

Abstract
The human heart sustains an exceptional energy transfer rate, consuming more energy per gram weight than any other organ system. The healthy heart can rapidly adapt to changes in demand, while the failing heart cannot. Cardiac energy flux systems falter in the failing heart. The purpose of this review is to characterize the fundamental role of mitochondria in this energy transfer system and describe our local research on mitochondrial respiratory capacity in failing human hearts.
Keywords: heart failure, myocardial energetics, mitochondria, oxidative phosphorylation, oximetry, respiratory capacity

Introduction
The healthy human heart consumes more energy per gram of tissue than any other organ.1 To feed this demand, the myocardium cycles an amount of adenosine triphosphate (ATP) that is 15 to 20 times its own weight, or 6 to 30 kg each day. The heart provides this remarkable flux while maintaining a small but constant ~10 mM ATP tissue concentration. This provides little reserve, only enough for a few beats.2 With increased power demands such as during exercise or fever, the healthy heart maintains cellular concentration. This underscores the vital dynamic link between beat-by-beat utilization and high-energy ATP production. The failing heart, however, is energy depleted.3
Cellular energy transfer occurs primarily in mitochondria. These organelles extract chemical bond energy, transpose it to an electrochemical gradient, and then channel it into a high-energy chemical bond, the third phosphate on ATP. The rate of transfer matches demand, and oxidative capacity is important. At maximum work load, energy transfer operates at 80–90% capacity; at rest it operates at 15–20%.4 The process of oxidative phosphorylation (OXPHOS) has been described and reviewed in biochemistry and cardiology publications.5, 6 Electrons mediate this flux. Transfer proceeds through a series of redox reactions along the electron transfer chain (ETC) and ultimately results in the electron reduction of atomic oxygen to water (H2O). In pathological states, reactive oxygen species (ROS) such as superoxide (O2•-) and hydroxyl radicals (•OH) are generated at a high rate. Excess ROS production is one mechanism by which mitochondria contribute to heart failure.7, 8
Mechanistic roles of mitochondria in the pathogenesis of heart failure have been the subject of widespread investigation. Oxygen stress, accelerated apoptosis, and altered calcium handling have been areas of focus. These are processes that not only impair energy flux but also result in cell and organ damage. Interrelated mitochondrial functions contribute to heart failure.
Oxidative Stress, Apoptosis, and Calcium Spikes
Oxidative stress, mediated by short-lived toxic radicals, induces DNA strand damage, oxidation of protein groups, and peroxidation of membrane lipid. ROS overproduction has been investigated and quantified in heart failure tissues. It also has been studied as a potential target for treatment.7, 8 Oxidative radicals are produced systemically and locally in failing hearts, and yet antioxidant treatment has failed to reduce hospitalization and mortality.9 Mitochondrial apoptotic pathways also represent pathological mechanisms potentially amenable to treatment in failing hearts. Apoptosis has been characterized,10, 11 and intrinsic and extrinsic pathways resulting in myocardial death have been described. While it appears likely that these processes play a mechanistic role in heart failure pathology, targeted therapy to attenuate them has not been developed. Mitochondrial regulation of intracellular calcium is implicated in both of these mechanisms.11, 12 Mitochondrial-derived ROS results in cellular calcium sparks that are buffered by the mitochondria matrix; when in excess, calcium triggers apoptotic pathways.13 In relation to energy production, ROS signaling triggers mitochondrial matrix calcium sparks.14 The channels for this include the mitochondrial calcium uniporter and Na+/Ca2+ exchanger.15 Rises in mitochondrial matrix calcium then activate dehydrogenases and enzymes of phosphorylation, thereby regulating respiration capacity and OXPHOS.16 While this fine-tunes the energy production system on a beat-to-beat basis, ROS excess contributes to cell damage. Mitochondrial dysfunction may contribute through several mechanisms in the failing, and ultimately energy supply is affected.
Mitochondria Energy Transmission in Heart Failure
Fueling the Heart
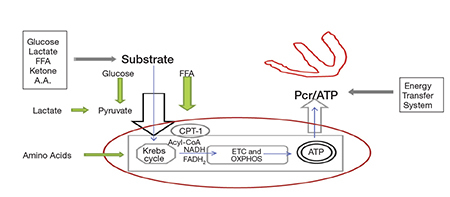
Fuel supply is important to this process. The initial mix includes combinations of fatty acids, ketone bodies, carbohydrates, lactate, and amino acids (Figure 1). Some mixtures are more efficient than others. At rest, the normal heart derives 60–70% of its energy need from fatty acid oxidation. With acute increased demand, as with exercise, fever, or arrhythmia, there is an adaptive shift to a more efficient energy combination with a higher percentage of carbohydrates. However, the relative quantities are determined not only by demand but also by the presence of comorbidities such as obesity, diabetes, and hypertension. In a patient with insulin resistance induced by obesity or chronic heart failure, fatty acids supply >90% of the energy at rest, and the percentage rises rather than shifts with physiological stress. In these circumstances, the heart cannot utilize more efficient fuel combinations.
Figure 1.
Schematic representation of the cardiac energy system. Three major components are denoted: substrate supply; Krebs cycle and oxidative phosphorylation (OXPHOS); the energy transfer system. The red oval demarks mitochondrial contained processes discussed in this review. Substrate combinations enter and are processed through the Krebs cycle. Electrons are delivered as NADH & FADH2 to the electron transport chain (ETC), along which oxidative phosphorylation (OXPHOS) is completed. Newly formed ATP is transferred to cytoplasmic usage sites by phosphocreatine energy transfer system. Measurement of the phosphocreatine/ATP (PCr/ATP) ratio quantifies a cell’s energy state.
Energy Transfer
Interrelated pathways transfer these organic substrates into the mitochondrial matrix and the Krebs cycle (Figure 1). These include glycolysis, pyruvate carrier proteins, carnitine palmitoyltransferase (CPT-1), beta-oxidation, and amino acid exchangers. Once in the matrix, the Krebs cycle oxidizes carbon bonds and releases electrons to two carriers, nicotinamide adenine dinucleotide (NAD+) and flavin adenine dinucleotide (FAD). The newly formed NADH and FADH2 introduce electrons onto the ETC to fuel OXPHOS. This electron energy transfer proceeds through a series of oxidation-reduction reactions along four complexes that comprise the ETC. During the process, hydrogen ions (H+) are pumped to generate a proton motive force across the inner mitochondrial membrane. This force then drives an embedded inner membrane generator, F1F0-ATPase, to phosphorylate ADP. Newly formed ATP is then shuttled to and stored in the cytoplasm by the creatine kinase energy shuttle. This is a dynamic interlinked process, and its failure to continuously supply adequate ATP results in energy depletion.3
Energy Measurement
The energy state in heart failure has been quantified. Phosphorus-31 NMR spectroscopy yields data that is used to calculate a phosphocreatine-to-ATP ratio (PCr/ATP). As such it provides an in vivo assessment of a tissue energy state.17, 18 This ratio is lower in failing hearts than in controls. Although both PCr and ATP compounds are reduced, consistent with impaired high energy phosphate metabolism, the decline in storage and transport molecule PCr is greater than that of ATP. This finding supports other evidence that heart failure is an energy depleted state, which has been documented as such for years.19 This could be the result of impairment in any of the processes — from substrate mix to ATP transfer (Figure 1). (See Lemieux and Hoppel for a review of mitochondrial function in the human heart.20)
Assessing Respiratory Capacity
There are very few direct recordings of the energy respiratory capacity of mitochondria from the ventricular wall of failing human hearts. Studies performed on heart tissue from animal models have provided important insights, but alterations in these models might not be directly applicable to mitochondrial changes that occur in chronic human heart failure. Figure 2 illustrates a tracing of isolates from the left ventricular wall of a nonfailing heart. As can be seen, oxygraph tracings from high-resolution respirometers such as the Oroboros Instrument provide graphic and quantitative data that can be used to assess respiratory capacity and control. Patient safety, tissue procurement procedures, and processing times present challenges when investigating human heart tissue. Respiratory measurements of human heart mitochondrial isolates such as those illustrated in Figures 2 and 3 from our laboratory necessitate procurement of 1–3 g of myocardial tissue, with isolation completed within 60 to 90 minutes. Similar measurements can be performed more easily using permeabilized fibers. The use of these saponin permeabilized fibers reduces sample size requirements from 1–3 g of myocardial tissue for isolations to 30–40 mg for fiber preparations. These findings must be interpreted within the limitations of this technique.21 As an alternative, left atrial appendage procurements are routinely accessible; however, like animal models, their energetic function might not correlate with left ventricle wall characteristics. Snap frozen tissue from transcutaneous biopsies cannot be analyzed for functionality, but they provide tissue for important assessments of gene expression, proteomics, metabolomics, and enzyme kinetics. These findings provide important complimentary information that relates to respiratory capacity. It remains that direct measurement of oxygen consumption from fresh tissue isolates provides specific information about mitochondrial functional integrity and energetic capacity.
Figure 2.
Representative oxygen tracing from isolated nonfailing human left ventricular wall mitochondria. Using high-resolution respirometry, the oxygen flux rates (red line) are normalized to milligram protein in the isolate. The first arrow denotes the rise in O2 flux (red line) following addition of fatty acid substrate palmitoylcarnitine (PALM CARN), then ADP and MALATE. This is the state 3 rate. The second arrow notes flux rise after uncoupling with FCCP. The ratio of the state 3 [approx. 92 pmol/(s*mg)] to uncoupled [approx. 99 pmol/(s*mg)] give a measure of flux control (approx. 0.92 here). The third arrow highlights the abrupt fall in O2 flux after adding the inhibitor antimycin A. This confirms a functionally intact electron transport chain.
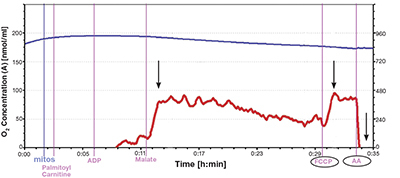
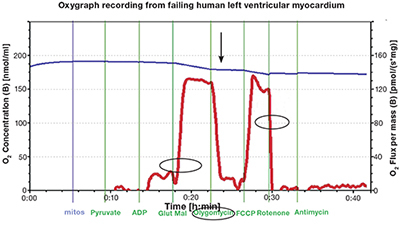
Figure 3.
Representative oxygen consumption tracing from isolated failing left ventricular human mitochondria. O2 flux recording (circled red line) following sequential addition of pyruvate, ADP, and glutamate-malate. This reflects the state 3 rate oxidative phosphorylation. Note the fall of O2 flux (arrow) after addition of oligomycin, an inhibitor of OXPHOS. The next steep rise reflects the addition of mitochondrial uncoupler FCCP (carbonylcyanide p-triflouromethoxyphenylhydrazone). Rotenone, an inhibitor of complex I, causes a rapid drop in the oxygen consumption rate (circled). This supports the conclusion that the mitochondria are coupled with intact ETC chains. Blue line: O2 concentration (nmol/ml); red line: rate of change in rate O2 consumption [pmol/(s*mg)].
Mitochondrial Respiratory Capacity in HF: TMH-based studies
Our research team at The Methodist Hospital in Houston undertook a series of translational investigations to build on available information from the above-mentioned techniques. Drawing on legacy experience from the hospital’s heart transplant program,22 we reestablished procedures for human heart mitochondrial isolation and used protocols developed during initial experiments with murine heart tissue to isolate mitochondria from fresh human ventricular tissue.
Our study involved patients with severe chronic heart failure resulting from both ischemic and nonischemic etiologies. By necessity, these samples are from patients with very advanced heart failure that requires either support or transplant. The findings from this tissue might not reflect earlier stages of heart failure. The large sample size requirement precluded percutaneous endomyocardial biopsy. Therefore, fresh ventricular tissue specimens were procured during left ventricular assist device surgery and orthotopic heart transplantation. We are also interrogating the metabolic effect of mechanical left ventricular support by investigating paired pre- and post-device heart samples. After surgical hand-off, 1–3 g of reddish scar-free tissue were dissected and promptly transported to nearby research laboratories. Preparation for mitochondrial isolation included mincing and homogenization in cold buffers that are designed to maintain mitochondrial function. These processed samples are centrifuged at differential speeds, rinsed, and respun to complete the isolation. Time is a limiting factor; functional mitochondria must be isolated and studied within 60 to 90 minutes. The time from surgical hand-off to mitochondrial isolation was reduced by 40 minutes on average, a distinct advantage of interdisciplinary teamwork combined with laboratory facilities minutes away from the operating suites.
A Population of Cardiac Mitochondria Retains Functional Capacity
Once isolation is completed, respiratory measurements are taken. The study protocols were initially developed using a Clark electrode in collaboration with Dr. William Widger at the University of Houston’s department of biochemistry. With these initial specimens, we directly measured membrane potential, phosphorous-to-oxygen ratios, and ATP production rates. Investigations were then complemented by high-resolution respirometry from Oroboros Instruments courtesy of the Methodist Hospital Research Institute’s diabetes research laboratory. Figures 2 and 3 show representative tracings of nonfailing and failing left ventricular tissue isolates.
Failing human heart isolates were first assessed without added substrate. They were found to consume low amounts of oxygen, indicating little oxygen wastage. As Figure 3 illustrates, the sequential addition of substrate pyruvate, ADP, then glutamate-malate rapidly stimulated OXPHOS and resulted in a steep rise in the oxygen consumption curve. The key findings in the figure are indicated by the arrow and circles. These include a rapid rise in oxygen consumption after pyruvate, ADP, and glutamate-malate (state 3 respiration) and the subsequent decline with OXPHOS inhibition, confirming intact phosphorylation apparatus. This is overcome by the addition of uncoupling agent carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone. Uncoupling unleashes respiratory control and allows electrons to flow at maximum ETC capacity. It requires an intact electron transport chain. As a final touch, complex I and complex III inhibitors (rotenone or antimycin A) were added. The findings illustrated in Figure 3 provide strong evidence for the survival of fully coupled and functioning mitochondria. This confirms electron transport-dependent oxygen consumption. Oxygen flux, as indicated by the last circle in Figure 3, rapidly drops. The elements of the OXPHOS are functioning in these isolates. This is in contrast to similar published respirometry analyses that used permeabilized left ventricular fibers.23, 21
The ratio of state 3 [~162 pmol/O2/(s*mg)] to uncoupled rate [~170 pmol/O2/(s*mg)] in the failing left ventricular isolate shown in Figure 3 is 0.95, higher than recently reported flux ratios of 0.42 and 0.41 from studies using permeabilized fibers from heart disease and heart failure samples, respectively.23 A lower coupling control ratio reflects limitation of OXPHOS by phosphorylation apparatus. Our finding supports integrity of this system, a finding we didn’t anticipate. The results also differ from the impaired respiratory function reported from animal models of heart failure and human left atrial appendage isolates.24
Limitations
We documented the presence of functioning, coupled, ATP-producing mitochondria in heart failure ventricular samples from more than 40 patients. Broader conclusions from this are complicated by the presence of cellular subpopulations and regional differences. Within each myocardiocyte, mitochondria reside in two strategic subpopulations that function differently under a variety of conditions.25 One subgroup, subsarcolemmal mitochondria, is positioned directly beneath membranes, and the other, interfibrillar mitochondria, is adjacent to myofibrils. In consideration of the evidence that these two subpopulations function differently in disease states, it is possible that we looked at surviving mitochondria isolated from only one of these populations.
Cardiac regional variation presents another variable. We compared mitochondrial function in isolates from different regions of the heart in a patient with hypertrophic cardiomyopathy. We published respiratory function assessments in tissue samples from the left ventricular (LV) apex, right ventricular (RV) free wall, and LV septum.26 Regional differences were evident. In Table 1, the respiratory control ratios (RCR), a measure of coupled mitochondrial function defined as state 3 divided by state 4, is lowest for all substrates in the hypertrophied LV septum. Regional differences also exist between the LV apex and RV free wall, a finding confirmed in a paired sample analysis from a patient with primary pulmonary hypertension that we presented at the 2012 Heart Failure Society of America Annual Scientific meeting.27 Conclusions about cardiac mitochondrial function drawn from investigations of atrial or left atrial tissue appendage samples as they apply to other cardiac regions must be interpreted in a narrow scope.24
Table 1.
Mitochondrial respiratory control ratios (RCR) taken from different regions of a heart from a patient with hypertrophic cardiomyopathy. RCR defined as state 3/state 4 represents a strong indicator of an intact coupled oxidative phosphorylation. Note lower numbers for all substrates from the septum isolates.
| Mitochondrial respiratory control ratio (RCR) by site for the different substrates* | ||||
| Site | RCR for Palmitoyl Carnitine | RCR for Pyruvate Malate | RCR for Glutamate Malate | RCR for Succinate |
| LV apex | 5.0616 | 3.0713 | 7.1832 | 1.5599 |
| RV free wall | 3.9818 | 3.4478 | 7.6043 | 1.5005 |
| LV Septum | 3.0000 | 2.1090 | 2.5667 | 1.0000 |
Printed with permission from Cordero-Reyes et al.26
Conclusions
Our findings support the conclusion that mitochondria capable of robust energy production exist within ventricular tissue of chronic failing human hearts. There is compelling evidence that in situ function is impaired, as reported with phosphorus-31 NMR spectroscopy. A question arises as to what is happening during isolation to suddenly restore their respiratory function. If reversibility exists, this would give hope for metabolic-targeted treatments that could be used to enhance current therapies.
Clinical Implications
Current treatments for heart failure are inadequate. Neurohormonal-based therapies have reduced mortality, but even today heart failure survival rates are no better than many cancers.28, 29 Hearts available for transplant are limited. To advance therapeutically, there is a compelling need to investigate underlying biological mechanisms, such as those involved in altered energy production. Attempts to treat energy deficiency have not been fully developed.30 Agents that alter myocardial metabolism by shifting to favor glucose metabolism include ranolazine, propionyl L-carnitine, dichloroacetate, etomoxir, trimetazidine, and GLP-1 agonists.31
If, as our findings reveal, there is a population of dormant in vivo mitochondria that can return to full respiratory function, then this potential for reversibility will kindle speculation about potential future therapeutics. These treatments might include devices, such as the early phase use of less invasive unloading devices as bridges to recovery. Gene and microRNA transfection, hormonal administrations (leptin, adiponection), and incretin-based therapies could be tested for therapeutic benefit. One line of investigation might target the liver and visceral adipose as a source of lipotoxic compounds that alter mitochondrial substrate supply. Various therapeutics or surgical procedures to alter nutrient supply might lead to innovative heart failure treatment. Exploring the mechanistic link between heart failure and impaired systemic and organ-specific insulin action might inspire opportunities for therapeutic innovation. Translational investigation is necessary to explore the underlying biological mechanisms.
Acknowledgments
Research team members include Methodist DeBakey Heart & Vascular Center (MDHVC) transplant surgeons Matthias Loebe, M.D., Ph.D. and Brian Bruckner, M.D., Guillermo Torre-Amione, M.D., Ph.D., chief of the MDHVC heart failure division, and Keith A. Youker, Ph.D. and Andrea M. Cordero-Reyes, M.D. from the MDHVC cardiac research laboratory; Willa A. Hsueh, M.D. and Anisha A Gupte, Ph.D. from The Methodist Hospital Research Institute Diabetes Research Center; and John H. Miller, Ph.D., William Widger, Ph.D. and graduate students Lucy E Vela and Heather Brasher from the University of Houston.
Funding Statement
Funding/Support: The author has received funding from the MDHVC Texans Grant; NIH R21 CA133153; TMH Foundation John Kotts Family, the Rodney Bradley Family, and Stedman-West foundation.
Footnotes
Conflict of Interest Disclosure: The author has completed and submitted the Methodist DeBakey Cardiovascular Journal Conflict of Interest Statement and none were reported.
References
- 1.Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal stressed, and failing human heart. Proc Natl Acad Sci U S A. 2005 Jan 18;102(3):808–13. doi: 10.1073/pnas.0408962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009 Feb 15;81(3):412–9. doi: 10.1093/cvr/cvn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007 Mar15;356(11):1140–51. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 4.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005 Jul;85(3):1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 5.Boyer PD, Chance B, Ernster L, Mitchell P, Racker E, Slater EC. Oxidative phosphorylation and photophosphorylation. Annu Rev Biochem. 1977;46:955–66. doi: 10.1146/annurev.bi.46.070177.004515. [DOI] [PubMed] [Google Scholar]
- 6.Stanley WC, Chandler MP. Energy metabolism in the normal and failing heart: potential for therapeutic interventions. Heart Fail Rev. 2002 Apr;7(2):115–30. doi: 10.1023/a:1015320423577. [DOI] [PubMed] [Google Scholar]
- 7.Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS. Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol. 2002 Apr;34(4):379–88. doi: 10.1006/jmcc.2002.1526. [DOI] [PubMed] [Google Scholar]
- 8.Giordano FJ. Oxygen oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005 Mar;115(3):500–8. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hare JM, Mangal B, Brown J, Fisher C, Jr, Freudenberger R, Colucci WS, et al. Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J Am Coll Cardiol. 2008 Jun 17;51(24):2301–9. doi: 10.1016/j.jacc.2008.01.068. [DOI] [PubMed] [Google Scholar]
- 10.Foo RS, Mani K, Kitsis RN. Death begets failure in the heart. J Clin Invest. 2005 Mar;115(3):565–71. doi: 10.1172/JCI24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2008 Jan 15;77(2):334–43. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 12.Lehnart SE, Maier LS, Hasenfuss G. Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail Rev. 2009 Dec;14(4):213–24. doi: 10.1007/s10741-009-9146-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol. 2003 Dec;5(12):1041–3. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
- 14.Yan Y, Liu J, Wei C, Li K, Xie W, Wang Y, et al. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc Res. 2008 Jan 15;77(2):432–41. doi: 10.1093/cvr/cvm047. [DOI] [PubMed] [Google Scholar]
- 15.Ruiz-Meana M, Fernandez-Sanz C, Garcia-Dorado D. The SR-mitochondria interaction: a new player in cardiac pathophysiology. Cardiovasc Res. 2010 Oct 1;88(1):30–9. doi: 10.1093/cvr/cvq225. [DOI] [PubMed] [Google Scholar]
- 16.Moreno-Sanchez R, Hogue BA, Hansford RG. Influence of NAD-linked dehydrogenase activity on flux through oxidative phosphorylation. Biochem J. 1990 Jun 1;268(2):421–8. doi: 10.1042/bj2680421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beer M, Seyfarth T, Sandstede J, Landschutz W, Lipke C, Kostler H, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002 Oct 2;40(7):1267–74. doi: 10.1016/s0735-1097(02)02160-5. [DOI] [PubMed] [Google Scholar]
- 18.Nakae I, Mitsunami K, Omura T, Yabe T, Tsutamoto T, Matsuo S, et al. Proton magnetic resonance spectroscopy can detect creatine depletion associated with the progression of heart failure in cardiomyopathy. J Am Coll Cardiol. 2003 Nov 5;42(9):1587–93. doi: 10.1016/j.jacc.2003.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Hardy CJ, Weiss RG, Bottomley PA, Gerstenblith G. Altered myocardial high-energy phosphate metabolites in patients with dilated cardiomyopathy. Am Heart J. 1991 Sep;122(3 Pt 1):795–801. doi: 10.1016/0002-8703(91)90527-o. [DOI] [PubMed] [Google Scholar]
- 20.Lemieux H, Hoppel CL. Mitochondria in the human heart. J Bioenerg Biomembr. 2009 Apr;41(2):99–106. doi: 10.1007/s10863-009-9211-0. [DOI] [PubMed] [Google Scholar]
- 21.Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol. 2000 Dec;32(12):2361–7. doi: 10.1006/jmcc.2000.1266. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz A, Sordahl LA, Entman ML, Allen JC, Reddy YS, Goldstein MA, et al. Abnormal biochemistry in myocardial failure. Am J Cardiol. 1973 Sep 20;32(4):407–22. doi: 10.1016/s0002-9149(73)80031-1. [DOI] [PubMed] [Google Scholar]
- 23.Lemieux H, Semsroth S, Antretter H, Hofer D, Gnaiger E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int J Biochem Cell Biol. 2011 Dec;43(12):1729–38. doi: 10.1016/j.biocel.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 24.Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009 Nov 10;54(20):1891–8. doi: 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoppel CL, Tandler B, Fujioka H, Riva A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int J Biochem Cell Biol. 2009 Oct;41(10):1949–56. doi: 10.1016/j.biocel.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cordero-Reyes AM, Youker K, Hamilton DJ, Torre-Amione G, Marian AJ, Nagueh SF. Molecular cellular, and functional characterization of myocardial regions in hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2012 May 1;5(3):419–22. doi: 10.1161/CIRCIMAGING.112.972802. [DOI] [PubMed] [Google Scholar]
- 27.Cordero-Reyes AM, Hamilton DJ, Gupte AA, Youker K, Loebe M, Estep J, et al. Mitochondrial function is preserved and enhanced in failing human myocardium: An analysis of paired samples obtained from failing right ventricle (RV) and normal left ventricle (LV) from patients with PPH. J Card Fail. 2012 Aug 1;18(8):S10. [Google Scholar]
- 28.Stewart S, Ekman I, Ekman T, Oden A, Rosengren A. Population impact of heart failure and the most common forms of cancer: a study of 1 162 309 hospital cases in Sweden (1988 to 2004). Circ Cardiovasc Qual Outcomes. 2010 Nov;3(6):573–80. doi: 10.1161/CIRCOUTCOMES.110.957571. [DOI] [PubMed] [Google Scholar]
- 29.Teng TH, Finn J, Hobbs M, Hung J. Heart failure: incidence case fatality, and hospitalization rates in Western Australia between 1990 and 2005. Circ Heart Fail. 2010 Mar;3(2):236–43. doi: 10.1161/CIRCHEARTFAILURE.109.879239. [DOI] [PubMed] [Google Scholar]
- 30.Gordon A, Hultman E, Kaijser L, Kristjansson S, Rolf CJ, Nyquist O, et al. Creatine supplementation in chronic heart failure increases skeletal muscle creatine phosphate and muscle performance. Cardiovasc Res. 1995 Sep;30(3):413–8. [PubMed] [Google Scholar]
- 31.Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure. Cardiovasc Res. 2011 May 1;90(2):202–9. doi: 10.1093/cvr/cvr038. [DOI] [PMC free article] [PubMed] [Google Scholar]