

Abstract
Heart failure is a complex disease that has great impact on morbidity and mortality in the general population. No recent therapies have proven to be effective; however, the discovery of new potential pathophysiological mechanisms involved in heart failure expression and progression could offer novel therapeutic strategies. A number of studies have shown that the immune system may be a central mediator in the development and progression of heart failure, and here we describe how the B-cell and B-cell-mediated pathways play specific roles in the heart failure state. Therapies aimed at B-cells, either blocking antibody production or inactivating B-cell function, may suggest potential new treatment strategies.
Keywords: B-cell, cardiomyopathy, immune system, auto-antibodies, humoral

Background
Heart failure is a complex disease with high morbidity and mortality.1 Currently available therapies, such as angiotensin converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), and beta blockers (BBs) are beneficial and have proven successful in treating heart failure. Yet these strategies that modify the neurohormonal system have reached a plateau, and end-stage heart failure requiring mechanical support or cardiac transplantation as therapy is still prevalent. Therefore, the need to investigate new pathogenic mechanisms involved in the course and progression of this devastating disease are urgent and must be explored in order to develop alternative therapeutic strategies.
Data to date have demonstrated that activation of the immune system has major effects on the heart failure state, whether by cytokine surge, antibody production, humoral responses, or other immune factors. The significance and possible implications that the immune response may have in disease progression and outcomes makes it an attractive area of research with potential for developing new therapeutic strategies. Specific subsets of the immune system that are of particular interest in heart failure are the B-cell and B-cell-mediated pathways.
B-Cell Maturation and Activation
The B-cell pathway has a major role in the development of adaptive immunity and cell-cell interactions. The B-cell maturation process originates in the bone marrow in early stage pro-B-cells; after a series of conformational changes, they are secreted into the circulation as immature B-cells. In this phase, they migrate to the secondary lymphoid tissue and/or spleen, where they become transitional B-cells and can be dormant for several years. Subsequently, B-cell activation and maturation take place once they encounter an antigenic stimulus and form a specific response. This specific response results in either isotype switching and antibody production, or presenting the foreign molecule to T cells via the major histocompatibility complex (MHC). As mature B-cells, they will express important molecules such as CD19, CD20, and CD22. Concomitantly, B-cells will interact with other components of the immune system (i.e., complement) to mount a specific immune reaction that will clear the system of the antigen. The B-cell interaction with several portions of the immune system represents an important natural defense mechanism. However, in the case of heart failure, it also can be a mediator of disease and disease severity when self proteins are recognized as foreign and an immune response is mounted. More importantly, existing data demonstrates that the manipulation of B-cell maturation, activation, and interaction processes can cause major effects in the cardiovascular system.
B-Cells and the Implications in Heart Failure
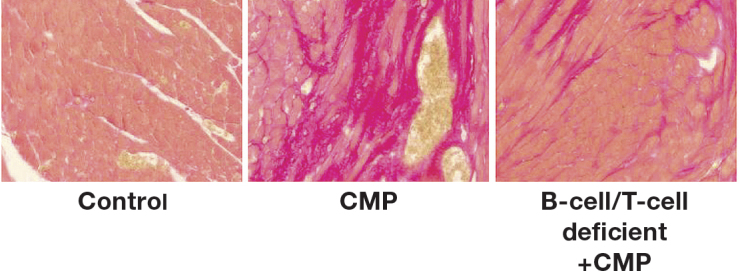
A link exists between the different arms of the immune system, specifically B-cells, and heart failure. As shown by Nishimura et al., mice lacking programmed cell death protein-1 (PD-1-/-), a key factor for B-cell differentiation, develop a severe form of spontaneous dilated cardiomyopathy (DCM) and express high levels of circulating IgG that binds specifically to cardiac myocytes.2 Furthermore, others have reported similar findings with the formation of antibodies against troponin I.3 However, this effect was not observed in PD-1-/- mice that also had defective T- and B-cells (RAG2-/-, Recombination Activation Gene). Similarly, unpublished data from our group demonstrates that SCID mice, which are T- and B-cell deficient due to a defective maturation process in V(D)J recombination, do not fully develop acute cardiomyopathy (CMP) in a nonischemic mouse model (Figure 1). This result is explained by the idea that absent or defective B-cells attenuate the expression of acute CMP. Similarly, Xiu et al. demonstrated a delay in disease progression with the depletion of B-cells in autoimmune illnesses, as in the case of autoimmune diabetes.4 These findings all support the idea that B-cells play a key role in immunity homeostasis, and alterations in B-cell expression can affect several systems, including the heart and its function.
Figure 1.
Absence of B- and T-cells prevents the development of fibrosis in a mouse model of acute cardiomyopathy. Staining is shown for fibrosis (red) in cardiac tissues from (left) control mice, (center) normal mice that underwent cardiomyopathy (CMP) induction, and (right) B-cell/T-cell deficient mice that underwent CMP induction.
B-cell interactions with the innate immune system, such as those mediated by the presence of toll-like receptors (TLRs), play a key role in CMP. This tight relationship is of great significance because data have shown that when there is over-activation of B-cell and TLR-mediated pathways, inflammation and pathogenesis develops — as demonstrated in atherosclerosis, viral myocarditis, and septic CMP5, 6 — as well as maladaptive ventricular remodeling after myocardial infarction in mice.7 Thirteen TLRs have been identified, and one of specific importance is TLR-4, which is up-regulated in heart failure.8, 9 Other significant interactions with the innate immune system occur via myeloid differentiation factor 88 (MyD88) and interleukin-1 receptor-associated kinase 4 (IRAK-4), which determines the formation of mature, antibody producing plasma cells.6 Maladaptive signaling mechanisms via this pathway also are linked to cardiac fibrosis during progression to heart failure.10 Another potential contributor from the innate immune system is interferon regulatory factor-3 (IRF-3), which is an important mediator of interferon gamma (IFN- ) synthesis. In an angiotensin-II-induced hypertension mouse model, decreased fibrosis was observed in mice lacking IRF-3 expression (IRF-3-/-) when compared to wild type.11
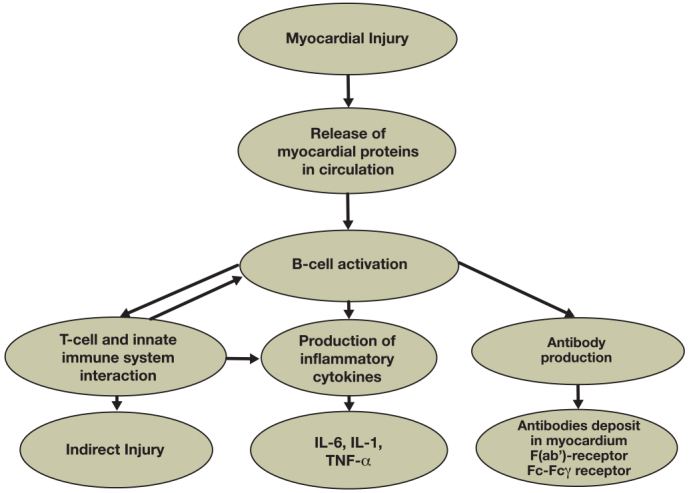
There are three roles that the B-cell performs during the immune response activation phase that also are relevant to the heart failure state. One role is the interaction with T-cells, specifically T helper (Th1) cells, to stimulate the production of circulating cytokines, which can affect contractility as well as adverse remodeling and have a great impact on prognosis and outcomes.12-15 Even though the interaction with T-cells remains important for the production of cytokines, B-cells also can act in a T-cell independent way. This observation is supported by recent findings from our group demonstrating that nude/athymic (nu-nu) mice, which lack T-cells, develop a severe, acute CMP similar to that observed in wild-type mice in a nonischemic CMP mouse model (unpublished data). The second role in heart failure, occurring when B-cells are activated, is to produce direct injury via apoptotic signaling pathways and complement-mediated cytotoxicity.16 This firmly correlates with the observation that activated B-cells are upregulated during episodes of acute decompensated heart failure and then return to basal levels once it resolves.14 The third role in heart failure occurs when B-cells are activated and can become memory B-cells. These memory B-cells form a secondary response when they encounter the same antigen; the resulting response is greater, much stronger, and can eventually cause more damage. In cases of multiple episodes of myocardial infarction where the immune system encounters myocardial proteins such as troponin, the memory B-cell response may lead to a persistent inflammatory state, enhancing myocardial cell death and injury (Figure 2).
Figure 2.
Flow chart showing the central role that B-cells may play in heart failure induction and progression.
During the B-cell activation process, CD19 phosphorylation triggers the activation of signaling pathways that have an effect on the progression of CMP. One important signaling regulator triggered by this process, phosphoinositide 3-kinase (PI3K), contributes to maladaptive remodeling in a transverse aortic constriction mouse model17 along with decreases in cardiac contractility and progression to hypertrophy.18 B-cells can also stimulate the secretion of proteins such as the enzyme matrix metalloproteinase-9 (MMP-9), a key factor in extracellular matrix (ECM) remodeling, which was shown to be upregulated in the failing heart, as well as contributing significantly to adverse remodeling in the myocardium.19 These key findings demonstrate that pathways leading to the activation of B-cells are important players in heart failure disease progression.20
Antibody Production and Heart Failure
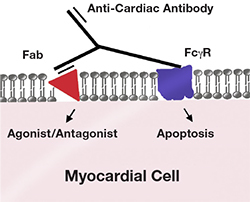
After activation, B-cells may transform into plasma cells and generate antibodies. In the CMP state, these antibodies can recognize cardiac-specific antigens and either deposit in the myocardium and bind through the F(ab’) region to specific proteins, or bind through the Fc fragment to the Fc gamma receptor (Fcγ) on cardiomyocytes.14, 21, 22 This binding can have a direct effect, causing cellular apoptosis, or an agonistic/antagonistic effect towards the specific protein/receptor (Figure 3).23 These antibodies can bind several different proteins/receptors such as the beta-1 adrenergic receptor.20 Beta-1 adrenergic receptor autoantibodies can induce apoptosis in isolated myocytes and exert a similar effect in vivo, causing myocardial dysfunction.24, 25 Antibodies against the Na+/K+-ATPase also have been demonstrated. Their presence seems to contribute to electrical instability in the heart, possibly making it prone to arrhythmias. This negative effect may be caused by binding of the antibody to the alpha subunit of the Na+/K+-ATPase.26 Finally, antibodies specifically targeting the Kv channel interacting protein (KChIP) also are associated with dilated CMP and can potentially cause cardiomyocyte death as shown in a rat model.27
Figure 3.
Effects of autoantibodies in the cardiomyocyte. Binding of the F (ab’) region to a specific receptor can cause an agonistic/antagonistic response, while binding of the Fc fragment to the Fcγ receptor can cause direct cell death.
Antibodies against intracellular proteins form after injury has exposed the circulation to these proteins that typically would not be recognized by the immune system.28 For example, antibodies against myosin and troponin I have been reported to be present in experimental models of autoimmune myocarditis,29, 30 in humans with dilated CMP, and in ischemic heart disease.31-33 Other important antibodies reported in the literature include antimuscarinic receptor 2, antimitochondrial M7, anti-actin, and anti-heat shock protein 60 (HSP-60) (Table 1).34-37 In heart failure, HSP-60 present in the mitochondrial matrix will undergo translocation to the cellular membrane, where antibodies will bind and cause increased rates of apoptosis. In mouse models of ischemic CMP, the expression of cytokines, IgM, and IgG were increased 3-fold in the post-ischemic state compared to controls.38 This association between myocardial cell death and immune system activation may indicate a major pathogenic role in the evolution of disease.
Table 1.
Presence of anti-cardiac antibodies in cardiomyopathy.
| Presence of Anti-Cardiac Antibodies in Cardiomyopathy | |||
| Antibody* | Comments | Etiology | Reference |
| Anti-B1-adrenergic receptor | cardiomyocyte apoptosis | DCM | Staudt et al.24 |
| agonist/antagonist effect, cell surface | DCM | Limas et al.20 | |
| Anti-M2-receptor | agonist/antagonist effect | DCM | Fu et al.34 |
| Anti-troponin I | Increased expression in sera | ICM/DCM | Doesch et al.28 |
| ------ | Myocarditis | Kaya et al.3 | |
| ------ | DCM | Okazaki et al.30 | |
| Anti-myosin | ------ | Myocarditis | Neu et al.29 |
| cardiomyocyte impaired contractility | DCM | Warraich et al.32 | |
| Anti-Na+/K+-ATPase | agonist/antagonist effect, electrical instability | DCM | Baba et al.26 |
| Anti-Kv channel | cardiomyocyte cell death | DCM/rat | Landsberger et al.27 |
| Anti-mitochondrial | M7, inner mitochondrial membrane | DCM | Klein et al.35 |
| Anti-actin | Increased expression in sera | DCM/ICM | Latif et al.36 |
| Anti-HSP-60 | abnormal localization to cell surface | DCM/ICM | Lin et al.37 |
*Other anti-cardiac reported in the literature include: anti-SR-Ca2+ATPase, anti-laminin, adenine-nucleotide transporter, anti-tropomyosin.
DCM: dilated cardiomyopathy; ICM: ischemic cardiomyopathy; HSP: heat shock protein.
The presence of autoantibodies and the subclass of immunoglobulin to which they belong are shown to be of great importance. IgG3 is the most abundant subclass present in sera of patients with dilated CMP. In these patients, the presence of IgG3 correlates with depressed cardiac function, poor exercise tolerance, and poor outcomes.39 This may be due to the presence of a hinge region in IgG3 that has increased affinity to the Fc receptor.40 Additionally, IgG3 has high reactivity against proteins as well as high specificity to activate the complement cascade.41 Recent findings demonstrate that IgG deposited in myocardial tissue from end-stage heart failure patients are predominantly subclass IgG3, corroborating the findings in sera. Moreover, the pattern of IgG3 deposition overlaps with complement (C3c) deposition (sarcolemmal pattern), supporting the theory of immune cascade activation and injury (unpublished data).
B-Cells, Autoimmune Diseases, and Heart Failure
B-cells are the core mediators of disease manifestation and progression for several autoimmune illnesses (e.g., rheumatoid arthritis, systemic lupus erythematosus, nephritis, autoimmune diabetes). There also appears to be a correlation between autoimmune illnesses and heart disease, since patients with rheumatoid arthritis have higher incidences of heart failure than the general population, and increased levels of c-reactive protein (CRP) are associated with heart failure progression.42, 43 In cases of nephritis, deposition of IgG is similar to that observed in end-stage heart failure, myocarditis, and dilated CMP.44 Therapeutic strategies used in these disease settings have been used to treat CMP with the notion that it will have similar effects on this disease alone.
Therapeutic Options and Future Directions
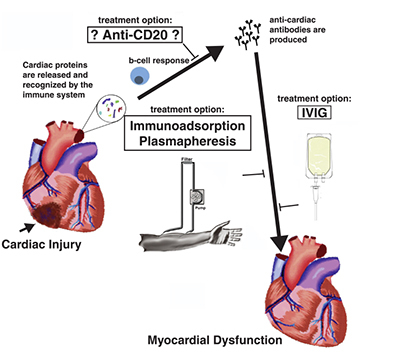
Several therapies that target the immune system have been tested. Therapies using TNF-α inhibitors, such as etanercept and infliximab, did not prove beneficial in the heart failure population and therefore are not currently used;45, 46 other therapies have shown promise but have not yet been fully tested. Immunomodulatory therapies such as Celecade (Vasogen Inc., Mississauga, ON, Canada) were successful in treating patients without ischemic etiology and patients within New York Heart Association (NYHA) class II.47 Currently, the removal of antibodies using intravenous immunoglobulin (IVIG) has been proposed to treat patients with dilated CMP and has had beneficial effects in several small clinical trials.48, 49 Immunoadsorption of IgG350, 51 and plasmapheresis also have been performed in small, single-center populations, with treatment resulting in beneficial effects and decreased antibody deposition in the myocardium.14 The current findings demonstrate a role for B-cells in the progression of heart failure and support potential therapeutic strategies that decrease B-cell function or eliminate B-cells from the periphery (Figure 4). Accordingly, potential new treatment strategies for heart failure would consist of antibody depletion or the utilization of molecules that block or inactivate B-cell function.
Figure 4.
Induction of myocardial dysfunction following cardiac injury and potential therapeutic targets.
Funding Statement
Funding/Support: The authors have no funding disclosures.
Footnotes
Conflict of Interest Disclosure: The authors have completed and submitted the Methodist DeBakey Cardiovascular Journal Conflict of Interest Statement and none were reported.
References
- 1.Jessup M, Brozena S. Heart failure. N Engl J Med. 2003 May 15;348(20):2007–18. doi: 10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- 2.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001 Jan 12;291(5502):319–22. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 3.Kaya Z, Katus HA, Rose NR. Cardiac troponins and autoimmunity: their role in the pathogenesis of myocarditis and of heart failure. Clin Immunol. 2012 Jan;134(1):80–8. doi: 10.1016/j.clim.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiu Y, Wong CP, Bouaziz JD, Hamaguchi Y, Wang Y, Pop SM, et al. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol. 2008 Mar 1;180(5):2863–75. doi: 10.4049/jimmunol.180.5.2863. [DOI] [PubMed] [Google Scholar]
- 5.Mann DL. The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res. 2011 Apr 29;108(9):1133–45. doi: 10.1161/CIRCRESAHA.110.226936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crampton SP, Voynova E, Bolland S. Innate pathways to B-cell activation and tolerance. Ann N Y Acad Sci. 2010 Jan;1183:58–68. doi: 10.1111/j.1749-6632.2009.05123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008 Feb 1;102(2):257–64. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- 8.Frantz S, Kobzik L, Kim YD, Fukazawa R, Medzhitov R, Lee RT, et al. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest. 1999 Aug;104(3):271–80. doi: 10.1172/JCI6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Birks EJ, Felkin LE, Banner NR, Khaghani A, Barton PJ, Yacoub MH. Increased toll-like receptor 4 in the myocardium of patients requiring left ventricular assist devices. J Heart Lung Transplant. 2004 Feb;23(2):228–35. doi: 10.1016/S1053-2498(03)00106-2. [DOI] [PubMed] [Google Scholar]
- 10.Blyszczuk P, Kania G, Dieterle T, Marty RR, Valaperti A, Berthonneche C, et al. Myeloid differentiation factor-88/interleukin-1 signaling controls cardiac fibrosis and heart failure progression in inflammatory dilated cardiomyopathy. Circ Res. 2009 Oct 23;105(9):912–20. doi: 10.1161/CIRCRESAHA.109.199802. [DOI] [PubMed] [Google Scholar]
- 11.Tsushima K, Osawa T, Yanai H, Nakajima A, Takaoka A, Manabe I, et al. IRF3 regulates cardiac fibrosis but not hypertrophy in mice during angiotensin II-induced hypertension. Faseb J. 2011 May;25(5):1531–43. doi: 10.1096/fj.10-174615. [DOI] [PubMed] [Google Scholar]
- 12.Cappuzzello C, Di Vito L, Melchionna R, Melillo G, Silvestri L, Cesareo E, et al. Increase of plasma IL-9 and decrease of plasma IL-5, IL-7, and IFN-gamma in patients with chronic heart failure. J Transl Med. 2011;9:28. doi: 10.1186/1479-5876-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aukrust P, Ueland T, Lien E, Bendtzen K, Muller F, Andreassen AK, et al. Cytokine network in congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1999 Feb 1;83(3):376–82. doi: 10.1016/s0002-9149(98)00872-8. [DOI] [PubMed] [Google Scholar]
- 14.Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD). J Am Coll Cardiol. 1996 Apr;27(5):1201–6. doi: 10.1016/0735-1097(95)00589-7. [DOI] [PubMed] [Google Scholar]
- 15.Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. 2001 Apr 24;103(16):2055–9. doi: 10.1161/01.cir.103.16.2055. [DOI] [PubMed] [Google Scholar]
- 16.Yasuda M, Takeuchi K, Hiruma M, Iida H, Tahara A, Itagane H, et al. The complement system in ischemic heart disease. Circulation. 1990 Jan;81(1):156–63. doi: 10.1161/01.cir.81.1.156. [DOI] [PubMed] [Google Scholar]
- 17.Damilano F, Franco I, Perrino C, Schaefer K, Azzolino O, Carnevale D, et al. Distinct effects of leukocyte and cardiac phosphoinositide 3-kinase gamma activity in pressure overload-induced cardiac failure. Circulation. 2011 Feb 1;123(4):391–9. doi: 10.1161/CIRCULATIONAHA.110.950543. [DOI] [PubMed] [Google Scholar]
- 18.Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, et al. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002 Sep 20;110(6):737–49. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 19.Reinhardt D, Sigusch HH, Hensse J, Tyagi SC, Korfer R, Figulla HR. Cardiac remodelling in end stage heart failure: upregulation of matrix metalloproteinase (MMP) irrespective of the underlying disease and evidence for a direct inhibitory effect of ACE inhibitors on MMP. Heart. 2002 Nov;88(5):525–30. doi: 10.1136/heart.88.5.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Limas CJ, Goldenberg IF, Limas C. Autoantibodies against beta-adrenoceptors in human idiopathic dilated cardiomyopathy. Circ Res. 1989 Jan;64(1):97–103. doi: 10.1161/01.res.64.1.97. [DOI] [PubMed] [Google Scholar]
- 21.She RC, Hammond EH. Utility of immunofluorescence and electron microscopy in endomyocardial biopsies from patients with unexplained heart failure. Cardiovasc Pathol. 2010 Jul-Aug;19(4):e99–105. doi: 10.1016/j.carpath.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 22.Staudt A, Eichler P, Trimpert C, Felix SB, Greinacher A. Fc(gamma) receptors IIa on cardiomyocytes and their potential functional relevance in dilated cardiomyopathy. J Am Coll Cardiol. 2007 Apr 24;49(16):1684–92. doi: 10.1016/j.jacc.2006.11.051. [DOI] [PubMed] [Google Scholar]
- 23.Kaya Z, Leib C, Katus HA. Autoantibodies in heart failure and cardiac dysfunction. Circ Res. 2012 Jan 6;110(1):145–58. doi: 10.1161/CIRCRESAHA.111.243360. [DOI] [PubMed] [Google Scholar]
- 24.Staudt Y, Mobini R, Fu M, Felix SB, Kuhn JP, Staudt A. Beta1-adrenoceptor antibodies induce apoptosis in adult isolated cardiomyocytes. Eur J Pharmacol. 2003 Apr 11;466(1-2):1–6. doi: 10.1016/s0014-2999(03)01431-6. [DOI] [PubMed] [Google Scholar]
- 25.Nussinovitch U, Shoenfeld Y. The Clinical Significance of Anti-Beta-1 Adrenergic Receptor Autoantibodies in Cardiac Disease. Clin Rev Allergy Immunol. 2010 Dec 29. doi: 10.1007/s12016-010-8228-9. [DOI] [PubMed] [Google Scholar]
- 26.Baba A, Yoshikawa T, Ogawa S. Autoantibodies produced against sarcolemmal Na-K-ATPase: possible upstream targets of arrhythmias and sudden death in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2002 Sep 18;40(6):1153–9. doi: 10.1016/s0735-1097(02)02075-2. [DOI] [PubMed] [Google Scholar]
- 27.Landsberger M, Staudt A, Choudhury S, Trimpert C, Herda LR, Klingel K, et al. Potential role of antibodies against cardiac Kv channel-interacting protein 2 in dilated cardiomyopathy. Am Heart J. 2008 Jul;156(1):92–9 e2. doi: 10.1016/j.ahj.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 28.Doesch AO, Mueller S, Nelles M, Konstandin M, Celik S, Frankenstein L, et al. Impact of troponin I-autoantibodies in chronic dilated and ischemic cardiomyopathy. Basic Res Cardiol. 2011 Jan;106(1):25–35. doi: 10.1007/s00395-010-0126-z. [DOI] [PubMed] [Google Scholar]
- 29.Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol. 1987 Dec 1;139(11):3630–6. [PubMed] [Google Scholar]
- 30.Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. 2003 Dec;9(12):1477–83. doi: 10.1038/nm955. [DOI] [PubMed] [Google Scholar]
- 31.Leuschner F, Li J, Goser S, Reinhardt L, Ottl R, Bride P, et al. Absence of auto-antibodies against cardiac troponin I predicts improvement of left ventricular function after acute myocardial infarction. Eur Heart J. 2008 Aug;29(16):1949–55. doi: 10.1093/eurheartj/ehn268. [DOI] [PubMed] [Google Scholar]
- 32.Warraich RS, Griffiths E, Falconar A, Pabbathi V, Bell C, Angelini G, et al. Human cardiac myosin autoantibodies impair myocyte contractility: a cause-and-effect relationship. Faseb J. 2006 Apr;20(6):651–60. doi: 10.1096/fj.04-3001com. [DOI] [PubMed] [Google Scholar]
- 33.Watanabe K, Sukumaran V, Veeraveedu PT, Thandavarayan RA, Gurusamy N, Ma M, et al. Regulation of inflammation and myocardial fibrosis in experimental autoimmune myocarditis. Inflamm Allergy Drug Targets. 2011 Jun;10(3):218–25. doi: 10.2174/187152811795564091. [DOI] [PubMed] [Google Scholar]
- 34.Fu LX, Magnusson Y, Bergh CH, Liljeqvist JA, Waagstein F, Hjalmarson A, et al. Localization of a functional autoimmune epitope on the muscarinic acetylcholine receptor-2 in patients with idiopathic dilated cardiomyopathy. J Clin Invest. 1993 May;91(5):1964–8. doi: 10.1172/JCI116416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klein R, Maisch B, Kochsiek K, Berg PA. Demonstration of organ specific antibodies against heart mitochondria (anti-M7) in sera from patients with some forms of heart diseases. Clin Exp Immunol. 1984 Nov;58(2):283–92. [PMC free article] [PubMed] [Google Scholar]
- 36.Latif N, Baker CS, Dunn MJ, Rose ML, Brady P, Yacoub MH. Frequency and specificity of antiheart antibodies in patients with dilated cardiomyopathy detected using SDS-PAGE and western blotting. J Am Coll Cardiol. 1993 Nov 1;22(5):1378–84. doi: 10.1016/0735-1097(93)90546-d. [DOI] [PubMed] [Google Scholar]
- 37.Lin L, Kim SC, Wang Y, Gupta S, Davis B, Simon SI, et al. HSP60 in heart failure: abnormal distribution and role in cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007 Oct;293(4):H2238–47. doi: 10.1152/ajpheart.00740.2007. [DOI] [PubMed] [Google Scholar]
- 38.Lachtermacher S, Esporcatte BL, Montalvao F, Costa PC, Rodrigues DC, Belem L, et al. Cardiac gene expression and systemic cytokine profile are complementary in a murine model of post-ischemic heart failure. Braz J Med Biol Res. 2010 Apr;43(4):377–89. doi: 10.1590/s0100-879x2010007500014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warraich RS, Dunn MJ, Yacoub MH. Subclass specificity of autoantibodies against myosin in patients with idiopathic dilated cardiomyopathy: pro-inflammatory antibodies in DCM patients. Biochem Biophys Res Commun. 1999 Jun 7;259(2):255–61. doi: 10.1006/bbrc.1999.0761. [DOI] [PubMed] [Google Scholar]
- 40.Canfield SM, Morrison SL. The binding affinity of human IgG for its high affinity Fc receptor is determined by multiple amino acids in the CH2 domain and is modulated by the hinge region. J Exp Med. 1991 Jun 1;173(6):1483–91. doi: 10.1084/jem.173.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tao MH, Smith RI, Morrison SL. Structural features of human immunoglobulin G that determine isotype-specific differences in complement activation. J Exp Med. 1993 Aug 1;178(2):661–7. doi: 10.1084/jem.178.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Myasoedova E, Crowson CS, Nicola PJ, Maradit-Kremers H, Davis JM, 3rd, Roger VL, et al. The influence of rheumatoid arthritis disease characteristics on heart failure. J Rheumatol. 2011 Aug;38(8):1601–6. doi: 10.3899/jrheum.100979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maradit-Kremers H, Nicola PJ, Crowson CS, Ballman KV, Jacobsen SJ, Roger VL, et al. Raised erythrocyte sedimentation rate signals heart failure in patients with rheumatoid arthritis. Ann Rheum Dis. 2007 Jan;66(1):76–80. doi: 10.1136/ard.2006.053710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Combes B, Shorey J, Barrera A, Stastny P, Eigenbrodt EH, Hull AR, et al. Glomerulonephritis with deposition of Australia antigen-antibody complexes in glomerular basement membrane. Lancet. 1971 Jul 31;2(7718):234–7. doi: 10.1016/s0140-6736(71)92572-4. [DOI] [PubMed] [Google Scholar]
- 45.Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. 2004 Apr 6;109(13):1594–602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 46.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003 Jul 1;107(25):3133–40. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 47.Torre-Amione G, Anker SD, Bourge RC, Colucci WS, Greenberg BH, Hildebrandt P, et al. Results of a non-specific immunomodulation therapy in chronic heart failure (ACCLAIM trial): a placebo-controlled randomised trial. Lancet. 2008 Jan 19;371(9608):228–36. doi: 10.1016/S0140-6736(08)60134-8. [DOI] [PubMed] [Google Scholar]
- 48.Gullestad L, Aass H, Andreassen AK, Ihlen H, Simonsen S, Kjekshus J, et al. [Immunomodulating treatment in advanced heart failure--effect of intravenous immunoglobulin]. Tidsskr Nor Laegeforen. 2001 Jun 20;121(16):1902–7. [PubMed] [Google Scholar]
- 49.Aukrust P, Yndestad A, Ueland T, Damas JK, Froland SS, Gullestad L. The role of intravenous immunoglobulin in the treatment of chronic heart failure. Int J Cardiol. 2006 Sep 10;112(1):40–5. doi: 10.1016/j.ijcard.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 50.Staudt A, Dorr M, Staudt Y, Bohm M, Probst M, Empen K, et al. Role of immunoglobulin G3 subclass in dilated cardiomyopathy: results from protein A immunoadsorption. Am Heart J. 2005 Oct;150(4):729–36. doi: 10.1016/j.ahj.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 51.Baba A, Akaishi M, Shimada M, Monkawa T, Wakabayashi Y, Takahashi M, et al. Complete elimination of cardiodepressant IgG3 autoantibodies by immunoadsorption in patients with severe heart failure. Circ J. 2010 Jul;74(7):1372–8. doi: 10.1253/circj.cj-09-0748. [DOI] [PubMed] [Google Scholar]