 1H), 2.97–3.07 (m, 1H), 3.82 (s, 3H), 3.90 (s, 3H), 5.41 (d, J = 14.8 Hz, 1H), 6.10 (s, 1H), 6.16 (s, 1H), 7.31–7.49 (m, 5H); 13C NMR (150 MHz, CDCl3) δ 45.6, 55.6, 56.2, 79.3, 93.2, 93.6, 106.0, 126.1, 128.7, 128.8, 138.8, 162.3, 165.0, 166.0, 189.2; HRMS (APCI) calcd for [M+H]+ C17H17O4 285.1121, found 285.1124. The 1H and 13C NMR spectral data are in good agreement with the literature data.53
Methyl 2-(2-methyl-3-oxo-2,3-dihydrobenzofuran-2-yl)acetate (45)
This compound was obtained as a yellow solid in a 79% yield starting from the monomethyl ester of itaconic acid: mp 127–130 °C; 1H NMR (400 MHz, CDCl3) δ 1.46 (s, 3H), 2.93 (d, J = 16.4 Hz, 1H), 3.02 (d, J = 16.4 Hz, 1H), 3.53 (s, 3H), 7.08 (t, J = 8.1 Hz, 2H), 7.59 (t, J = 7.8 Hz, 1H), 7.69 (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 22.6, 41.5, 52.1, 86.5, 113.5, 120.6, 122.2, 124.8, 138.0, 169.3, 171.1, 202.8; HRMS (APCI) calcd for [M+H]+ C12H13O4 221.0808, found 221.0809.
4.4. Procedure for the reaction of 2-alkynoic acids with arynes
The aryne precursor (1.5 equiv) was added to a mixture of the 2-alkynoic acid (0.25 mmol) and TBAT (2.0 equiv) in 5 mL of anhydrous toluene, and the reaction mixture was then stirred in a closed vial at 60 °C for 24 h. After the reaction mixture was allowed to cool to room temperature, the reaction mixture was then poured into brine (15 mL) and the organic layer was separated. The aqueous layer was extracted with ethyl acetate (2 × 15 mL) and the organic layers were combined and concentrated in vacuo. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc as the eluent to afford the desired chromone derivatives.
Chromone (50)
This compound was obtained as a yellow semisolid in a 71% yield running the reaction in 15 mL of THF at 65 °C, instead of toluene at 60 °C. The same compound was also obtained in an 18% yield starting from 2-bromoacrylic acid (51) using the CsF/THF protocol at 125 °C: 1H NMR (400 MHz, CDCl3) δ 6.34 (d, J = 6.0 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.67 (t, J = 7.8 Hz, 1H), 7.85 (d, J = 6.0 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 113.3, 118.4, 125.1, 125.5, 126.0, 134.0, 155.5, 156.7, 177.8; HRMS (EI) calcd for C9H6O2 146.0368, found 146.0370. The 1H and 13C NMR spectral data are in good agreement with the literature data.54
3,4-Dihydro-1H-xanthen-9(2H)-one (53)
This compound was obtained as a white solid in an 85% yield starting from 2-bromocyclohexenoic acid (52) using the CsF/THF protocol at 125 °C: mp 87–88 °C (lit.55 mp 91–92 °C); 1H NMR (400 MHz, CDCl3) δ 1.71–1.91 (m, 4H), 2.56–2.68 (m, 4H), 7.29–7.39 (m, 2H), 7.59 (ddd, J = 8.6, 7.1, 1.7 Hz, 1H), 8.19 (dd, J = 8.0, 1.7 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 21.0, 21.7, 21.9, 28.2, 117.6, 118.4, 123.2, 124.4, 125.7, 132.9, 155.9, 163.9, 177.8; HRMS (APCI) calcd for [M+H]+ C13H13O2 201.0910, found 201.0916. The 1H and 13C NMR spectral data are in good agreement with the literature data.56
4.5. Procedure for the preparation of methyl (E)-2-benzyl-3-phenylacrylate (57)
o-(Trimethylsilyl)phenyl triflate (3.0 equiv) was added to a mixture of methyl methacrylate (0.25 mmol) and CsF (6.0 equiv) in 5 mL of MeCN, and the reaction mixture was stirred at room temperature for 18 h. The reaction mixture was then eluted through a plug of silica gel with ethyl acetate and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel using hexanes/EtOAc as the eluent to afford the desired diarylated product 57 as a colorless oil in an 80% yield: 1H NMR (400 MHz, CDCl3) δ 3.76 (s, 3H), 3.97 (s, 2H), 7.17–7.41 (m, 10H), 7.95 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 33.4, 52.3, 126.3, 128.1, 128.7, 128.8, 129.0, 129.4, 130.9, 135.6, 139.6, 141.2, 168.8; HRMS (EI) calcd for C17H16O2 252.1150, found 252.1170. The 1H and 13C NMR spectral data are in good agreement with the literature data.43b
Supplementary Material
Acknowledgments
We thank the National Science Foundation, the National Institute of General Medical Sciences (GM079593) and the National Institutes of Health Kansas University Center of Excellence in Chemical Methodology and Library Development (P50 GM069663) for their generous financial support. We also thank Dr. Feng Shi, while at Iowa State University, for the preparation of some aryne precursors.
Footnotes
Supplementary data related to this article can be found online at DOI.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Liu Z, Larock RC. J Am Chem Soc. 2005;127:13112. doi: 10.1021/ja054079p. [DOI] [PubMed] [Google Scholar]
- 2.Pintori DG, Greaney MF. Org Lett. 2010;12:168. doi: 10.1021/ol902568x. [DOI] [PubMed] [Google Scholar]
- 3.Tambar UK, Stoltz BM. J Am Chem Soc. 2005;127:5340. doi: 10.1021/ja050859m. [DOI] [PubMed] [Google Scholar]
- 4.Sato Y, Kobayashi Y, Sugiura M, Shirai H. J Org Chem. 1978;43:199. [Google Scholar]
- 5.Yoshida H, Honda Y, Shirakawa E, Hiyama T. Chem Commun. 2001:1880. doi: 10.1039/b103745p. [DOI] [PubMed] [Google Scholar]
- 6.Yoshida H, Mimura Y, Ohshita J, Kunai A. Chem Commun. 2007:2405. doi: 10.1039/b701581j. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida H, Watanabe M, Fukushima H, Ohshita J, Kunai A. Org Lett. 2004;6:4049. doi: 10.1021/ol048298b. [DOI] [PubMed] [Google Scholar]
- 8.Liu Z, Larock RC. Org Lett. 2004;6:99. doi: 10.1021/ol0361406. [DOI] [PubMed] [Google Scholar]
- 9.Dubrovskiy AV, Larock RC. Org Lett. 2010;12:3117. doi: 10.1021/ol101017z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chai G, Qiu Y, Fu C, Ma S. Org Lett. 2011;13:5196. doi: 10.1021/ol202076c. [DOI] [PubMed] [Google Scholar]
- 11.Himeshima Y, Sonoda T, Kobayashi H. Chem Lett. 1983:1211. [Google Scholar]
- 12.Slana GBCA, de Azevedo MS, Lopes RSC, Lopes CC, Cardoso JN. Beilstein J Org Chem. 2006;2:1. doi: 10.1186/1860-5397-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For a related transformation, see: Zhao J, Larock RC. J Org Chem. 2007;72:583. doi: 10.1021/jo0620718. See the supplementary data for spectral evidence for the formation of 5.
- 14.Kotali A, Harris PA. Org Prep Proc Int. 1994;26:159. [Google Scholar]
- 15.(a) Zhang L, Zhang JY. J Comb Chem. 2006;8:361. doi: 10.1021/cc0501007. [DOI] [PubMed] [Google Scholar]; (b) Piccolo O, Filippini L, Tinucci L, Valoti E, Citterio A. Tetrahedron. 1986;42:885. [Google Scholar]
- 16.Rozenberg V, Danilova T, Sergeeva E, Vorontsov E, Starikova Z, Lysenko K, Belokon’ Y. Eur J Org Chem. 2000:3295. [Google Scholar]
- 17.See Katritzky AR, Le KNB, Mohapatra PP. Synthesis. 2007:3141. and references therein for the biological activity of o-hydroxyaryl ketones.
- 18.(a) Merza J, Aumond MC, Rondeau D, Dumontet V, Le Ray AM, Séraphin D, Richomme P. Phytochemistry. 2004;65:2915. doi: 10.1016/j.phytochem.2004.06.037. [DOI] [PubMed] [Google Scholar]; (b) Mors WB, Gottlieb OR, Djerassi C. J Am Chem Soc. 1957;79:4507. [Google Scholar]; (c) Nanda B, Patwardhan SA, Gupta AS. Ind J Chem, Sec B. 1983;22:185. [Google Scholar]
- 19.Okuma K, Nojima A, Matsunaga N, Shioji K. Org Lett. 2009;11:169. doi: 10.1021/ol802597x. [DOI] [PubMed] [Google Scholar]
- 20.Wu CP, van Schalkwyk DA, Taylor D, Smith PJ, Chibale K. Int J Antimicrob Agents. 2005;26:170. doi: 10.1016/j.ijantimicag.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Chibale K, Visser M, van Schalkwyk D, Smith PJ, Saravanamuthu A, Fairlamb AH. Tetrahedron. 2003;59:2289. [Google Scholar]
- 22.Burmester JK. US Patent WO 2006102102. 2006
- 23.Tadross PM, Gilmore CD, Bugga P, Virgil SC, Stoltz BM. Org Lett. 2010;12:1224. doi: 10.1021/ol1000796. [DOI] [PubMed] [Google Scholar]
- 24.Joshi BS, Ravindranath KW. J Chem Soc, Perkin Trans 1. 1977:433. [Google Scholar]
- 25.(a) Na Y. J Pharm Pharmacol. 2009;61:707. doi: 10.1211/jpp/61.06.0002. [DOI] [PubMed] [Google Scholar]; (b) El-Seedi HR, El-Ghorab DMH, El-Barbary MA, Zayed MF, Goeransson U, Larsson S, Verpoorte R. Curr Med Chem. 2009;16:2581. doi: 10.2174/092986709788682056. [DOI] [PubMed] [Google Scholar]; (c) Diderot NT, Silvere N, Etienne T. Adv Phytomed. 2006;2:273. [Google Scholar]
- 26.Examples of Heck and Suzuki-Miyaura coupling processes for 2-bromoxanthone have been demonstrated by the Larock group in: Zhao Z, Larock RC. J Org Chem. 2007;72:583. doi: 10.1021/jo0620718.
- 27.(a) Jeganmohan M, Bhuvaneswari S, Cheng CH. Chem Asian J. 2010;5:153. doi: 10.1002/asia.200900324. [DOI] [PubMed] [Google Scholar]; (b) Rogness DC, Markina NA, Waldo JP, Larock RC. J Org Chem. 2012;77:2743. doi: 10.1021/jo2025543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pedro M, Cerqueira F, Sousa ME, Nascimento MSJ, Pinto M. Bioorg Med Chem. 2002;10:3725. doi: 10.1016/s0968-0896(02)00379-6. [DOI] [PubMed] [Google Scholar]
- 29.See Peres V, Nagem TJ, de Oliveira FF. Phytochem. 2000;55:683. doi: 10.1016/s0031-9422(00)00303-4. and references therein.
- 30.Chen CH, Lin JY, Lin CN, Hsu SY. J Nat Prod. 1992;55:691. doi: 10.1021/np50083a025. [DOI] [PubMed] [Google Scholar]
- 31.Wang JP, Raung SL, Lin CN, Teng CM. Eur J Pharmacol. 1994;251:35. doi: 10.1016/0014-2999(94)90440-5. [DOI] [PubMed] [Google Scholar]
- 32.Gnerre C, Thull U, Gaillard P, Carrupt PA, Testa B, Fernandes E, Silva F, Pinto M, Pinto MMM, Wolfender JL, Hostettmann K, Cruciani G. Helv Chim Acta. 2001;84:552. [Google Scholar]
- 33.For similar reactivity of 3-methoxybenzyne, see: Dubrovskiy AV, Larock RC. Org Lett. 2010;12:1180. doi: 10.1021/ol902921s.
- 34.(a) Brahmachari G. Nat Prod Comm. 2008;3:1337. [Google Scholar]; (b) Horton DA, Bourne GT, Smythe ML. Chem Rev. 2003;103:893. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; (c) Birt DF, Hendrich S, Wang W. Pharm Ther. 2001;90:157. doi: 10.1016/s0163-7258(01)00137-1. [DOI] [PubMed] [Google Scholar]; (d) Kim HP, Son KH, Chang HW, Kang SS. J Pharmacol Sci. 2004;96:229. doi: 10.1254/jphs.crj04003x. [DOI] [PubMed] [Google Scholar]
- 35.Running the reaction with TBAT in THF ensures the complete conversion of the acid, but also results in higher yields of undesired overarylated products (analogous to products 5 and 6).
- 36.Apparently, an excess of the benzyne intermediate is more likely to engage in the reaction with the phenoxide formed as a result of the opening of a 4-membered ring in an intermediate like 9, rather than with the starting, less reactive carboxylic acid. Ideally, the slow addition technique could be beneficial to the yield of the reaction. However, running the reaction in an overheated solvent in a pressurized vial makes this hypothesis difficult to test.
- 37.Tanaka K, Sugino T. Green Chem. 2001;3:133. [Google Scholar]
- 38.The only detected by-product was the O-monoarylation product.
- 39.Bowers W, Ohta T, Cleere J, Marsella P. Science. 1976;193:542. doi: 10.1126/science.986685. [DOI] [PubMed] [Google Scholar]
- 40.Rao YK, Fang SH, Tzeng YM. J Ethnopharm. 2005;100:249. doi: 10.1016/j.jep.2005.02.039. [DOI] [PubMed] [Google Scholar]
- 41.Le Bail JC, Aubourg L, Habrioux G. Cancer Lett. 2000;156:37. doi: 10.1016/s0304-3835(00)00435-3. [DOI] [PubMed] [Google Scholar]
- 42.Martin R. Org Prep Proced Int. 1992;24:369. [Google Scholar]
- 43.(a) Navarre L, Darses S, Genet JP. Adv Synth Catal. 2006;348:317. [Google Scholar]; (b) Kantam ML, Kumar KBS, Sreedhar B. J Org Chem. 2008;73:320. doi: 10.1021/jo701982m. [DOI] [PubMed] [Google Scholar]
- 44.Canter FW, Curd FH, Robertson A. J Chem Soc. 1931:1245. [Google Scholar]
- 45.Dhar SN. J Chem Soc. 1920;117:1053. [Google Scholar]
- 46.Wang Z, Zhou LJ, Wang YL, Weng YB, He J, Nie K. J Chem Res. 2011;35:373. [Google Scholar]
- 47.Zhao J, Yue D, Campo MA, Larock RC. J Am Chem Soc. 2007;129:5288. doi: 10.1021/ja070657l. [DOI] [PubMed] [Google Scholar]
- 48.Gindy M. Nature. 1949;164:577. [Google Scholar]
- 49.Hassall CH, Lewis JR. J Chem Soc. 1961:2312. [Google Scholar]
- 50.Ueda S, Kurosawa K. Bull Chem Soc Jpn. 1977;50:193. [Google Scholar]
- 51.Hu H, Liao H, Zhang J, Wu W, Yan J, Yan Y, Zhao Q, Zou Y, Chai X, Yu S, Wu Q. Bioorg Med Chem Lett. 2010;20:3094. doi: 10.1016/j.bmcl.2010.03.101. [DOI] [PubMed] [Google Scholar]
- 52.Biddle MM, Lin M, Scheidt KA. J Am Chem Soc. 2007;129:3830. doi: 10.1021/ja070394v. [DOI] [PubMed] [Google Scholar]
- 53.Yenjai C, Wanich S. Bioorg Med Chem Lett. 2010;20:2821. doi: 10.1016/j.bmcl.2010.03.054. [DOI] [PubMed] [Google Scholar]
- 54.Okuma K, Matsunaga N, Nagahora N, Shioji K, Yokomori Y. Chem Commun. 2011;47:5822. doi: 10.1039/c1cc11234a. [DOI] [PubMed] [Google Scholar]
- 55.Watanabe T, Katayama S, Nakashita Y, Yamauchi M. Chem Pharm Bull. 1977;25:2778. [Google Scholar]
- 56.Turner PA, Griffin EM, Whatmore JL, Shipman M. Org Lett. 2011;13:1056. doi: 10.1021/ol103103n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.