

Abstract
Nanoparticle encapsulation has been used as a means to manipulate the pharmacokinetic (PK) and safety profile of drugs in oncology. Using pegylated liposomal doxorubicin (PLD) vs. conventional doxorubicin as a model system, we developed and experimentally validated a multiscale computational model of liposomal drug delivery. We demonstrated that, for varying tumor transport properties, there is a regimen where liposomal and conventional doxorubicin deliver identical amounts of doxorubicin to tumor cell nuclei. In mice, typical tumor properties consistently favor improved delivery via liposomes relative to free drug. However, in humans, we predict that some tumors will have properties wherein liposomal delivery delivers the identical amount of drug to its target relative to dosing with free drug. The ability to identify tumor types and/or individual patient tumors with high degree of liposome deposition may be critical for optimizing the success of nanoparticle and liposomal anticancer therapeutics.
Doxorubicin is a chemotherapeutic that is a backbone of cancer therapy, but its clinical utility can be hindered due to dose-limiting cardiotoxicity.1,2 Liposomal encapsulation of doxorubicin, known as pegylated liposomal doxorubicin (PLD), has improved the cardiac safety profile.3 Preclinical studies have consistently shown PLD to outperform conventional doxorubicin at equivalent doses in mouse tumor models.4,5 However, improvement in clinical efficacy has been less clear across solid tumor types evaluated. For example, in breast cancer, there was no difference in outcome for patients treated with liposomal vs. conventional doxorubicin3 whereas in Kaposi sarcoma, PLD showed markedly increased response rates relative to treatment with conventional doxorubicin.6 This work aims to systematically examine the effect of liposomal encapsulation of doxorubicin on its delivery to solid tumors and the potential relationship between deposition and efficacy in both mice and humans.
Liposomal encapsulation alters the pharmacokinetic (PK) profile of doxorubicin, resulting in a dramatically longer half-life relative to free doxorubicin. PLD liposomes are ~100 nm in diameter and surface coated with polyethylene glycol to minimize detection and elimination by the reticulo-endothelial system.7 Liposomes accumulate in tissues with functionally porous vasculature such as liver and spleen and with leaky vasculature such as tumors. By contrast, the free drug freely distributes in the body. For the doxorubicin within PLD to reach its intracellular target and be cytoreductive, it must first be released from the liposomes.
There is evidence for PLD that variation in tumor delivery is a primary factor controlling performance in preclinical and clinical studies. A study with 111In-labeled PLD in human subjects with a variety of solid tumor types showed liposome deposition that varied from undetectable to 53% injected dose per kg of tumor.8 In patients with nonsmall cell lung cancer, 99Tc-labeled PLD showed variable tumor deposition that correlated with microvessel density (MVD).9
Emerging evidence supports the hypothesis that differences in tumor deposition of PLD can alter efficacy. Karathanasis et al. demonstrated that variation in liposomal delivery to a tumor could be directly linked to the level of tumor response to PLD.10 The extent of liposome deposition, quantified using iodixanol-loaded liposomes and mammography, was predictive of antitumor response to subsequent treatment with PLD in a rat xenograft model.
It is well documented that drug delivery by nanoparticles or even free drugs have limitations.11,12 Transport of macromolecules across the vasculature is a complex function of vessel perfusion, surface area, permeability, and tumor and drug characteristics.13,14 For nanoparticles, transport across the vasculature is largely driven via convection, and subsequent penetration into the tumors is limited by their slow diffusion in interstitial matrix.15 Earlier work has built compartmental models for scaling between mice and human's16 as well as models with mechanistic representations of drugs interacting with cell surface receptors and ligands.17 There has been limited work modeling liposomal therapeutics,18,19 and it has not incorporated mechanistic cell-level detail in terms of delivery of doxorubicin to its target.
We hypothesize that clinically observed differences in cytoreduction between conventional vs. liposomal doxorubicin may be driven by differences in tumor properties that alter the deposition of drug into tumor cells. We used the literature and experimental data to develop a robust computational model of liposomal drug delivery of doxorubicin to tumor cells. With this model, we gain insight into transport kinetics in the mouse tumor and enable prediction in humans.
Results
Computational model
We constructed a computational model of the kinetic steps in the transport of doxorubicin into tumor cells to quantify the role of liposome and tumor-specific parameters on liposomal drug delivery (Figure 1).
Figure 1.
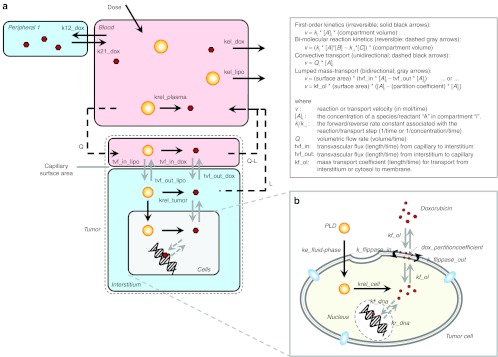
Computational model diagram. (a) The physiologically based drug delivery model is shown in cartoon form. The model consists of a one-compartment pharmacokinetic (PK) model for liposomes (yellow circles) superimposed on a pseudo two-compartment model for doxorubicin (red hexagons). The two models are connected by the release of doxorubicin from the liposomes. A physiologically based tumor compartment (outlined with dashed line) is connected to the PK model. Doxorubicin must be released from liposomes to be active and once it is released, it is assumed to behave identical to free doxorubicin. (b) A cartoon of the detailed model of doxorubicin transport into and out of cells is shown. Doxorubicin partitions into the cell membrane and then is reversibly transported from the outer to inner leaflet of the membrane via flippases, dissociates into the cytosol, and then reversibly binds to DNA in the nucleus. The cellular model for doxorubicin transport is independently applied to cell line data and is also embedded within the larger mechanism-based model in a. Complete parameter descriptions and their values are shown in Table 1. The kinetic rate laws used for each reaction and/or transport step are shown in the box in the upper right. The complete model, including all reaction rate laws and parameter values is available in Supplementary Data online as well as a parameter and reaction list in Supplementary Tables S1 and S2 online. PLD, pegylated liposomal doxorubicin.
Cellular doxorubicin transport
To understand the kinetics of uptake, 21 cancer cell lines were used to generate time courses following incubation with doxorubicin (Figure 2a). Cell lines were selected to provide a variation in cellular uptake, with particular emphasis on breast cancer, an indication where anthracyclines are heavily used. Our modified Eytan and Kuchel20 model was used to estimate the rate of doxorubicin association with the cell, kf_ol, and the rate of outward flippase activity, kflippase_out, for each cell line. In addition to being highly sensitive parameters at the cellular level, kflippase_out and kf_ol were chosen for estimation because it may reflect variation in transporter activity across cell lines and capture differences in total membrane area that cannot be easily measured. Shown in Figure 2b is a representative fit of the model to the cellular uptake data. The variation in estimated values for parameters kf_ol (coefficient of variation, CV = 86%) and kflippase_out (CV = 270%) is presented in Figure 2c,d, respectively.
Figure 2.

Total cellular doxorubicin uptake was measured in multiple cell lines (AdRr, OVCAR8, MCF7, HeLa, MKN-45, IGROV1, ZR75-1, MDA-MB-361, MDA-MB-453, 4T1-clone-12W7, OVCAR8-Her2, HCC1954, AdRr-Her2, JIMT-1, MCF7-c18, Calu-3, MKN-7, NCI-N87, SkBr3, SKOV3, and BT474-M3) following incubation with 3, 15, or 75 µg/ml of doxorubicin (squares, circles, and triangles, respectively) for up to 3 hours. (a) Time courses for 21 different cell lines following incubation with 15 µg/ml doxorubicin. The kinetic model for doxorubicin transport into and out of cells (see Figure 1b) was fit to the experimental data for each to estimate rates of doxorubicin-cell association (kf_ol) and rate of outward flippase/efflux activity (kflippase_out). (b) A representative model fit (solid lines) to data for the OVCAR8-Her2 cell line for 3, 15, and 75 µg/ml data (circles, squares, triangles, respectively). (c,d) Histograms of the fitted parameter values for kf_ol and kflippase_out, respectively.
PKs
The PK of intravenous doxorubicin in the mouse was compiled from the literature (Figure 3a) and normalized in terms of % ID/ml to enable comparison. The data show variability across studies, but did not exhibit any clear dose-dependence (data not shown). Shown in Figure 3a is the mean fit of the model to the entire data set, and Figure 3c shows the variability of parameter estimates from fitting the elimination rate, kel_dox to each data set individually.
Figure 3.

Model training with literature data. (a,b) Doxorubicin and pharmacokinetic (PK) and tumor deposition data in mice were gathered from the literature (Supplementary References online), normalized to percent injected dose per ml and is shown in panels a and b (circles). Doxorubicin doses ranged from 0.5 to 20 mg/kg. In all studies, doxorubicin was quantified via high-performance liquid chromatography (HPLC). Tumor models included HepG2, Li-7, BT-474, 4T1, and NCI-N87. Similarly, tumor deposition data for conventional doxorubicin and pegylated liposomal doxorubicin (PLD) were extracted from the literature, normalized to percent injected dose/g of tumor tissue is shown in d and e (circles). Liposome data were restricted to pegylated liposomes, ~100 nm in diameter and containing doxorubicin. Liposome doses ranged from 3 to 20 mg/kg (equivalent doxorubicin dose). Xenograft models included BT-474, NCI-N87, KB, A375, B16F10, HepG2, Li-7, 4T1, and M190-FR (data not shown). Multiple detection methods were used for tracking liposomes: encapsulated doxorubicin (measured by HPLC) or radiolabeled lipids (3H, 67Ga, 111In, or 125I). The kinetic model from Figure 1 was fit to each set of data in panels a,b, d, and e (solid lines), as described in Methods section. Individual parameter estimates for doxorubicin (red) and PLD (blue) elimination from the central compartment (kel_dox and kel_lipo, respectively) are shown as a histogram in c. Individual parameter estimates for conventional doxorubicin (dashed lines) and PLD (solid lines) transvascular flux (tvf_in_dox and tvf_in_lipo respectively) from capillary to interstitial space in the tumor are shown as a histogram in f. (g) Human plasma PK data were compiled from the literature and as normalized to percent injected dose/ml (circles). Doxorubicin PK parameters (k12_dox, k21_dox, kel_dox) were fit to the data with the mean fit to the data shown in red. (h) Representative PLD plasma PK data from Harrington et al.8 is shown with the corresponding model fit, estimating kel_lipo, shown in red. A single data set is shown for clarity, due to the varying need for one vs. two-compartment models to describe human liposome PK. (i) The variability in estimated elimination rates for doxorubicin and PLD (kel_dox and kel_lipo), is shown. (j) A representative fit of the human model, fitting tvf_out_dox, to conventional doxorubicin tumor deposition data for 10 mg/m2 doxorubicin is shown from a patient with Kaposi sarcoma from the study of Northfelt et al.21 (k) A representative fit of the human model (fitting tvf_in_lipo) to PLD tumor deposition data is shown for a patient with breast cancer from the study of Harrington et al.8 treated with 111In-labeled PLD. (j,k) Representative fits for clarity of presentation, the PLD tumor deposition data are also shown in Supplementary Figure S1b online. (l) A summary of the variability of doxorubicin and PLD deposition parameters (tvf_in_dox and tvf_in_lipo) for human patients is shown. tvf_in_dox values were estimated from dynamic contrast enhanced-magnetic resonance imaging data in human tumors, as described in Results section. tvf_in_lipo values were estimated from patients with various tumor types from the study of Harrington et al. and Northfelt et al.8,21
The literature data for the PK of liposomes in mouse is shown in Figure 3b. As with doxorubicin, there was no distinct dose- dependence across the different studies (data not shown). Liposome clearance in mouse is markedly slower than that of conventional doxorubicin and displays mono-exponential kinetics.
The mean model fit to the collection of data is shown in Figure 3b, and Figure 3c shows the variability of the elimination rate, kel_lipo, from fitting data sets individually. Liposomes are cleared from the general circulation ~100-fold slower than doxorubicin. There appears to be more variation in doxorubicin clearance than liposome clearance.
Tumor deposition
Data for the time course of deposition of doxorubicin in mouse xenograft tumors were compiled from the literature and are shown as percent injected dose/g (Figure 3d).
The doxorubicin PK, characterized by our model, provides an estimate of the input function driving the extent of tumor deposition. Furthermore, the cellular uptake model (Figures 1b and 2) provides an independently validated description of doxorubicin interaction with tumor cells. We then estimated the rates of doxorubicin transport from the tumor capillaries into and out of the interstitial space. The model was separately fit to each tumor deposition data set. The mean model fit to the data is shown in Figure 3d, and the variability of estimated values of tvf_in_dox (CV = 270%) in Figure 3f. tvf_out_dox values were of similar magnitude and had similar variability as tvf_in_dox (data not shown).
Tumor deposition for PLD in mouse xenograft tumors is shown in Figure 3e. There was no significant dose dependence in deposition across the ranges tested (data not shown); however, there was significant variability in deposition both within and across different xenograft models (Supplementary Figure S1a online).
Using the liposome PK model and the model for doxorubicin, we have a description of the driving force for liposome transport into and out of the tumors, taking into account the behavior of the doxorubicin once it has been released from the liposomes. The transvascular flux per surface area for liposome transport from the capillary into and out of the interstitial space of the tumor (tvf_in_lipo and tvf_out_lipo, respectively) were estimated for each data set. The mean model fit to the data is shown in Figure 3e and the variability in deposition rate (CV = 130%) in Figure 3f. The mean liposome transport rate into the tumor is ~100-fold slower than that of doxorubicin.
Human scale-up
Because our model was based on a mechanistic understanding of drug delivery, it was possible to scale the model to reflect human physiology. Parameters describing human PK for free doxorubicin and PLD were estimated from clinical data in a manner identical to that used for mice (Figure 3g,h and Table 1). The elimination of doxorubicin is slower in humans than mice (approximately fourfold), whereas the half-life of PLD in human patients is longer than that of mouse (2–3 days vs. 10 h, respectively). The variability in estimated values for kel_dox (CV = 81%) and kel_lipo (CV = 40%) is shown in (Figure 3i).
Table 1. Complete parameter descriptions and their values.
Two key studies provide tumor data in human patients dosed with either doxorubicin or PLD. The first is a study by Harrington et al.8 in which patients with a variety of tumor types were dosed with 111In-labeled liposomes, and then PK and tumor deposition were measured via SPECT imaging. The second is a study by Northfelt et al.21 in which PK and doxorubicin tumor accumulation were measured in patients with Kaposi sarcoma who were dosed with doxorubicin or PLD.
To estimate transport coefficients in human tumors, we took advantage of the fact that doxorubicin has a molecular weight similar to that of gadolinium-based contrast agents used in dynamic contrast-enhanced magnetic resonance imaging; and therefore it is hypothesized to have similar diffusive transport characteristics. We compiled “Ktrans” (a net tumor uptake constant) values from human studies22,23,24 and back-calculated tvf_in_dox based on our model's capillary surface area. tvf_in_dox was fixed at the mean value based on Ktrans and tvf_out_dox was estimated from the doxorubicin data in the study by Northfelt et al.21 A representative model fit is shown in Figure 3j, with the range tvf_in_dox (CV = 102%) as shown in Figure 3l.
For estimating liposome transport, it was necessary to assume that tvf_out_lipo was the same for humans and mice, and then tvf_in_lipo could be estimated for each patient8,21 (Table 1). A representative model fit is shown in Figure 3k. The variability of estimated tvf_in_lipo values (CV = 78%) is shown in Figure 3l. Estimation of liposome deposition parameters from breast cancer tumors indicates a lower degree of leakiness than that of more vascular tumors, such as Kaposi sarcoma.
The timescale for each transport step and simulated time courses of free and liposomal doxorubicin for both models are summarized in Supplementary Figure S2 online. To relate time courses of doxorubicin exposure to an overall response, we chose to use the area under the curve of DNA-bound doxorubicin. Doxorubicin may exert its effects through multiple mechanisms beyond its interaction with DNA and topo2a; however, the use of area under the curve was further supported by in vitro studies (Supplementary Figure S3 online).
Sensitivity analysis
We then performed a local sensitivity analysis on the model to gain insight into the relative importance of different kinetic steps regulating DNA-bound doxorubicin following liposomal delivery (Figure 4) for the mouse and human models.
Figure 4.

A local sensitivity analysis of the mouse (top panel) and human (bottom panel) model was performed by varying each parameter by a small amount and calculating the relative change in the area under the curve of DNA-bound doxorubicin. Positive-sensitivity values indicate that an increase in that parameter would result in an increase in doxorubicin exposure at the tumor DNA, whereas negative values predict a decrease in exposure.
The sensitivity analysis indicates a complex interplay between vascular and transport parameters for doxorubicin and PLD. The parameters can be roughly characterized into five categories: PK, tumor permeability, tumor vascularity, drug release, and cell parameters. The two most sensitive parameters are the elimination rate of liposomes and the permeability of the tumor to liposomes. Longer circulation times of liposomes can compensate for lower permeability and vice versa in terms of total tumor cell exposure. Increasing blood flow or the MVD (thereby increasing interfacial surface area) is also predicted to positively affect tumor cell exposure. The release rate of drug in the various compartments may also have a modest effect, although increased drug release will be countered by the faster clearance of free drug relative to liposomal drug. Cellular parameters such as the rate of fluid-phase uptake have a modest effect. As expected, increased drug efflux also had a potent effect on decreasing overall exposure. The sensitivity to transvascular flux of free doxorubicin differed between mice and humans. These results prompted further investigation into the role of the transvascular flux of doxorubicin and PLD (tvf_in_dox and tvf_in_lipo).
Conventional vs. liposomal doxorubicin
Using the mouse model, we simulated 3 mg/kg of either doxorubicin or PLD across a grid of tvf_in_dox and tvf_in_lipo values and calculated the area under the curve of DNA-bound doxorubicin. The relative doxorubicin delivery of PLD vs. doxorubicin was calculated (Figure 5a). The estimates of tvf_in_dox and tvf_in_lipo define a rectangle of physiologically meaningful parameter space. For the majority of the possible scenarios, liposomes outperform free drug in the mouse tumor. This is consistent with preclinical results showing dramatically improved efficacy in mice for liposomal vs. conventional delivery for equivalent doxorubicin doses.4,5
Figure 5.
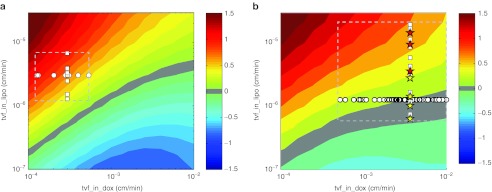
Relative delivery of doxorubicin for liposomal vs. conventional administration. (a) Mouse model simulations were performed for 3 mg/kg of conventional doxorubicin or pegylated liposomal doxorubicin (PLD) across a grid of tumor permeability values (tvf_in_dox and tvf_in_lipo). For each simulation, the area under the curve (AUC) of DNA bound in the tumor was determined over a 1 week period. The relative performance of conventional doxorubicin vs. PLD is plotted in the contours as log10(PLD AUC/conventional doxorubicin AUC). Positive values indicate increased doxorubicin delivery via PLD and negative values indicate increased exposure via conventional doxorubicin. Individual parameter estimates for tvf_in_dox and tvf_in_lipo determined from model training are plotted as circles and squares for reference. The plotted values outline a region of physiologically relevant parameter space (dashed rectangle). (b) The model was scaled to reflect human physiology and an analysis similar to that of the mouse was performed. Simulations of the human model were performed for 60 mg/m2 of conventional doxorubicin every 3 weeks or 50 mg/m2 of PLD every 4 weeks mimicking the clinical study of O'Brien, et al. 3 across a grid of tumor permeability values (tvf_in_dox and tvf_in_lipo). The AUC of tumor bound doxorubicin over 3 weeks was calculated for each simulation and is plotted in the contours as log10(PLD AUC/conventional doxorubicin AUC). Estimates of tvf_in_dox values back-calculated from dynamic contrast enhanced-magnetic resonance imaging Ktrans values are plotted as circles and the estimates of tvf_in_lipo from solid tumors from Harrington et al. and Northfelt et al.8,21 are plotted as squares. tvf_in_lipo values for breast cancer and Kaposi sarcoma tumors are plotted as yellow and red stars, respectively. The dashed rectangle outlines a region of physiologically relevant parameter space. Note that a value of 1 on the contour indicates a 10-fold increase in total exposure.
We next performed a similar analysis using the human model using 60 mg/m2 of doxorubicin every 3 weeks or 50 mg/m2 of PLD every 4 weeks, mimicking the clinical study of O'Brien et al.3 The relative area under the curves of DNA-bound doxorubicin upon liposomal vs. free drug treatment were calculated (Figure 5b). Our model predicts that for human tumors, there is a significant regimen where equal delivery of doxorubicin to its target is achieved by conventional and liposomal delivery.
Other factors influencing liposome performance
To explore the effect of vascular parameters on delivery, we simultaneously varied blood flow rate and MVD. At very low blood flow rates, there is an optimal MVD for delivery (Figure 6a). At very low MVD, the interfacial surface area is greatly reduced and the liposomes do not extravasate; whereas at very high MVD, doxorubicin clearance is enhanced by increased interfacial surface area.
Figure 6.
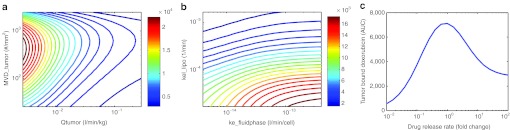
Investigation of the effect of select model parameters on doxorubicin delivery. (a) Simulations of the human model were performed following a 40 mg/m2 dose of PLD while systematically varying both the tumor blood flow rate (Qtumor) and microvessel density (MVD) (MVD_tumor) 0.1–10x from their nominal value. The area under the curve of DNA-bound doxorubicin ((µg doxorubicin)/(g tumor)-min) was calculated and is plotted as a contour plot. (b) Simulations were performed as in a, except now varying parameters for liposome clearance (kel_lipo) and cellular uptake (ke_fluidphase) 0.1–10x from their nominal value. (c) Simulations were performed as in a except the release rates of drug from liposome (krel_plasma, krel_tumor, krel_cell) was systematically varied 0.01–100x from their nominal value. The relative values for the different release rates were fixed and all three parameters varied in concert.
We also explored the use of the human model for evaluating parameters that might be liposome specific and possibly used to establish design criteria: liposome PK (kel_lipo), cellular uptake (ke_fluidphase), and liposome drug release (krel_plasma, krel_tumor, krel_cell). Figure 6b demonstrates that enhancing cellular uptake can be compromised if accompanied by an increase in liposome clearance. Figure 6c shows the effect of varied release rates, assuming that plasma, tumor, and cellular release rates move in concert. At low-release rates, liposomes are cleared from circulation before drug can be released, and at very high release rates, it behaves like free drug. This demonstrates that the release rate from PLD is optimal.
Discussion
In this work, aspects of PK, physiologically based modeling, tumor physiology, and cell-level mechanistic modeling were combined to construct a model for liposomal drug delivery.
We found variable uptake of doxorubicin at the cellular level in vitro, probably due to differences in uptake and/or efflux transporter activity as well as varying extents of sequestration in subcellular organelles such as lysosomes. Using the literature data, we demonstrate significant variability for PK and tumor deposition of conventional and liposomal doxorubicin.
Variability in tumor deposition of PLD was believed to reflect differences in the enhanced permeability and retention effect across different models and could be quantified via the transvascular flux parameters. As expected, liposome transport into and out of the tumors was significantly slower than that of free doxorubicin. Estimated values for tvf_in_dox and tvf_in_lipo were compared with values from other work for similarly sized agents.25 Of note, the CV for tvf_in_dox was much greater than that of tvf_in_lipo (270% vs. 130%), possibly indicative of a greater variability of how free drug interacts with biological tissue vs. the encapsulated form. Using a sensitivity analysis, we identified transvascular flux parameters as key in controlling the liposome-mediated delivery of doxorubicin to its target within the tumor cell. The transvascular flux parameters, tvf_in_dox and tvf_in_lipo, are empirically derived parameters that likely exhibit some covariance and are determined by a collection of tumor and drug-specific parameters. Liposome deposition rates are likely affected by the pore size and distribution in the vasculature as well as liposome size, surface composition, and charge. In addition, the long-circulating nature of liposome PK was also critical in enabling effective drug target exposure. By constructing a computational model of the competing kinetic processes, we have made an important first step at elucidating the complex relationship between tumor transport parameters, liposome parameters, and delivery.
In mouse models, liposomal doxorubicin consistently and dramatically outperforms conventional doxorubicin;4,5 however, in humans, this has not always been the case.3 Our analysis suggests that this may be due to differences in local tumor cell delivery that are controlled by PKs and tumor transport properties. Of note, we demonstrate that some human breast cancer tumors may fall in the regimen of equal delivery. This suggests that the lack of difference in efficacy between doxorubicin and PLD in human breast cancer is quantitatively consistent with the hypothesis that there was equal exposure of doxorubicin at the level of the tumor cell. The model suggests that there may be individual variation in tumor deposition that favors enhanced activity with PLD. Sensitivity analyses indicate that liposome PK and deposition are the most important parameters controlling drug delivery. This is consistent with preclinical work in which imaging liposome deposition predicted efficacy in a rat model10 and corresponds to the validity of our modeling approach. It also suggests PK and deposition as the most efficient means to maximize patient responses to liposomal therapeutics. Noninvasive imaging with radiolabeled liposomes may provide such an avenue.26,27,28 The role of nonspecific uptake of liposomes, as indicated by ke_fluidphase, also indicates the possible advantage of receptor-mediated targeting strategies as a means to increase local delivery to tumor cells as well.29
It is important to note some limitations inherent in the literature studies used. The use of mean data from multiple mice probably averages out some of the individual tumor heterogeneity. Animal and regional differences in the extraction efficiency of doxorubicin from the different tumors may also affect the ability to precisely quantify permeability. Most importantly, these data were used to obtain order-of-magnitude estimates of the rate of liposome deposition and identify the physiologically relevant range.
As with any large model, validity and parameter identifiability are concerns. We addressed model viability by constructing our model based on known physiology and proposed mechanism of action derived from a rich body of experimental literature. Parameter identifiability was addressed by training the model in a hierarchical fashion and minimizing the number of parameters to be estimated. The cellular transport model was trained first, followed by the larger systemic model for doxorubicin, and finally PLD.
An important model limitation is the assumption of a well-mixed tumor. This assumption enabled direct comparison with available experimental data, but PLD has transport limitations and does not distribute evenly throughout the tumor tissue. Relationships between vascularity, tumor size, liposome size, and penetration would be logical areas for future refinement. Relatedly, the presence of necrotic cores, as may occur in larger tumors, could skew some of the estimated values on a per cell basis. Incorporating a spatial component into the model would address this issue; however, significantly more complex experimental data would be required.
This work has also been extended to examine liposome deposition in cardiac tissue to study the effect of liposome encapsulation on doxorubicin delivery to cardiomyocyte nuclei,30 the key dose-limiting organ for doxorubicin. Together, these create a model for the therapeutic index of doxorubicin delivery. Comparison of parameter values for the tumor vs. the heart indicate that transvascular flux of liposomes in the tumor is 10-fold greater than that of the heart, whereas similar rates were obtained for doxorubicin transvascular flux. To generalize this work to other systems, it may be necessary to expand to include other organs such as the spleen, liver, or bone marrow that may be involved in dose-limiting toxicities. In these cases, we anticipate that the model structure can be re-used and only its parameterization altered.
Finally, we demonstrated the use of this model optimizing liposome design parameters, such as PK, release rate, and cellular uptake. We showed that there are optimal tumor and liposome properties for maximizing delivery that could be used for designing related liposomes. How the optimal kinetic parameters relate to physicochemical liposome properties, such as size or ζ potential, may require further investigation in order to translate into practice. One could also study the PK and transport behavior of small molecules to identify optimal candidates for liposomal encapsulation.
Methods
Doxorubicin cellular uptake. Cells were obtained from American Type Culture Collection and handled according to the manufacturer's instructions. Cells were treated with doxorubicin (Sigma-Aldrich, St. Louis, MO) in RPMI as indicated. Total cell-associated doxorubicin was quantified by high-performance liquid chromatography following washing, as previously described.30
Combined PK model. The PKs of doxorubicin were captured with a two-compartment model and the PK of PLD with a one-compartment model (Figure 1). To allow superposition of the two models, they were constrained to share a common central compartment. The central compartment volume was set to the blood volume to give it physiological meaning (consistent with the volume of distribution of liposomes). Following intravenous dosing, doxorubicin is transported from the central to the peripheral compartment and back again at rates k12_dox and k21_dox. Doxorubicin is eliminated from the central compartment in a first-order manner, at rate kel_dox. Similarly, PLD is dosed intravenously and eliminated at rate kel_lipo.
The two PK models are linked via the release of doxorubicin from the liposomes (stoichiometry = 20,000:1). Doxorubicin release was estimated from data tracking the relative distribution of radiolabeled liposomes and doxorubicin,31 exhibiting first-order kinetics and rates of 5.47e-5 min−1 and 1.77e-4 min−1 in the plasma and capillary (krel_plasma) and tumor (krel_tumor), respectively. These estimates may incorporate release mediated by liposome breakdown that is catalyzed by multiple mechanisms, including nonspecific uptake by macrophages.
Tumor deposition model. Doxorubicin and liposome transport in the tumor model is linked to the PK model via tumor blood flow, Qtumor (Figure 1). The tumor compartment is comprised of capillary, interstitial, and cellular space, characterized by a fractional vascular volume (frxn_vascularvolume) and the remaining cellular and interstitial space characterized by their volume ratio (cell2interstitium_ratio). The interfacial area for drug transport from capillary to interstitium was estimated from MVD and capillary radius in the tumor (MVD_tumor and capillaryradius_tumor, respectively), assuming cylindrical capillaries. The number of tumor cells was estimated based on spherical cells with radius, cellradius_tumor. The tumor is assumed to be “well mixed” and lymphatic drainage (L) negligible.
The rate of transport of doxorubicin from capillary to interstitium and vice versa was described as a transvascular flux per unit surface area (are tvf_in_dox and tvf_out_dox, respectively). The transvascular flux is a combination of diffusive and convective transport. For small molecules like doxorubicin, diffusive transport dominates whereas for larger species, such as liposomes, convective transport dominates. The use of two different rates for transport into and out of the interstitial space was required to account for multiple poorly characterized mechanisms of retention in the tissue. The transport of liposomes was modeled in an analogous manner with parameters, tvf_in_lipo and tvf_out_lipo.
Pegylation of liposomes greatly reduces their nonspecific uptake by multiple cell types, including macrophages.32 Koning et al.32 showed the rate of uptake of pegylated liposomes to be roughly linear with dose, consistent with a nonspecific fluid-phase uptake mechanism. The rate of uptake of liposomes (ke_fluidphase) was estimated to be 4e-4 l/min/cell and is implemented in the model.33,34 We conservatively estimated the half-life of intracellular doxorubicin release to be 12 h (release rate, krel_cell = 9.6e-4 1/min) (data not shown).
Doxorubicin cellular transport model. The kinetics of doxorubicin transport into and out of cells was described with a modified version of the model from Eytan and Kuchel20 (Figure 1b).
Doxorubicin in the external environment reversibly partitions into the outer leaflet of the cell membrane, characterized by a mass transfer coefficient (kf_ol), cell surface area (assuming spherical cells), and a partition coefficient (partitioncoefficient). Flippases within the cell membrane shuttle doxorubicin between the inner and outer leaflets. Because transport across the membrane is relatively fast, it is the ratio of inward to outward flippase activity (kflippase_in and kflippase_out) that plays a key role in the transport. The parameter kflippase_in was set using the value from Eytan and Kuchel20 and kflippase_out was estimated. P-glycoprotein activity was not explicitly represented in the model because of insufficient data to uniquely identify parameters. Doxorubicin transport between the inner leaflet of the membrane and the cytosol is characterized similarly by kf_ol.
Cytosolic doxorubicin reversibly binds DNA, characterized by dissociation rate constant, Kd_DNA and off-rate, kr_DNA, with roughly 0.18 binding sites per nucleotide.14,35,36,37,38,39 On the basis of the measurements of the total nuclear doxorubicin, one can estimate the “effective” number of nucleotides available for doxorubicin binding in the cell to be 4.6 × 107 1/cell.40 This is less than the total nucleotides within a mammalian cell, probably due to higher order DNA structure that obscures binding sites. The cellular model for doxorubicin transport was implemented separately for quantifying the uptake into cell lines and within the cellular compartment of the tumor model.
Human scale-up. The model was scaled to human by adjusting the PK parameters for free and liposomal doxorubicin derived from clinical data. Parameters specific to human physiology—blood volume, body weight, tumor flow rate, and drug dose—were adjusted. All other parameters were held fixed. The PK data from Harrington et al.8 was adequately described with a one-compartment model although a two-compartment model was required to capture data from Northfelt et al.21 consistent with other work.7
Implementation. The model was implemented in SimBiology (The Mathworks, Natick, MA) and parameter estimation was performed using the Optimization Toolbox (The Mathworks, Natick, MA).
Author Contributions
B.S.H. and T.J.W. wrote the manuscript; B.S.H., J.G.R., S.G.K., E.G., H.L., D.G., C.W.E., U.B.N., and T.J.W. designed the research; B.S.H., J.G.R., S.G.K., E.G., H.L., D.G., and C.W.E. performed the research; B.S.H., J.G.R., S.G.K., E.G., H.L., S.C.L., D.G., and C.W.E. analyzed the data; and B.S.H. contributed new reagents/analytical tools.
Conflict of Interest
All authors are employed by Merrimack Pharmaceuticals.
Study Highlights

Acknowledgments
The authors acknowledge the contributions of Isabelle Eckelhofer, Daryl Drummond, Dmitri Kirpotin, Victor Moyo, Matt Onsum, Sam Agresta, Chris Benz, and John Park. This work was funded by Merrimack Pharmaceuticals.
Supplementary Material
References
- Lefrak E.A., Pitha J., Rosenheim S., &, Gottlieb J.A. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32:302–314. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Von Hoff D.D.et al. Risk factors for doxorubicin-induced congestive heart failure Ann. Intern. Med 91710–717.1979 [DOI] [PubMed] [Google Scholar]
- O'Brien M.E.et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (Caelyx/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer Ann. Oncol 15440–449.2004 [DOI] [PubMed] [Google Scholar]
- Colbern G.T., Hiller A.J., Musterer R.S., Pegg E., Henderson I.C., &, Working P.K. Significant increase in antitumor potency of doxorubicin Hc1 by its encapsulation in pegylated liposomes. J. Liposome Res. 1999;9:523. [Google Scholar]
- Hong R.L.et al. Direct comparison of liposomal doxorubicin with or without polyethylene glycol coating in C-26 tumor-bearing mice: is surface coating with polyethylene glycol beneficial Clin. Cancer Res 53645–3652.1999 [PubMed] [Google Scholar]
- Northfelt D.W.et al. Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi's sarcoma: results of a randomized phase III clinical trial J. Clin. Oncol 162445–2451.1998 [DOI] [PubMed] [Google Scholar]
- Doxil package insert . www.doxil.com .
- Harrington K.J.et al. Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes Clin. Cancer Res 7243–254.2001 [PubMed] [Google Scholar]
- Koukourakis M.I.et al. Liposomal doxorubicin and conventionally fractionated radiotherapy in the treatment of locally advanced non-small-cell lung cancer and head and neck cancer J. Clin. Oncol 173512–3521.1999 [DOI] [PubMed] [Google Scholar]
- Karathanasis E.et al. Imaging nanoprobe for prediction of outcome of nanoparticle chemotherapy by using mammography Radiology 250398–406.2009 [DOI] [PubMed] [Google Scholar]
- Minchinton A.I., &, Tannock I.F. Drug penetration in solid tumours. Nat. Rev. Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- Jain R.K., &, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R.K., Tong R.T., &, Munn L.L. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: insights from a mathematical model. Cancer Res. 2007;67:2729–2735. doi: 10.1158/0008-5472.CAN-06-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunino F., Gambetta R., Di Marco A., &, Zaccara A. Interaction of daunomycin and its derivatives with DNA. Biochim. Biophys. Acta. 1972;277:489–498. doi: 10.1016/0005-2787(72)90092-5. [DOI] [PubMed] [Google Scholar]
- Jain R.K. Transport of molecules in the tumor interstitium: a review. Cancer Res. 1987;47:3039–3051. [PubMed] [Google Scholar]
- Baxter L.T., Zhu H., Mackensen D.G., Butler W.F., &, Jain R.K. Biodistribution of monoclonal antibodies: scale-up from mouse to human using a physiologically based pharmacokinetic model. Cancer Res. 1995;55:4611–4622. [PubMed] [Google Scholar]
- Mac Gabhann F., &, Popel A.S. Targeting neuropilin-1 to inhibit VEGF signaling in cancer: Comparison of therapeutic approaches. PLoS Comput. Biol. 2006;2:e180. doi: 10.1371/journal.pcbi.0020180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harashima H., Iida S., Urakami Y., Tsuchihashi M., &, Kiwada H. Optimization of antitumor effect of liposomally encapsulated doxorubicin based on simulations by pharmacokinetic/pharmacodynamic modeling. J. Control. Release. 1999;61:93–106. doi: 10.1016/s0168-3659(99)00110-8. [DOI] [PubMed] [Google Scholar]
- El-Kareh A.W., &, Secomb T.W. A mathematical model for comparison of bolus injection, continuous infusion, and liposomal delivery of doxorubicin to tumor cells. Neoplasia. 2000;2:325–338. doi: 10.1038/sj.neo.7900096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eytan G.D., &, Kuchel P.W. Mechanism of action of P-glycoprotein in relation to passive membrane permeation. Int. Rev. Cytol. 1999;190:175–250. doi: 10.1016/s0074-7696(08)62148-8. [DOI] [PubMed] [Google Scholar]
- Northfelt D.W.et al. Doxorubicin encapsulated in liposomes containing surface-bound polyethylene glycol: pharmacokinetics, tumor localization, and safety in patients with AIDS-related Kaposi's sarcoma J. Clin. Pharmacol 3655–63.1996 [DOI] [PubMed] [Google Scholar]
- Baek H.M., Yu H.J., Chen J.H., Nalcioglu O., &, Su M.Y. Quantitative correlation between (1)H MRS and dynamic contrast-enhanced MRI of human breast cancer. Magn. Reson. Imaging. 2008;26:523–531. doi: 10.1016/j.mri.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ah-See M.L.et al. Early changes in functional dynamic magnetic resonance imaging predict for pathologic response to neoadjuvant chemotherapy in primary breast cancer Clin. Cancer Res 146580–6589.2008 [DOI] [PubMed] [Google Scholar]
- Yankeelov T.E.et al. Integration of quantitative DCE-MRI and ADC mapping to monitor treatment response in human breast cancer: initial results Magn. Reson. Imaging 251–13.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan V.P.et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner Nat. Nanotechnol 7383–388.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen A.L.et al. 64Cu loaded liposomes as positron emission tomography imaging agents Biomaterials 322334–2341.2011 [DOI] [PubMed] [Google Scholar]
- Kubo A.et al. Indium-111-labelled liposomes: dosimetry and tumour detection in patients with cancer Eur. J. Nucl. Med 20107–113.1993 [DOI] [PubMed] [Google Scholar]
- Koukourakis M.I.et al. High intratumoural accumulation of stealth liposomal doxorubicin (Caelyx) in glioblastomas and in metastatic brain tumours Br. J. Cancer 831281–1286.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirpotin D.B.et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models Cancer Res 666732–6740.2006 [DOI] [PubMed] [Google Scholar]
- Reynolds J.G.et al. HER2-targeted liposomal doxorubicin (MM-302) displays enhanced anti-tumorigenic effects without associated cardiotoxicity Toxicol Appl Pharmacol 2621–10.2012 [DOI] [PubMed] [Google Scholar]
- Charrois G.J., &, Allen T.M. Drug release rate influences the pharmacokinetics, biodistribution, therapeutic activity, and toxicity of pegylated liposomal doxorubicin formulations in murine breast cancer. Biochim. Biophys. Acta. 2004;1663:167–177. doi: 10.1016/j.bbamem.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Koning G.A., Morselt H.W., Kamps J.A., &, Scherphof G.L. Uptake and intracellular processing of PEG-liposomes and PEG-immunoliposomes by kupffer cells in vitro 1 *. J. Liposome Res. 2001;11:195–209. doi: 10.1081/LPR-100108462. [DOI] [PubMed] [Google Scholar]
- Lee K.D., Nir S., &, Papahadjopoulos D. Quantitative analysis of liposome-cell interactions in vitro: rate constants of binding and endocytosis with suspension and adherent J774 cells and human monocytes. Biochemistry. 1993;32:889–899. doi: 10.1021/bi00054a021. [DOI] [PubMed] [Google Scholar]
- Lauffenburger D.A., &, Linderman J.J. Oxford University Press, New York; 1993. Receptors. [Google Scholar]
- Schneider Y.J., Baurain R., Zenebergh A., &, Trouet A. DNA-binding parameters of daunorubicin and doxorubicin in the conditions used for studying the interaction of anthracycline-DNA complexes with cells in vitro. Cancer Chemother. Pharmacol. 1979;2:7–10. doi: 10.1007/BF00253097. [DOI] [PubMed] [Google Scholar]
- Frezard F., &, Garnier-Suillerot A. Comparison of the binding of anthracycline derivatives to purified DNA and to cell nuclei. Biochim. Biophys. Acta. 1990;1036:121–127. doi: 10.1016/0304-4165(90)90023-p. [DOI] [PubMed] [Google Scholar]
- Tarasiuk J., Frézard F., Garnier-Suillerot A., &, Gattegno L. Anthracycline incorporation in human lymphocytes. Kinetics of uptake and nuclear concentration. Biochim. Biophys. Acta. 1989;1013:109–117. doi: 10.1016/0167-4889(89)90038-4. [DOI] [PubMed] [Google Scholar]
- Rizzo V., Sacchi N., &, Valentini L. Fluorescence stopped-flow kinetic analysis of anthracyclines–DNA interaction. Biochem. Pharmacol. 1988;37:1819–1820. doi: 10.1016/0006-2952(88)90462-5. [DOI] [PubMed] [Google Scholar]
- Zunino F., Di Marco A., Zaccara A., &, Gambetta R.A. The interaction of daunorubicin and doxorubicin with DNA and chromatin. Biochim. Biophys. Acta. 1980;607:206–214. doi: 10.1016/0005-2787(80)90073-8. [DOI] [PubMed] [Google Scholar]
- Gigli M., Doglia S.M., Millot J.M., Valentini L., &, Manfait M. Quantitative study of doxorubicin in living cell nuclei by microspectrofluorometry. Biochim. Biophys. Acta. 1988;950:13–20. doi: 10.1016/0167-4781(88)90068-1. [DOI] [PubMed] [Google Scholar]
- Spoelstra E.C., Westerhoff H.V., Dekker H., &, Lankelma J. Kinetics of daunorubicin transport by P-glycoprotein of intact cancer cells. Eur. J. Biochem. 1992;207:567–579. doi: 10.1111/j.1432-1033.1992.tb17083.x. [DOI] [PubMed] [Google Scholar]
- Rizzo V., Sacchi N., &, Menozzi M. Kinetic studies of anthracycline–DNA interaction by fluorescence stopped flow confirm a complex association mechanism. Biochemistry. 1989;28:274–282. doi: 10.1021/bi00427a038. [DOI] [PubMed] [Google Scholar]
- Gandecha B.M., Brown J.R., &, Crampton M.R. Dissociation kinetics of DNA-anthracycline and DNA–anthraquinone complexes determined by stopped-flow spectrophotometry. Biochem. Pharmacol. 1985;34:733–736. doi: 10.1016/0006-2952(85)90751-8. [DOI] [PubMed] [Google Scholar]
- Yang M., Chan H.L., Lam W., &, Fong W.F. Cytotoxicity and DNA binding characteristics of dextran-conjugated doxorubicins. Biochim. Biophys. Acta. 1998;1380:329–335. doi: 10.1016/s0304-4165(97)00161-x. [DOI] [PubMed] [Google Scholar]
- Peterson C., &, Trouet A. Transport and storage of daunorubicin and doxorubicin in cultured fibroblasts. Cancer Res. 1978;38:4645–4649. [PubMed] [Google Scholar]
- Laboratory, T.J. Animal resources body weight study of 1993JAX® NOTES, , http://jaxmice.jax.org/jaxnotes/archive/457a.html ( 1994
- Laboratory, T.J. MGI-Mouse Facts. . http://www.informatics.jax.org/mgihome/other/mouse_facts1.shtml .
- Baxter L.T., Zhu H., Mackensen D.G., &, Jain R.K. Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res. 1994;54:1517–1528. [PubMed] [Google Scholar]
- Charrois G.J., &, Allen T.M. Rate of biodistribution of STEALTH liposomes to tumor and skin: influence of liposome diameter and implications for toxicity and therapeutic activity. Biochim. Biophys. Acta. 2003;1609:102–108. doi: 10.1016/s0005-2736(02)00661-2. [DOI] [PubMed] [Google Scholar]
- Stefanini M.O., Wu F.T., Mac Gabhann F., &, Popel A.S. A compartment model of VEGF distribution in blood, healthy and diseased tissues. BMC Syst. Biol. 2008;2:77. doi: 10.1186/1752-0509-2-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.