

The role of modeling and simulation (M&S) in early drug development was discussed in a break out session of the European Medicines Agency/European Federation of Pharmaceutical Industries and Associations (EMA/EFPIA) M&S workshop held in December 2011 in London, UK. The discussions were focused on two themes: (i) M&S in nonclinical and early clinical drug development and (ii) M&S for first-in-human (FIH) dose selection. Illustrative case studies were presented and links are listed in Table 1. Common objectives and next steps were identified, discussed, and agreed.
M&S in Nonclinical and Early Clinical Drug Development
M&S in preclinical drug development focuses on the translation of preclinical data into quantitative predictions of the pharmacokinetics, pharmacology (proof of mechanism and concept) and safety in man. This enables selection of drug candidates with the best efficacy–safety profile for clinical development and optimization of first-in-human clinical trial designs.1,2 Both the existing field of mechanism-based pharmacokinetic–pharmacodynamic (PKPD) modeling and the emerging field of Systems Pharmacology offer important tools and concepts for the prediction of drug efficacy and safety in humans.3,4,5 The key elements in the development of mechanism-based models are: (i) understanding and mathematical modeling of the functioning of the underlying biological system and (ii) quantification and interspecies extrapolation of the systems- and drug-parameters that determine the time course of the drug effect.3 Typically, for extrapolation from animal to man, interspecies scaling of system-specific properties is required.3 On the other hand, Systems Pharmacology is based on the analysis of drug action and target activation in integrated networks. It integrates experimental and computational approaches to study and understand and predict biological processes in cells, tissues, and organisms.6
Quantitative Systems Pharmacology (QSP) is an emerging discipline which is based on the integration of (i) mechanism-based PKPD modeling concepts and (ii) systems pharmacology concepts.4,5,6 A unique feature of QSP is that it significantly improves our ability to quantitatively understand and characterize pathways of disease and to predict the efficacy compounds with (novel) mechanism of action.4 This multidisciplinary approach enables forward integration of information along the value chain and can therefore be used to rationalize decision making.4 Apart from therapeutic effects, it is envisaged that QSP can also assist in delineation of the adverse effects of new drugs such as QT prolongation, liver injury, hormone deregulation and other safety issues thereby resulting in improved assessment of the efficacy–safety profile of clinical candidates.4 Comparison and analysis of existing public domain data on standard of care or comparator compounds (both preclinical and clinical) is of crucial interest for the development of translational PKPD or QSP models to allows for predictions on novel drugs. Within the industry M&S in early clinical development is used for go–no go decisions, the selection of doses and dosing schedules and the selection of clinical trial designs (see Table 1 for examples)7,8.It is, however, a misconception that the risk in early drug discovery and development is solely for the Industry. Regulators have an important role in addressing the problem of high attrition rates by facilitating the use of novel approaches, such as M&S, to drug development and engaging in early dialogue with drug developers.
Table 1. Presentations break out session 1: M&S in early development.
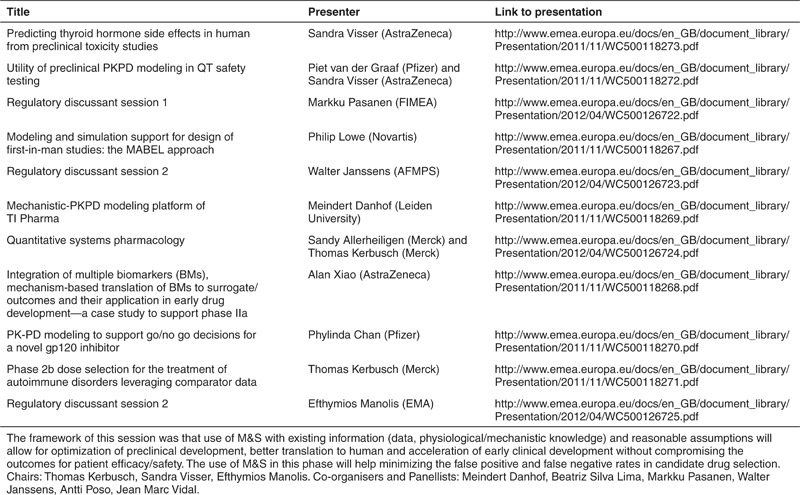
M&S For FIH Dose Selection
Traditionally, the maximum recommended starting dose for FIH trials is selected using the no observed adverse effect level (NOAEL) that is allometrically scaled to the human equivalent dose multiplied with a safety scaling factor. Limitations of this method are that it relies on allometric scaling, which may not be a valid approach in all cases, and arbitrary safety factors to ensure safety of the starting dose. Therefore, a PKPD-guided approach to determine the minimal anticipated biological effect level (MABEL) has been recommended to provide a more mechanistic rationale for starting dose selection by considering the human predicted PK, PD, and safety. This approach is also supported by the EMA guideline on FIH (see Supplementary Data online). It has been advocated that FIH trials should not necessary have safety and tolerability as primary outcome but rather should focus on multiple objectives related to understand the PK, PD, and safety of the novel entities.8
Availability of a quantitative prediction model during dose escalation that is updated with emerging clinical data from early dose panels may give increased confidence in predicting pharmacology and safety. This may yield situations where dose escalation steps may be larger than originally anticipated. However, this also requires adequate description of the assumptions made, and preferably a sensitivity analysis which allows for quantifying the risks of the assumptions. Regulators expressed, therefore, the need for quantitative comprehensive models for extrapolation and prediction to adhere to a process of model-development and qualification.9 An important issue in this regard is that model qualification by regulators is not only model validation per se but an evaluation of the modeling and simulation approach as an integral part of drug development. This would involve development and evaluation of a “comprehensive” model in the context of inclusion of multiple compounds, occasions and mechanisms to identify the underlying system parameters. A positive qualification opinion would result in increased confidence in the proposed methodology which would be reflected also in the regulatory requirements for drug development.
Common Objectives and Proposed Next Steps
Challenges in implementing M&S in drug development research and in regulatory documentation are not only the limited number of trained M&S scientists within Industry, that can utilize these methods to their full potential, but also the heterogeneity in the level of expertise at Regulatory Agencies to assess M&S documentation. To date, M&S contributions in regulatory submission are mainly limited to (population) PK. However, during the workshop there was a common view that extending M&S contributions towards the development of mechanistic models focusing on predicting human pharmacology and safety is important for both regulatory and industry decision-making. This advocates that academia, in collaboration with industry and regulators increase training activities and interactions within all aspects of M&S and quantitative systems pharmacology.4
Another challenge is information sharing of M&S efforts and advances. Despite increased numbers of scientific publications in the M&S field, much of the work is not shared internally and externally with a wider audience in a timely manner. Within industry, there are still hurdles between preclinical and clinical and between exploratory and confirmatory development with regard to the hand-over of modeling activities and/or the ownership of the data. In addition, many companies develop similar M&S approaches in parallel for predicting safety and efficacy profiles of new drug candidates. Therefore, all parties agreed that a multi-way scientific interaction would be beneficial for all stakeholders and predicted that M&S techniques will see an increased used in drug development and regulatory submissions. Early discussions around model-based development approaches will streamline the scientific advances on M&S with the regulatory requirements, and facilitate regulatory competence building on M&S and acceptance at FIH clinical trial applications, but also at later stages of development when regulatory interactions occur.9,10 There was some apprehension that there is a risk that regulators may put additional hurdles for industry if getting involved in exploratory development. Regulators anticipate this risk to be low based on the proposed framework for M&S review,10 and are committed at least not to exceed the standard regulatory requirements when evaluating approaches based on M&S.
The need for sharing data, models and best practices among industry, academia, and regulators has been highlighted previously.4 A common ground between regulators, academia, and industry was the wish to share precompetitive data and to build databases, enabling the development of comprehensive quantitative systems pharmacology models for a range of pharmacological mechanisms, for efficacy and in particular for safety. This would require an agreement on the prioritization of modeling platforms to be developed (e.g., pharmacological pathways, toxicity signals, DILI, QT, others) and an agreement on the scientific and regulatory aims (e.g., FIH dose justification and drug monitoring). A successful example of precompetitive sharing of data to develop systems models is the Industry–Academia consortium “TI Pharma Mechanism-based PKPD Modeling Platform” (see Table 1). The Drug Disease Model Resources initiative (http://www.ddmore.eu/) is another example of a precompetitive consortium focusing developing new standards via developing a common definition language for data, models, and workflows, along with ontology-based standard for storage and transfer of models and associated metadata.
Industry, regulators, and academia agreed that M&S is an essential tool in preclinical and early clinical drug development. The ultimate goal is to use M&S and in vitro experiments to predict accurately the activity and toxicity profile of a new compound with minimal animal experimentation. Applying M&S could facilitate the rational selection of new drug candidates and provide a quantitative prediction of anticipated human PK, pharmacology and safety in FIH trials. Integration of early clinical results with available preclinical, literature and competitor information using quantitative systems pharmacology concepts or semi-mechanistic models will improve the efficiency of early clinical development and will allow informed decisions for both industry and regulators.
A number of proposed next steps were identified and agreed. The first one was to develop best practices (or Points-to-Consider) on the use of model-based (PKPD) approaches (in parallel to NOAEL) for dose escalation in FIH trials including a focus on MABEL via collection of case studies and identification of common themes. Second, to increase capability of EMA to assess model-based approaches beyond population PK through focused training conducted in partnership with EFPIA and academia via training sessions based on shared presentations on case studies and best practices. A third agreed area was to provide guidance on preferred location and format of reporting of model-based approaches in regulatory documents (e.g., common technical document (CTD) modules). This could potentially be done through an EMA guidance document providing suggestions for typical locations in CTD modules for typical model-based approaches (FIH dose escalation steps, phase II design and dose-selection, covariate effects and impact on dosing, etc). The last proposal was to initiate focused effort on precompetitive sharing of systems data and toxicology signals via prioritization and identification of key gaps and opportunities for precompetitive sharing of data (e.g., DILI, QTc) based on need, feasibility, and availability.
Conflict of Interest
The authors declared no conflict of interest.
Acknowledgments
The authors thank the individuals who served with them on the programming committee and those who contributed to the panel discussions and presentations in break out session 1: Beatriz Silva Lima (University of Lisbon), Markku Pasanen (FIMEA), Walter Janssens (AFMPS), Antti Poso (University of Finland), Jean Marc Vidal (EMA), Philip Lowe (Novartis), Alan Xiao (AstraZeneca), Phylinda Chan (Pfizer), Sandy Allerheiligen (Merck), and Piet van der Graaf (Pfizer). In addition, the authors also thank the co-chairs Solange Rohou (AstraZeneca, EFPIA) and Rob Hemmings (MHRA/EMA) for their excellent leadership.
Supplementary Material
References
- Gabrielsson J., Dolgos H., Gillberg P.G., Bredberg U., Benthem B., &, Duker G. Early integration of pharmacokinetic and dynamic reasoning is essential for optimal development of lead compounds: strategic considerations. Drug Discov. Today. 2009;14:358–372. doi: 10.1016/j.drudis.2008.12.011. [DOI] [PubMed] [Google Scholar]
- Morgan P.et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival Drug Discov. Today 17419–424.2012 [DOI] [PubMed] [Google Scholar]
- Danhof M., de Jongh J., De Lange E.C., Della Pasqua O., Ploeger B.A., &, Voskuyl R.A. Mechanism-based pharmacokinetic-pharmacodynamic modeling: biophase distribution, receptor theory, and dynamical systems analysis. Annu. Rev. Pharmacol. Toxicol. 2007;47:357–400. doi: 10.1146/annurev.pharmtox.47.120505.105154. [DOI] [PubMed] [Google Scholar]
- Sorger P.K.et al. Quantitative and systems pharmacology in the post-genomic era: new approaches to discovering drugs and understanding therapeutic mechanisms NIHWhite Paper by the QSP Workshop Group (2011
- Van der Graaf P.H. CPT: Pharmacometrics and Systems Pharmacology. CPT Pharmacometrics Syst. Pharmacol. 2012;1:8. doi: 10.1038/psp.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J.M., Rogers M.E., &, Lyster P.M. Systems biology and pharmacology. Clin. Pharmacol. Ther. 2010;88:17–19. doi: 10.1038/clpt.2010.69. [DOI] [PubMed] [Google Scholar]
- Lowe P.J., Hijazi Y., Luttringer O., Yin H., Sarangapani R., &, Howard D. On the anticipation of the human dose in first-in-man trials from preclinical and prior clinical information in early drug development. Xenobiotica. 2007;37:1331–1354. doi: 10.1080/00498250701648008. [DOI] [PubMed] [Google Scholar]
- Cohen A. Pharmacokinetic and pharmacodynamic data to be derived from early-phase drug development: designing informative human pharmacology studies. Clin. Pharmacokinet. 2008;47:373–381. doi: 10.2165/00003088-200847060-00002. [DOI] [PubMed] [Google Scholar]
- Manolis E., Vamvakas S., &, Isaac M. New pathway for qualification of novel methodologies in the European Medicines Agency. Proteomics. Clin. Appl. 2011;5:248–255. doi: 10.1002/prca.201000130. [DOI] [PubMed] [Google Scholar]
- Manolis E.et al. How to use Modeling and Simulation in Clinical Development: Output from the EFPIA/EMA Modeling and Simulation Workshop CPT Pharmacometrics Syst. Pharmacol. 2e31; 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.