

Abstract
A study of the nucleophilic addition of amines to 2,3-pyridyne has been carried out. 2-Aminopyridines have been generated exclusively. A series of benzonaphthyridinones have been synthesized by reacting 2,3-pyridyne and o-aminobenzoates.
Keywords: 2,3-Pyridyne; Amination; Annulation; Benzonaphthyridinone; Heterocycle
1. Introduction
Heterocyclic compounds have attracted significant attention for decades, because of their considerable biological activity and potential medicinal applications. Among the various heterocycles, benzonaphthyridinones, and dibenzonaphthyridinones (Fig. 1) are known as antimicrobial alkaloids,1 anticancer agents,2 and compounds that can reverse multidrug resistance.3
Fig. 1.
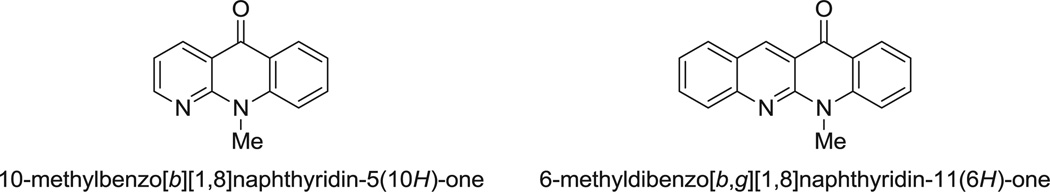
A representative benzonaphthyridinone and a dibenzonaphthyridinone.
However, synthetic approaches to these heterocycles remain limited. Quéguiner reported a process involving nucleophilic aromatic substitution to afford the benzonaphthyridinone core (Scheme 1).4 This ring system can also be prepared from 2-(phenylamino)nicotinic acid using either the strong acid PPA5 or microwave irradiation6 (Scheme 2). Dibenzonaphthyridinones have also been synthesized using PPA and a high temperature.7
Scheme 1.

Synthesis of benzonaphthyridinone involving nucleophilic aromatic substitution.
Scheme 2.

Synthesis of benzonaphthyridinone under harsh conditions.
Benzyne, a highly reactive intermediate, was first proposed by Wittig8 in 1942 and confirmed by Roberts9 in 1956. Kobayashi in 1983 reported a novelway to generate arynes from silylaryl triflates in the presence of fluoride.10 The Larock group has had an extensive ongoing research program in aryne chemistry in recent years.11 We have reported nucleophilic addition reactions of arynes,12 and later, a synthesis of xanthones, thioxanthones, and acridones from substituted benzoates involving nucleophilic attack on arynes, followed by ring closure.13 The exciting possibility of extending this methodology to hetarynes14 to generate polyheterocycles has now been examined.
2,3-Pyridyne has been the target of considerable research for decades.15 Classic approaches to this interesting intermediate include oxidation of an aminotriazolopyridine16 and a halogen-metal exchange-elimination sequence starting from 3-bromo-2-chloropyridine and employing a lithium reagent.17 3-(Trimethylsilyl)pyridin-2-yl triflate (1a) has been prepared by Effenberger and its fluoride-induced desilylation–elimination process generating 2,3-pyridyne has been reported.18 In a classic elimination–addition reaction of 3-halopyridine using KNH2/NH3,15a both 2- and 3-adducts have been reported with the former predominating. Fleming reported one example of the addition of acetic acid to 2,3-pyridyne to form the 2-substituted product.19 One example of the amination of 2,3-pyridyne has been reported in 2009, but not fully investigated.20 Herein, we report a study of the nucleophilic addition reactions of 2,3-pyridyne and amines, and a synthesis of benzonaphthyridinones from 2,3-pyridyne and o-aminobenzoates.
2. Results and discussion
Our initial work began with a study of the nucleophilic addition of N-methylaniline to 2,3-pyridyne. The 2,3-pyridyne precursor, 3-(trimethylsilyl)pyridin-2-yl triflate, was prepared using a literature procedure18 and the method was extended to the preparation of the 2,3-quinolyne precursor 3-(trimethylsilyl)quinolin-2-yl triflate (Scheme 3). We first allowed 3-(trimethylsilyl)pyridin-2-yl triflate to react with 2.0 equiv of CsF and 1.0 equiv of N-methylaniline in acetonitrile (MeCN) at room temperature for 24 h (Eq. 1). The tertiary amine N-methyl-N-phenylpyridin-2-amine was obtained in a 33% yield. Although two possible regioisomers could be formed in this reaction, we found that nucleophilic attack occurs exclusively at the 2-position of the 2,3-pyridyne, and the 2-pyridinylamine was the only observed product, albeit in only a relatively low yield.
Scheme 3.

Preparation of the 2,3-pyridyne and 2,3-quinolyne precursors.
Computational chemists have reported years ago that the cumulenic structure (structure B, Fig. 2) is favored for 2,3-pyridyne,21 which makes the 2-position more susceptible to be attacked by nucleophiles. This cumulenic structure also makes pyridyne extraordinarily reactive, which apparently results in unexpected side reactions leading to a low yield. We have actually stirred the pyridyne precursor with the fluoride source in the absence of any other reagents to check the reactivity and relative stability of pyridyne. It appears that the precursor disappears within an hour, and the pyridyne generated immediately turns into intractable tars, which we believe are the major by-products of the pyridyne chemistry.
Fig. 2.

The cumulenic structure of 2,3-pyridyne.
We have examined a number of different reaction conditions in an attempt to eliminate any undesired side reactions and improve the yield of the aminopyridine (Table 1). The yield was not improved when we changed the solvent (entries 2–6). The product was, in fact, totally different when we used tetrahydrofuran (THF) as the solvent. In THF, the THF acted as the nucleophile apparently attacking the pyridyne first to afford the final ring-opened, amine-containing product 3aa′ in up to a 58% yield (entries 7–9, Scheme 4). The yield was also not improved when we tried a different fluoride/base system (entry 10) or reaction temperature (entry 11). We did find that higher yields can be achieved when we changed the stoichiometry of the reactants 1a and 2a. A yield of 50% has been achieved when employing twice as much of the pyridyne precursor 1a (entry 12). The yield also improved when we reversed the ratio of 1a and 2a (entry 13), although the improvement was not as large as in the previous case (44% vs 50% in entry 12). Since the pyridyne precursor is less readily available than the amine, we decided to employ an excess of the amine in all further studies. However, the yield did not increase further when we used 3 equiv and 4 equiv of 2a (entries 14 and 15). The yield did, however, reach 49% when we diluted the reaction solution (entries 16 and 17). It appears that the pyridyne itself is so reactive that it causes side reactions. Thus, a lower concentration of pyridyne appears to reduce unwanted side reactions.
Table 1.
Optimization of the nucleophilic amination of 2,3-pyridynea
 | ||||||
|---|---|---|---|---|---|---|
| Entry | 1a (equiv) |
2a (equiv) |
Fluoride source (equiv) |
Solvent (mL) | T (°C)/ time (h) |
Yieldb (%) |
| 1 | 1 | 1 | CsF (2) | MeCN (4) | rt, 24 | 33 |
| 2 | 1 | 1 | CsF (2) | DME (4) | rt, 24 | 18 |
| 3 | 1 | 1 | CsF (2) | DMF (4) | rt, 24 | Trace |
| 4 | 1 | 1 | CsF (2) | MeCN (3)/Tol (1) | rt, 24 | 32 |
| 5 | 1 | 1 | CsF (2) | MeCN (2)/Tol (2) | rt, 24 | 30 |
| 6 | 1 | 1 | CsF (2) | MeCN (1)/Tol (3) | rt, 24 | 29 |
| 7 | 1 | 1 | CsF (2) | THF (4) | rt, 48c | 36d |
| 8 | 1 | 1 | CsF (2) | THF (4) | 50, 24 | 58d |
| 9 | 1 | 1 | TBAF (2) | THF (4) | rt, 24 | Trace |
| 10 | 1 | 1 | CsF (2)/Cs2CO3 (2) | MeCN (4) | rt, 24 | 35 |
| 11 | 1 | 1 | CsF (2) | MeCN (4) | 0→rt, 24 | 31 |
| 12 | 2 | 1 | CsF (4) | MeCN (4) | rt, 24 | 50 |
| 13 | 1 | 2 | CsF (2) | MeCN (4) | rt, 24 | 44 |
| 14 | 1 | 3 | CsF (2) | MeCN (4) | rt, 24 | 43 |
| 15 | 1 | 4 | CsF (2) | MeCN (4) | rt, 24 | 43 |
| 16 | 1 | 2 | CsF (2) | MeCN (8) | rt, 24 | 49 |
| 17 | 1 | 4 | CsF (2) | MeCN (8) | rt, 24 | 49 |
All reactions were carried out on a 0.25 mmol scale.
Isolated yield.
The pyridyne precursor remained unreacted.
Product 3aa′.
Scheme 4.

Nucleophilic attack of THF and the formation of compound 3aa′.
 |
(1) |
A second round of optimization has been carried out based on the idea that a lower concentration of pyridyne reduces side reactions (Table 2). We prepared a solution of 1a in 4 mL of MeCN (solution A) and a solution of 2a and CsF in 4 mL of MeCN (solution B). Solution A was slowly added to solution B by using a syringe pump over a long period of time. We found that the yield improved significantly (compare entry 1 and entry 2), because of the slow addition. The loading of 2a was subsequently reduced to 1 equiv, which did not affect the yield very much (entry 3). A longer addition time also did not improve the yield (entry 4). Therefore, we decided to use this optimized slow addition approach (entry 3) in all further investigations of this process.
Table 2.
Further optimization of the nucleophilic amination of 2,3-pyridynea
 | ||||||
|---|---|---|---|---|---|---|
| Entry | 1a (equiv) | 2a (equiv) | Solvent (mL) | Time (h) | Yieldb (%) | |
| 1 | 1 | 2 | MeCN (8) | 24 | 49 | |
| 2 | 1 | 2 | MeCN (4+4)c | 8+16d | 66 | |
| 3 | 1 | 1 | MeCN (4+4)c | 8+16d | 65 | |
| 4 | 1 | 1 | MeCN (4+4)c | 12+12d | 65 | |
All reactions were carried out on a 0.25 mmol scale with 2 equiv of CsF at room temperature.
Isolated yield.
A solution of 1a in 4 mL of MeCN (solution A) and a solution of 2a and CsF in 4 mL of MeCN (solution B) were prepared separately.
Solution A was slowly added to solution B with a syringe pump over a period of time; the resulting solution was stirred for an additional period of time.
A number of 2-pyridinylamines have been synthesized by reacting 2,3-pyridyne with a number of simple amines (Table 3). As can be seen, both secondary and tertiary amines have been obtained. However, tertiary amine products have not been observed in the reaction of 2,3-pyridyne and primary amines (entries 2–8). Halogens, such as iodine, can be tolerated under our reaction conditions to afford the corresponding halogenated products (entries 3 and 4). Both an electron-rich (entry 5) and an electron-deficient aniline (entry 6) have reacted well with pyridyne, although the electron-deficient aniline led to a lower yield. The yield was somewhat lower when the amine bears a sterically bulky group next to the amino group (entry 7). Aliphatic amines react as well under our reaction conditions to afford the corresponding products (entries 8 and 9). The quinolyne precursor 1b reacted in the same fashion, again tolerating a halogen (entry 10). Unfortunately, we failed to achieve ethers in a satisfactory yield by reacting 2,3-pyridyne and phenols due to the lower nucleophilicity of oxygen than nitrogen.
Table 3.
Synthesis of 2-aminopyridines from 2,3-pyridyne and various aminesa
 | ||||
|---|---|---|---|---|
| Entry | Aryne precursor | Amine | Product | Yieldb (%) |
| 1 |  |
PhMeNH 2a |
 |
65 |
| 2 | PhNH2 2b |
 |
58 | |
| 3 |  |
 |
57 | |
| 4 |  |
 |
64 | |
| 5 |  |
 |
61 | |
| 6 |  |
 |
47 | |
| 7 |  |
 |
46 | |
| 8 |  |
 |
60 | |
| 9 | Bn2NH 2i |
 |
57 | |
| 10 |  |
 |
 |
36 |
All reactions were carried out on a 0.25 mmol scale with 2 equiv of CsF at room temperature. A solution of 1 in 4 mL of MeCN (solution A) and a solution of 2 and CsF in 4 mL of MeCN (solution B) were prepared separately. Solution A was slowly added to solution B with a syringe pump over 8 h, the resulting solution was stirred for an additional 16 h.
Isolated yield.
Next, a polycyclic system has been achieved by reacting 2,3-pyridyne and o-aminobenzoates. In this process, a plausible mechanism is proposed in Scheme 5. The deprotonated (Scheme 5, Route A) or neutral amino group (Scheme 5, Route B) attacks the 2-position of the pyridyne, generating a carbanion, which is then trapped intramolecularly by the ester group of the o-amino-benzoate to form the benzonaphthyridinone.
Scheme 5.

Proposed mechanism for the formation of benzonaphthyridinone.
Using this basic process, a variety of benzonaphthyridinones and a dibenzonaphthyridinone have been synthesized from 2,3-pyridyne and 2,3-quinolyne using our slow addition reaction conditions (Table 4). It is worth noting that primary amines led to a complex mixture in this chemistry; therefore, only secondary amines have been employed for this synthesis. N-Methylbenzonaphthyridinone has been synthesized in a modest 56% yield by reacting 2,3-pyridyne and methyl 2-(methylamino)benzoate (entry 1). N-Allylbenzonaphthyridinone has been generated from the corresponding N-(allylamino)benzoate in a 48% yield (entry 2). Halides from chloride to iodide are tolerated under our reaction conditions (entries 2–6). The yields are moderate in these cases. It is interesting to compare entries 3–5 and entry 6, where we observe that the yield is higher when the halogen atom is para to the ester group of the benzoate. This trend is more obvious in entry 7, where an electron-withdrawing group is present para to the ester group, which makes the ester group more electrophilic, leading to a higher yield. A tertiary amine (entry 8) reacted as well as the secondary amines, affording the same product 5aa as in entry 1. This example shows that the neutral amine itself is nucleophilic enough for this transformation to take place (Scheme 6), suggesting that Route B in Scheme 5 is perhaps more favorable. 2,3-Quinolyne reacts the same way as 2,3-pyridyne, and the dibenzonaphthyridinone 5ba has been formed by reacting the quinolyne with 4a (entry 9).
Table 4.
Synthesis of benzonaphthyridinones and a dibenzonaphthyridinonea
 | ||||
|---|---|---|---|---|
| Entry | Aryne precursor |
Amine | Product | Yieldb (%) |
| 1 |  |
 |
 |
56 |
| 2 |  |
 |
48 | |
| 3 |  |
 |
53 | |
| 4 |  |
 |
52 | |
| 5 |  |
 |
48 | |
| 6 |  |
 |
66 | |
| 7 |  |
 |
72 | |
| 8 |  |
 |
50 | |
| 9 |  |
 |
 |
32 |
All reactions were carried out on a 0.25 mmol scale with 2 equiv of CsF at room temperature. A solution of 1 in 4 mL of MeCN (solution A) and a solution of 4 and sF in 4 mL of MeCN (solution B) were prepared separately. Solution A was slowly added to solution B with a syringe pump over 8 h, the resulting solution was stirred for an additional 16 h.
Isolated yield.
Scheme 6.

The reaction of 2,3-pyridyne and methyl 2-(dimethylamino)benzoate.
We have also tested a 2-hydroxybenzoate and a 2-mercaptobenzoate in this annulation process (Scheme 7). Again, the phenol group did not react well with the pyridyne apparently due to its lower nucleophilicity, while the sulfur-containing polyheterocycle 5ai has been formed from the 2-mercaptobenzoate, but in only a 34% yield.
Scheme 7.

The reaction of 2,3-pyridyne and 2-hydroxy- and 2-mercaptobenzoate.
3. Conclusions
Some nucleophilic amination reactions of 2,3-pyridyne and 2,3-quinolyne have been studied. We have found that nucleophilic attack of the amine occurs at the 2-position of 2,3-pyridyne/2,3-quinolyne exclusively, which is consistent with previous computational conclusions. 2,3-Pyridyne and 2,3-quinolyne appear to be so reactive that they cause unwanted side reactions. A slow addition procedure has thus been employed to reduce the side reactions. A variety of 2-pyridinylamines have thus been prepared using primary and secondary amines. When secondary and tertiary o-aminobenzoates are employed in this process, benzonaphthyridinones can be synthesized in reasonable yields.
4. Experimental section
4.1. General information
All reagents purchased from commercial sources were used as received. The solvent THF was distilled over Na/benzophenone. Anhydrous toluene and MeCN were used as received. The aryne precursors were prepared according to a literature procedure.18 Silica gel for column chromatography was supplied as 230–400 mesh from commercial source. Powdered CsF was used as received and stored in a desiccator.
All melting points were measured and are uncorrected. The 1H and 13C NMR spectra were recorded and are referenced to the residual solvent signals (7.26 ppm for 1H in CDCl3 and 77.2 ppm for 13C in CDCl3).
All aryne nucleophilic addition reactions and annulations were carried out in oven-dried glassware and were magnetically stirred. A nitrogen atmosphere was not used, except that a balloon of nitrogen was attached to the reaction flask to allow for the extra volume of added solution. A syringe pump was employed for the slow addition reaction conditions.
4.2. Preparation of pyridyne and quinolyne precursors
These aryne precursors were prepared following the literature procedure.18
4.2.1. 3-(Trimethylsilyl)pyridin-2-yl triflate (1a)
Colorless oil: 1H NMR (300 MHz, CDCl3) δ 8.33 (dd, J=4.8, 2.1 Hz, 1H), 7.92 (dd, J=7.2, 2.1 Hz, 1H), 7.31 (dd, J=7.2, 5.1 Hz, 1H), 0.37 (s, 9H).
4.2.2. 3-(Trimethylsilyl)quinolin-2-yl triflate (1b)
Colorless oil: Rf=0.33 (20:1 petroleum ether/EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 7.98 (d, J=8.4 Hz, 1H), 7.85 (d, J=8.4 Hz, 1H), 7.76 (td, J=7.6, 0.8 Hz, 1H), 7.59 (t, J=7.2 Hz, 1H), 0.43 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 158.6, 148.8, 146.3, 131.4, 128.5, 127.7, 127.5, 127.4, 124.4, 118.8 (CF3, 123.6, 120.4, 117.2, 114.1), −1.2; LRMS (EI) 349 (M); HRMS (EI) calcd for C13H14F3NO3SSi (M) 349.0416, found 349.0422; IR (KBr, cm−1) 3064 (w), 2959 (m), 2904 (w), 1590 (m).
4.3. General procedure for the synthesis of 2-pyridinylamines
To an oven-dried 4 dram vial equipped with a stir bar were added 0.25 mmol of amine and 76 mg of CsF (0.5 mmol, 2.0 equiv). MeCN (4 mL) was added and the vial was sealed with a rubber septum equipped with a balloon of nitrogen. A solution of 0.25 mmol of aryne precursor in 4 mL of MeCN was prepared separately, and was added to the reaction vial slowly by using a syringe pump while stirring. After the additionwas complete, the reaction mixture was allowed to stir for additional time and quenched with water, and extracted with EtOAc three times. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under vacuum. The residue was purified by column chromatography (petroleum ether/EtOAc) to afford the 2-pyridinylamine.
4.3.1. N-Methyl-N-phenylpyridin-2-amine (3aa)
Following the general procedure, this product was isolated as a yellow oil: Rf=0.24 (20:1 petroleum ether/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.23 (dd, J=4.8, 0.9 Hz, 1H), 7.40 (m, 2H), 7.30 (m, 2H), 7.21 (m, 2H), 6.60 (td, J=6.0, 0.9 Hz, 1H), 6.53 (d, J=8.4 Hz, 1H), 3.48 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 159.0, 148.0, 147.0, 136.7, 129.9, 126.5, 125.6, 113.3, 109.4, 38.6; LRMS (EI) 184 (M), 183 (M–H); HRMS (EI) calcd for C12H11N2 (M–H) 183.0922, found 183.0928. The structure of this compound was assigned based on the 1H NMR coupling pattern and was confirmed by comparison with the reported 1H and 13C NMR spectral data.22
4.3.2. N-Phenylpyridin-2-amine (3ab)
Yellow oil: Rf=0.11 (20:1 petroleum ether/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.21 (dt, J=5.1, 0.9 Hz, 1H), 7.49 (td, J=7.8, 1.2 Hz, 1H), 7.33 (d, J=4.5 Hz, 4H), 7.05 (m, 1H), 6.88 (d, J=8.4 Hz, 1H), 6.82 (s, 1H), 6.73 (td, J=3.6, 0.9 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 156.2, 148.6, 140.7, 137.9, 129.5, 123.0, 120.5, 115.2, 108.4; LRMS (EI) 170 (M), 169 (M–H); HRMS (EI) calcd for C11H10N2 (M) 170.0844, found 170.0847.
4.3.3. N-(2-Iodophenyl)pyridin-2-amine (3ac)
Yellow solid: mp 65–67 °C; Rf=0.35 (5:1 petroleum ether/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.25 (dd, J=5.1,1.2 Hz, 1H), 7.83 (td, J=7.2,1.2 Hz, 2H), 7.52 (td, J=7.2, 1.8 Hz, 1H), 7.31 (td, J=7.8, 1.2 Hz, 1H), 6.80 (m, 3H), 6.65 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 155.4, 148.6, 141.3, 139.6,138.0,129.2,124.4,120.4,116.1,109.7, 91.6; LRMS (EI) 296 (M); HRMS (EI) calcd for C11H9IN2 (M) 295.9810, found 295.9816; IR (KBr, cm−1) 3380 (s), 3192 (w), 3012 (m), 2954 (m), 2925 (m), 2866 (m), 1599 (s), 1573 (m), 1515 (s).
4.3.4. N-(3-Iodophenyl)pyridin-2-amine (3ad)
Yellow oil: Rf=0.33 (5:1 petroleum ether/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J=4.5 Hz, 1H), 7.75 (s, 1H), 7.52 (td, J=7.8, 1.5 Hz, 1H), 7.33 (t, J=8.4 Hz, 2H), 7.02 (m, 2H), 6.85 (d, J=8.4 Hz, 1H), 6.77 (dd, J=6.9, 5.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 155.4, 148.5, 142.2, 138.0, 131.5,130.8,128.4,119.0,115.8,109.1, 94.8; LRMS (EI) 296 (M); HRMS (EI) calcd for C11H9IN2 (M) 295.9810, found 295.9816; IR (KBr, cm−1) 3378 (s), 3190 (w), 3012 (m), 2929 (m), 2860 (m), 1595 (s), 1573 (m).
4.3.5. N-(2,4-Dimethylphenyl)pyridin-2-amine (3ae)
Yellow oil: Rf=0.22 (2:1 petroleum ether/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.15 (d, J=4.2 Hz, 1H), 7.41 (td, J=7.2, 1.8 Hz, 1H), 7.25 (t, J=3.6 Hz, 1H), 7.08 (s, 1H), 7.01 (d, J=7.8 Hz, 1H), 6.60 (m, 2H), 6.31 (s, 1H), 2.32 (s, 3H), 2.23 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 157.5, 148.6, 137.8, 135.8,134.7,131.9,131.3,127.6,124.2,114.4,107.2, 21.1,18.1; LRMS (EI) 198 (M); HRMS (EI) calcd for C13H14N2 (M) 198.1157, found 198.1159.
4.3.6. N-(4-Nitrophenyl)pyridin-2-amine (3af)
Yellow solid: mp 167–168 °C (lit.23 174–176 °C); Rf=0.41 (1:1 petroleum ether/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.34 (d, J=4.2 Hz, 1H), 8.20 (d, J=9.3 Hz, 2H), 7.64 (m, 3H), 6.94 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 153.9, 148.5, 147.1, 141.4, 138.3, 125.9, 117.6, 116.9, 111.5; LRMS (EI) 215 (M), 214 (M–H); HRMS (EI) calcd for C11H9N3O2 (M) 215.0695, found 215.0699.
4.3.7. N-(2-tert-Butylphenyl)pyridin-2-amine (3ag)
White solid: mp 122–123 °C; Rf=0.18 (5:1 petroleum ether/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.16 (d, J=4.2 Hz, 1H), 7.46 (d, J=7.5 Hz, 1H), 7.40 (t, J=7.2 Hz, 1H), 7.32 (d, J=7.5 Hz, 1H), 7.20 (m, 2H), 6.66 (t, J=6.0 Hz, 1H), 6.45 (d, J=8.4 Hz, 1H), 6.30 (s, 1H), 1.41 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 158.3, 148.7, 145.9, 138.8, 137.8, 128.8, 127.4, 127.3, 126.1, 114.3, 107.2, 35.2, 30.8; LRMS (EI) 226 (M); HRMS (EI) calcd for C15H18N2 (M) 226.1470, found 226.1475.
4.3.8. N-[2-(Cyclohex-1-en-1-yl)ethyl]pyridin-2-amine (3ah)
Yellow oil: Rf=0.13 (5:1 petroleum ether/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.08 (dd, J=4.5,1.2 Hz, 1H), 7.42 (td, J=7.2,1.8 Hz, 1H), 6.55 (t, J=5.7 Hz, 1H), 6.37 (d, J=8.4 Hz, 1H), 5.53 (s, 1H), 4.50 (s, 1H), 3.30 (dd, J=12.3, 6.9 Hz, 2H), 2.27 (t, J=6.6 Hz, 2H),1.98 (m, 4H), 1.60 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 159.0, 148.4, 137.6, 134.8, 124.0, 112.8, 106.6, 39.9, 37.8, 28.0, 25.4, 23.0, 22.6; LRMS (EI) 202 (M); HRMS (EI) calcd for C13H18N2 (M) 202.1470, found 202.1474.
4.3.9. N,N-Dibenzylpyridin-2-amine (3ai)
Yellow oil: Rf=0.27 (20:1 petroleum ether/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.20 (dt, J=5.1, 0.9 Hz, 1H), 7.31 (m, 11H), 6.58 (td, J=5.1, 0.9 Hz, 1H), 6.45 (d, J=8.7 Hz, 1H), 4.79 (s, 4H); 13C NMR (75 MHz, CDCl3) δ 158.8, 148.2, 138.6, 137.6, 128.8, 127.2, 127.1, 112.4, 106.1, 51.0; LRMS (EI) 274 (M); HRMS (EI) calcd for C19H18N2 (M) 274.1470, found 274.1473.
4.3.10. N-(4-Bromophenyl)quinolin-2-amine (3bj)
Brown solid: mp 143–145 °C; Rf=0.56 (2:1 petroleum ether/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J=8.8 Hz, 1H), 7.80 (d, J=8.0 Hz, 1H), 7.61 (m, 4H), 7.46 (d, J=8.8 Hz, 2H), 7.32 (t, J=7.2 Hz, 1H), 6.90 (d, J=8.8 Hz, 1H), 6.75 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 153.8, 147.6, 139.6, 138.0, 132.2, 130.1, 127.6, 127.1, 124.4,123.7, 121.6, 115.2,112.2; LRMS (EI) 299 (M); HRMS (EI) calcd for C15H11BrN2 (M) 298.0106, found 298.0111.
4.3.11. N-Methyl-N-[4-(pyridin-2-yloxy)butyl]aniline (3aa′)
Yellow oil: Rf=0.49 (20:1 petroleum ether/EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J=4.0 Hz, 1H), 7.54 (td, J=10.0, 4.0 Hz, 1H), 7.20 (m, 2H), 6.84 (m, 1H), 6.69 (m, 4H), 4.31 (t, J=6.0 Hz, 2H), 3.38 (t, J=8.0 Hz, 2H), 2.93 (s, 3H), 1.77 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 164.1, 149.5, 147.0, 138.7, 129.3, 116.7, 116.2, 112.4, 111.3, 65.7, 52.7, 38.5, 26.9, 23.7; LRMS (ESI) 257 (M+H); HRMS (ESI) calcd for C16H21N2O (M+H) 257.1648, found 257.1654; IR (KBr, cm−1) 3091 (w), 2945 (m), 2884 (m), 2867 (m), 1596 (s), 1569 (m).
4.4. General procedure for the synthesis of benzonaphthyridinones
The benzonaphthyridinones were prepared from the corresponding o-aminobenzoates and aryne precursors following the same procedure used for the synthesis of 2-pyridinylamines. The residue was purified by column chromatography (petroleum ether/EtOAc+1% TEA) to afford the benzonaphthyridinones.
4.4.1. 10-Methylbenzo[b][1,8]naphthyridin-5(10H)-one (5aa)
Yellow solid: mp 214–215 °C; Rf=0.24 (3:1 petroleum ether/EtOAc+1% TEA); 1H NMR (300 MHz, CDCl3) δ 8.79 (m, 2H), 8.55 (dd, J=8.1, 1.5 Hz, 1H), 7.79 (td, J=7.2, 1.5 Hz, 1H), 7.63 (d, J=8.4 Hz, 1H), 7.35 (t, J=7.5 Hz, 1H), 7.25 (m, 1H), 4.15 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 178.6, 153.5, 151.3, 142.6, 137.0, 134.6, 127.8, 122.8, 122.1, 117.6, 117.4, 115.6, 31.0; LRMS (EI) 210 (M), 209 (M–H); HRMS (EI) calcd for C13H10N2O (M) 210.0793, found 210.0796.
4.4.2. 10-Allyl-8-bromobenzo[b][1,8]naphthyridin-5(10H)-one (5ab)
Yellow solid: mp 145–146 °C; Rf=0.25 (5:1 petroleum ether/EtOAc+1% TEA); 1H NMR (400 MHz, CDCl3) δ 8.78 (d, J=6.4 Hz, 2H), 8.38 (d, J=8.4 Hz, 1H), 7.71 (s, 1H), 7.43 (d, J=8.4 Hz, 1H), 7.29 (m, 1H), 6.08 (m, 1H), 5.41 (s, 2H), 5.26 (d, J=10.0 Hz, 1H), 5.07 (d, J=17.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 178.0, 154.0, 150.9, 142.6, 137.0, 131.7, 129.7, 129.4, 125.6, 121.5, 119.1, 118.5, 117.34, 117.32, 46.0; LRMS (EI) 314 (M); HRMS (EI) calcd for C15H11BrN2O (M) 314.0055, found 314.0053; IR (KBr, cm−1) 3084 (w), 2954 (m), 2924 (m), 2870 (w), 1639 (m), 1587 (s).
4.4.3. 7-Iodo-10-methylbenzo[b][1,8]naphthyridin-5(10H)-one (5ac)
Yellow solid: mp 198–199 °C; Rf=0.13 (8:1 petroleum ether/EtOAc+1% TEA); 1H NMR (400 MHz, CDCl3) δ 8.78 (m, 3H), 7.99 (d, J=9.2 Hz, 1H), 7.38 (d, J=9.2 Hz, 1H), 7.25 (m, 1H), 4.11 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 177.2, 153.8, 151.1, 142.8, 141.9, 137.0, 136.4, 124.4, 118.0, 117.8, 117.4, 85.5, 31.1; LRMS (EI) 335 (M); HRMS (EI) calcd for C13H9IN2O (M) 335.9760, found 335.9765; IR (KBr, cm−1) 3083 (w), 2954 (m), 2915 (m), 2869 (w), 1635 (m), 1585 (s).
4.4.4. 7-Bromo-10-methylbenzo[b][1,8]naphthyridin-5(10H)-one (5ad)
Yellow solid: mp 202–203 °C; Rf=0.18 (5:1 petroleum ether/EtOAc+1% TEA); 1H NMR (400 MHz, CDCl3) δ 8.73 (m, 2H), 8.55 (s, 1H), 7.78 (d, J=8.8 Hz, 1H), 7.46 (d, J=8.8 Hz, 1H), 7.26 (m, 1H), 4.09 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 177.3, 153.8, 151.1, 141.3, 137.2, 136.9, 130.1, 123.9, 118.0, 117.6, 117.2, 115.6, 31.1; LRMS (EI) 288 (M); HRMS (EI) calcd for C13H9BrN2O (M) 287.9898, found 287.9904.
4.4.5. 7-Chloro-10-methylbenzo[b][1,8]naphthyridin-5(10H)-one (5ae)
Yellow solid: mp 210–212 °C; Rf=0.5 (2:1 petroleum ether/EtOAc+1% TEA); 1H NMR (400 MHz, CDCl3) δ 8.76 (m, 2H), 8.45 (s, 1H), 7.68 (d, J=9.2 Hz, 1H), 7.57 (d, J=9.2 Hz, 1H), 7.25 (m, 1H), 4.13 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 177.5, 153.8, 151.1, 141.0, 137.0, 134.6, 128.2, 126.9, 123.6, 118.0,117.4, 117.2, 31.2; LRMS (EI) 244 (M); HRMS (EI) calcd for C13H9ClN2O (M) 244.0403, found 244.0408.
4.4.6. 8-Bromo-10-methylbenzo[b][1,8]naphthyridin-5(10H)-one (5af)
Yellow solid: mp 234–235 °C; Rf=0.4 (3:1 petroleum ether/EtOAc+1% TEA); 1H NMR (400 MHz, CDCl3) δ 8.77 (m, 2H), 8.37 (d, J=8.8 Hz, 1H), 7.80 (s, 1H), 7.44 (d, J=8.4 Hz, 1H), 7.28 (t, J=6.0 Hz, 1H), 4.12 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 178.0, 153.7, 151.2, 143.3, 136.9, 129.8, 129.3, 125.5, 121.4, 118.5, 118.1, 117.5, 31.1; LRMS (EI) 288 (M); HRMS (EI) calcd for C13H9BrN2O (M) 287.9898, found 287.9901; IR (KBr, cm−1) 3084 (w), 2951 (w), 2917 (m), 2870 (m), 2849 (w), 1637 (m), 1587 (m).
4.4.7. Methyl 10-methyl-5-oxo-5,10-dihydrobenzo[b][1,8]naphthyridine-8-carboxylate (5ag)
Yellow solid: mp 225–226 °C; Rf=0.12 (5:1 petroleum ether/EtOAc+1% TEA); 1H NMR (400 MHz, CDCl3) δ 8.78 (m, 2H), 8.56 (d, J=8.0 Hz, 1H), 8.34 (s, 1H), 7.92 (d, J=8.4 Hz, 1H), 7.27 (m,1H), 4.20 (s, 3H), 4.02 (s, 3H); 13CNMR(100 MHz, CDCl3) δ 178.2, 166.4, 153.9, 151.4, 142.1, 136.9, 135.0, 128.1, 125.0, 122.0, 118.0, 117.6, 117.5, 52.9, 31.2; LRMS (EI) 268 (M); HRMS (EI) calcd for C15H12N2O3 (M) 268.0848, found 268.0854; IR (KBr, cm−1) 3082 (w), 2954 (m), 2923 (m), 2871 (w), 1730 (s), 1638 (s), 1614 (m), 1593 (s).
4.4.8. 6-Methyldibenzo[b,g][1,8]naphthyridin-11(6H)-one (5ba)
Yellow solid: mp 229–231 °C; Rf=0.27 (5:1 petroleum ether/EtOAc+1% TEA); 1H NMR (400 MHz, CDCl3) δ 9.32 (s, 1H), 8.53 (d, J=7.6 Hz, 1H), 8.00 (m, 2H), 7.80 (m, 2H), 7.60 (d, J=8.4 Hz, 1H), 7.47 (t, J=7.6 Hz, 1H), 7.31 (t, J=7.6 Hz, 1H), 4.22 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 179.7, 157.1, 150.1, 143.7, 139.2, 135.2, 132.8, 129.7, 128.1, 127.8, 124.8, 122.4, 121.6, 121.5, 117.8, 115.4, 31.0; LRMS (EI) 260 (M); HRMS (EI) calcd for C17H12N2O (M) 260.0950, found 260.0955; IR (KBr, cm−1) 3064 (w), 2954 (w), 2917 (m), 2871 (m), 2850 (w), 1608 (s).
4.4.9. 5H-Thiochromeno[2,3-b]pyridin-5-one (5ai)
White solid: mp 236–238 °C (lit.24 233–234 °C); Rf=0.24 (5:1 petroleum ether/EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.83 (m, 2H), 8.60 (d, J=8.0 Hz, 1H), 7.70 (m, 2H), 7.53 (td, J=8.0, 1.6 Hz, 1H), 7.46 (dd, J=8.0, 4.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 180.8, 158.9, 153.5, 138.0, 137.7, 133.2, 130.1, 129.0, 127.0, 126.65, 126.62, 121.8; LRMS (EI) 213 (M); HRMS (EI) calcd for C12H7NOS (M) 213.0248, found 213.0253.
Acknowledgements
We thank the National Science Foundation and the National Institutes of Health Center of Excellence for Chemical Methodology and Library Development at the University of Kansas (P50 GM069663 to R.C.L.) for financial support and Dr. Kermal Harrata (Iowa State University) for his help with the spectroscopic analysis.
References and notes
- 1.Mink K, Bracher F. Arch. Pharm. (Weinheim, Ger.) 2007;340:429. doi: 10.1002/ardp.200700064. [DOI] [PubMed] [Google Scholar]
- 2.Koch M, Tillequin F, Michel S, Hickman J, Pierre A, Leonce S, Pfeiffer B, Renard P. U.S. Patent Appl. Publ. 2005;24 [Google Scholar]
- 3.Misbahi H, Brouant P, Hever A, Molnar AM, Wolfard K, Spengler G, Mefetah H, Molnar J, Barbe J. Anticancer Res. 2002;22:2097. [PubMed] [Google Scholar]
- 4.Trécourt F, Marsais F, Güngör T, Quéguiner G. J. Chem. Soc. Perkin Trans. 1990;1:2409. [Google Scholar]
- 5.Denny WA, Atwell GJ, Cain BF. J. Med. Chem. 1977;20:1242. doi: 10.1021/jm00220a003. [DOI] [PubMed] [Google Scholar]
- 6.Afloroaei C, Vlassa M, Becze A, Brouant P, Barbe J. Heterocycl. Commun. 1999;5:249. [Google Scholar]
- 7.(a) Manoj M, Prasad KJR. J. Synth. Commun. 2010;40:3290. [Google Scholar]; (b) Manoj M, Prasad KJR. J. Chem. Res. 2009:720. [Google Scholar]
- 8.For a review, see: Sander W. Acc. Chem. Res. 1999;32:669. Wittig G. Naturwissenschaften. 1942;30:696..
- 9.Roberts JD, Semenow DA, Simmons HE, Jr, Carlsmith LA. J. Am.Chem. Soc. 1956;78:601. [Google Scholar]
- 10.Himeshima Y, Sonoda T, Kobayashi H. Chem. Lett. 1983:1211. [Google Scholar]
- 11.For selected recent Larock group aryne chemistry, see: Fang Y, Wu C, Larock RC, Shi FJ. Org. Chem. 2011;76:8840. doi: 10.1021/jo201605v. Rogness DC, Larock RC. J. Org. Chem. 2011;76:4980. doi: 10.1021/jo200651b. Kivrak A, Larock RC. J. Org. Chem. 2010;75:7381. doi: 10.1021/jo101656c. Dubrovskiy AV, Larock RC. Org. Lett. 2010;12:3117. doi: 10.1021/ol101017z. Wu C, Fang Y, Larock RC, Shi F. Org. Lett. 2010;12:2234. doi: 10.1021/ol100586r. Rogness DC, Larock RC. J. Org. Chem. 2010;75:2289. doi: 10.1021/jo1000687..
- 12.(a) Liu Z, Larock RC. Org. Lett. 2003;5:4673. doi: 10.1021/ol0358612. [DOI] [PubMed] [Google Scholar]; (b) Liu Z, Larock RC. Org. Lett. 2004;6:99. doi: 10.1021/ol0361406. [DOI] [PubMed] [Google Scholar]; (c) Liu Z, Larock RC. J. Org. Chem. 2006;71:3198. doi: 10.1021/jo0602221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Zhao J, Larock RC. Org. Lett. 2005;7:4273. doi: 10.1021/ol0517731. [DOI] [PubMed] [Google Scholar]; (b) Zhao J, Larock RC. J. Org. Chem. 2007;72:583. doi: 10.1021/jo0620718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Kauffmann T. Angew. Chem. Int. Ed. Engl. 1965;4:543. [Google Scholar]; (b) Adam W, Grimison A, Hoffmann RJ. Am. Chem. Soc. 1969;91:2590. [Google Scholar]; (c) Pellissier H, Santelli M. Tetrahedron. 2003;59:701. [Google Scholar]; (d) Bronner SM, Goetz AE, Garg NK. Synlett. 2011:2559. [Google Scholar]; (e) Garr AN, Luo D, Brown N, Cramer CJ, Buszek KR, VanderVelde D. Org. Lett. 2010;12:96. doi: 10.1021/ol902415s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Díaz M, Cobas A, Guitián E, Castedo L. Eur. J. Org. Chem. 2001:4543. [Google Scholar]
- 15.For the preparation of stabilized 2,3-pyridynes, see: den Hertog HJ, van der Plas HC. In: Advances in Heterocyclic Chemistry. Katritzky AR, editor. Vol. 4. New York, NY: Academic; 1965. p. 121e198. Reinecke MG. Tetrahedron. 1982;38:427. van der Plas HC, Roeterdink F. In: The Chemistry of Triple-Bonded Functional Groups Supplement C. Patai S, Rappoport Z, editors. Chichester, UK: John Wiley & Sons; 1983. p. 421e511. Hart H. In: The Chemistry of Triple-bonded Functional Groups Supplement C2. Patai S, editor. Vol. 2. John Wiley & Sons: Chichester, UK; 1994. pp. 1017–1134. Connon SJ, Hegarty AF. Tetrahedron Lett. 2001;42:735. Connon SJ, Hegarty AF. Eur. J Org. Chem. 2004:3477..
- 16.(a) Fleet GWJ, Fleming IJ. Chem. Soc. C. 1969:1758. [Google Scholar]; (b) Kurita J, Kakusawa N, Yasuike S, Tsuchiya T. Heterocycles. 1990;31:1937. [Google Scholar]
- 17.(a) Mallet M, Queguiner G, Pastour P. Comp. Rend. 1972;274:719. [Google Scholar]; (b) Cook JD, Wakefield BJ. J. Chem. Soc. C. 1969:1973. [Google Scholar]; (c) Martens RJ, den Hertog HJ. Tetrahedron Lett. 1962:643. [Google Scholar]; (d) Martens RJ, den Hertog HJ. Recl. Trav. Chim. Pays-Bas. 1964;83:621. [Google Scholar]
- 18.(a) Effenberger F, Daub W. Chem. Ber. 1991;124:2119. [Google Scholar]; (b) Walters MA, Shay JJ. Synth. Commun. 1997;27:3573. [Google Scholar]
- 19.Fleet GWJ, Fleming IJ. Chem. Soc. C. 1971:3948. [Google Scholar]
- 20.Cant AA, Bertrand GHV, Henderson JL, Roberts L, Greaney MF. Angew. Chem. Int. Ed. 2009;48:5199. doi: 10.1002/anie.200901410. [DOI] [PubMed] [Google Scholar]
- 21.(a) Radom L, Nobes R, Underwood D, Li W. Pure Appl. Chem. 1986;58:75. [Google Scholar]; (b) Liu R, Tate DR, Clark JA, Moody PR, Van Buren AS, Krauser JA. J. Phys. Chem. 1996;100:3430. [Google Scholar]; (c) Mariet N, Ibrahim-Ouali M, Parrain J, Santelli MJ. Mol. Struct. 2004;679:53. [Google Scholar]
- 22.Marion N, Ecarnot EC, Navarro O, Amoroso D, Bell A, Nolan SP. J. Org. Chem. 2006;71:3816. doi: 10.1021/jo060190h. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Lin Y, Lin Y, Liao FJ. Org. Chem. 2004;69:3517. doi: 10.1021/jo049902z. [DOI] [PubMed] [Google Scholar]
- 24.Li J, Jin C, Su W. Heterocycles. 2010;81:2555. [Google Scholar]